

Abstract
Capillary electrochromatography (CEC) of oligosaccharides in porous polyacrylamide monoliths has been explored. While it is possible to alter separation capacity for various compounds by copolymerization of suitable separation ligands in the polymerization backbone, “blank” acrylamide matrix is also capable of sufficient resolution of oligosaccharides in the hydrophilic interaction mode. The “blank” acrylamide network, formed with a more rigid crosslinker, provides maximum efficiency for separations (routinely up to 350,000 theoretical plates/m for fluorescently-labeled oligosaccharides). These columns yield a high spatial resolution of the branched glycan isomers and large column permeabilities. From the structural point of view, some voids are observable in the monoliths at the mesoporous range (mean pore radius ca. 35 nm, surface area of 74 m2/g), as measured by intrusion porosimetry in the dry state.
Keywords: polyacrylamide monoliths, analytical glycobiology, capillary electrochromatography, branched oligosaccharides
1. INTRODUCTION
Glycosylation is one of the most important post-translational modifications of biomolecules found in Nature. Characterization of a glycan residue covalently bound to a protein or lipid is very important, since such structures are crucial for antibody recognition and serve as a biological label (“postcode”) throughout enzymatic pathways in eukaryotic cells. The different analytical approaches utilized for the ultimate glycan profiling have been extensively reviewed [1], while a complete analytical solution for carbohydrate analysis has been summarized in a comprehensive book [2].
Capillary electrochromatography (CEC) is a separation approach combining some advantages of both liquid chromatography and capillary electrophoresis, offering the separation selectivity (through a stationary-phase design) and efficiency due to the electroosmotic flow [3,4]. Although “slurry-packed” columns still dominate in liquid chromatography, monolithic columns could supplement this conventional type of packings due to several advantages which include the ease of preparation, fritless design, and more efficient and permeable, non-particulate features [5,6]. After incorporation of suitable ionic species into the polymer backbone, the monoliths serve greatly as stationary phases for CEC, filling into narrow-bore capillaries more easily and reproducibly [7,8,9,10,11,12,13].
Continuous rods made from a crosslinked polyacrylamide were initially the first monolithic material rendered for chromatography purposes [14,15]. Svec and co-workers introduced the phase separation process for the pore formation in rigid acrylamide networks [16]. The utilization of organic solvents facilitates dissolution of more hydrophobic monomers in the polymerization feed, allowing a preparation of monoliths with suitable functionalities [17,18,19,20]. However, the main realm of acrylamide networks still rests in electroseparation methods, mainly in SDS-PAGE for slightly crosslinked gels [21], and electrochromatography for highly crosslinked gels [22,23].
Several laboratories have explored the favorable properties of acrylamide-based monomers, such as their hydrophilicity, crystallinity, versatility in chromatographic behavior, solubility in various solvents including water, propitious copolymerization parameters, simple chemical modification and a possibility to induce polymerization with all main types of initiators. The fundamental polymerization procedure and its kinetics in acrylamide porous systems have been extensively discussed [24,25,26]. Another approach to create a macroporous acrylamide monolith was studied by Hoegger and Freitag [27]. The addition of a lyotropic salt to aqueous polymerization feed manifested itself in the phase separation during a redox-initiated polymerization [28,29,30]. Maruska also developed a sturdier acrylamide monolithic material [31] and demonstrated its application in chiral ligand-exchange CEC separations [32,33,34,35]. A novel superporous acrylamide network has been recently launched by Plieva et al. who harnessed radical cryo-polymerization in order to reach up to 100 μm pore sizes in a polymer network [36,37]. More recently, the utility of DMF/alkanol solvents, such as a porogen mixture in acrylamide-based monoliths, was demonstrated for CEC [38].
The utility of acrylamide monoliths for the separation of different analytes was initially reported by Palm and Novotny [39]. Their monolith was prepared using buffered water/formamide/PEG mixtures to dissolve a mixture of acrylamide monomers with an alkyl acrylate as a separation ligand, employing the resulting monolith as a CEC reversed stationary phase with exceptional separation efficiencies. The efficiency of this monolith was later validated by adjustment of the amount of ionic monomer integrated into the polymer backbone for generating EOF [40]. The use of a hydrophilic separation ligand in acrylamide monolithic stationary phase allowed the structural characterization of neutral oligosaccharides in conjunction with mass spectrometry [41,42]. The hydrophilic interaction mode was found very advantageous for mass-spectrometric detection of carbohydrates [43] and permitted profiling of glycans at high efficiencies [44].
Although this type of monolith has been very successful in several CEC applications [41,42,43,44,45,46,47], no detailed structural investigation of this matrix has been reported thus far. The present study discusses improvements in the polymerization procedure toward achieving higher separation efficiencies. It also includes a detailed structural visualization of this porous matrix.
2. MATERIALS AND METHODS
2.1. Chemicals
Acrylamide (AAm) and N,N’-methylenebisacrylamide (Bis) were obtained from Bio-Rad (Hercules, CA), while N,N’-diacryloylpiperazine (PDA) was acquired from PolySciences (Warrington, PA). Sodium ethenesulfonate 30 % aq. (ESA), 2-cyanoethyl acrylate (CEA), formamide, N-methylformamide, and 2-aminobenzamide (2-AB) were purchased from Aldrich (Milwaukee, WI). Ammonium persulfate (APS), polyethylene glycol (PEG), N-[tris(hydroxymethyl)methyl]acrylamide (TRIS-AAm), all oligosaccharide standards and all other common chemicals were acquired from Sigma Co. (St. Louis, MO). Milli-Q water was from Millipore (Bedford, MA).
2.2. Column preparation
A fused silica tubing (100 μm i.d., 360 μm o.d.) from Polymicro Technologies (Phoenix, AZ) was pre-treated according to a previously published procedure [39] to modify the silica surfaces with pendant vinyl groups. The polymer mixture for “blank” acrylamide columns contained T 5%, C 60%, ESA 10% (according to Hjerten’s designation [48]) as described before [41,42]. Briefly, 40 mg of monomers AAm, 60 mg of crosslinker and 21-μl aliquot of ESA were dissolved in a porogenic mixture of 1875 μl N-methylformamide, 100 μl H2O and 60 mg PEG. In the case of columns with a separation ligand, an appropriate molar amount of AAm was replaced by TRIS-AAm. A 200-μl aliquot of the polymerization mixture was degassed with nitrogen for 15 min. Next, 3.2 μl of 10 % APS were added, while the polymerization mixture was filled into freshly pre-treated capillaries. The ends of capillaries were sealed with rubber septa and the polymerization was allowed to proceed in water bath at 55 °C for 8 h. The monolith was purged with running buffer for 2 h, while a detection window was created by pyrolyzing the monolith. Finally, the capillary was conditioned by applying a voltage (in small increments) prior to use.
2.3. Sample preparation
The oligosaccharide mixture (Glc4 up to Glc10) was tagged with 2-aminobenzamide (2-AB) according to the procedure described previously [39], then dialyzed through Spectra/Por (Mw cut-off 1000) to remove all salts. As a model glycoprotein, ribonuclease B was digested with N-glycosidase (PNGase F from Prozyme, Novato, CA) [49]. The deglycosylated proteins were removed from the reaction mixture by a C18 cartridge (Waters, Milford, MA). The released glycans were next derivatized with 2-AB as previously described [50], and dialyzed prior to analysis.
2.4. Instruments
All running buffers were filtered through SteriCup 0.22 μm filters (Millipore, Billerica, MA). The electrophoresis measurements were performed with a CE3D instrument from Agilent Technologies (Santa Clara, CA) controlled through ChemStation software. CEC-LIF experiments were performed with a homemade set-up equipped with a He/Cd Melles Griot laser (Series 56) [51]. MS characterization was conducted using MALDI TOF Perseptive Biosystems Voyager DE-RP mass spectrometer. Dihydroxybenzoic acid was utilized as a matrix at 10 mg/ml concentration. The electron microscopy measurements were performed on SEM Tescan TS5130 Vega (Brno, Czech Rep.) at magnification of 20 000× at 30 kV and on Aquasem Tescan (Brno, Czech Rep.) at magnification of 1 000× at 15 kV.
2.5. Porosity measurements
Porosimetry measurements were performed on mercury porosimeter (Pascal 440, ThermoFinnigan, Rodano, Italy) and nitrogen Brunauer-Emmett-Teller method for pore size estimation (BET) on Quantasorb Quantachrome (Boynton Beach, FL). Total porosity of the studied material was calculated using equation (1), as previously described [26]. The true polymer density (d) was set to 1.30 g/ml, formerly obtained by pycnometry in cyclohexane. Total cumulative pore volume (Vg) was obtained from mercury intrusion porosimetry (HgIP) measurements.
| (1) |
The equilibrium weight swelling ratio qw (2) includes the amount of solvent taken by both network chains and a pore system inside the structure, while the equilibrium volume swelling ratio qv (3) is caused by solvation of network only [52].
| (2) |
| (3) |
The symbols mswollen and mdry represent the mass of polymer in swollen and dry states, respectively. The diameters of the equilibrium swollen and dry gels are represented by D and Ddry, respectively. The porosity in the swollen state, Ps, can be calculated from equation (4), where d2 is the density of polymer in the swollen sample, d1 is the density of solvent and d0 is the bulk (mercury) density [53].
| (4) |
3. RESULTS AND DISCUSSION
3.1. Optimization of porogenic mixture
Various organic phases permitting effective polymerization included polar aprotic solvents, such as DMSO, N-methylformamide, or formamide. They serve, in combination with water, as a poorer solvent of porogenic mixture, while water is the best solvent for the acrylamide chains with the polymer/solvent interaction parameter (χ ≈ 0.48 [54]). The most promising porogenic pair consisted of N-methylformamide and water at a maximum possible content of an organic phase. Although the polymerization kinetics is fairly slow in comparison with traditional and a more hydrophilic formamide solvent [42], the resulting porous networks still hold neat opacity and, as we noticed, a significant increase in the column separation efficiency for tagged oligosaccharides (data not shown). The positive influence of a more hydrophobic co-solvent probably comes from enhanced phase separation process, lesser reswelling, and larger lateral aggregation effects. The lateral aggregation is a special case of porosity formation during gelation which is very unique for polyacrylamides. Their growing chains are forced to aggregate around the suitable macromolecules present in porogenic media via multiple interchain hydrogen bond formation [55]. At higher contents of a hydrophobic porogenic co-solvent, the lateral aggregation is even more accentuated. The maximum applicable content of organic phase is 95 % (v/v), and beyond this value, the opacity of gel disappears, indicating a greater role of water and hydrogen-bonding during polymerization. As far as the syntheses reproducibility concerned, the capillaries from the same batch yield a column-to-column percent relative standard deviation (%RSD) of 3.4 %, calculated for the migration times of the EOF marker (n = 3). The batch-to-batch reproducibilites, calculated similarly, are slightly higher, reaching a %RSD value of 6.4 %. These results are fully comparable to what has been previously reported [39]. The stability of the prepared columns is discussed in section 3.2.
3.2. Chromatographic features of acrylamide monoliths
The initial recipe for the creation of hydrophilic stationary phase (“cyano column”) involved a tetrapolymerization of four monomeric components - acrylamide and N,N’-methylenebisacrylamide (together shaping macroporous polymer backbone) with suitable ligands, such as 2-cyanoethyl acrylate (hydrophilic separation factor) and ionic ethenesulfonic acid (needed for EOF generation) [41,42]. The structures of different monomers utilized in this study are depicted in Figure 1. Generally, the total concentration of monomers and the ratio of crosslinkers are mainly responsible for the structural features and separation efficiency of continuous rods. On the other hand, the ratio between a monomeric ligand and monofunctional acrylamide is responsible for the capacity factors of eluting analytes.
Figure 1.
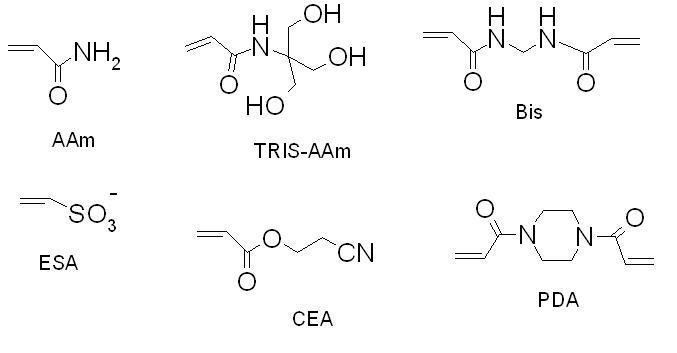
Structures of the different monomers utilized. AAm - acrylamide, ESA - ethenesulfonic acid for the EOF generation; crosslinkers: Bis - N,N’-methylenebisacrylamide, PDA - N,N’-diacryloylpiperazine; ligands: CEA - 2-cyanoethyl acrylate, TRIS-AAm - N-[tris(hydroxymethyl)methyl]acrylamide
Several types of hydrophilic ligands have been used in hydrophilic-interaction liquid chromatography, with largely those bearing aminoalkyl, cyano or diol functional groups [2]. However, in order to increase the separation capacity, we switched to more hydrophilic acrylamide derivatives. The outward advantages over the acrylate derivatives lie in their better copolymerization characteristics with acrylamide backbone, their crystallinity (easy purification and no inhibitor content) and their nitrogen link which should exert better lateral aggregation effects. We used N-[tris(hydroxymethyl)methyl]acrylamide (“TRIS-AAm” in Figure 1) because it is known to be the most hydrophilic acrylamide derivative available [56]. Ultimately, TRIS-AAm allowed us to exchange acrylamide to more than 50 % while retaining sufficient column permeability with a minor effect on gel opacity. The effect of organic solvent and crosslinkers on the efficiency of separation is illustrated in Figure 2. Optimum separation efficiency was attained using a PDA crosslinker prepared in 95% N-methylformamide (353,000 theoretical plates/m, Figure 2A). The separation efficiency was slightly decreased as a result of using Bis crosslinker (Figure 2B), while a substantial decrease in efficiency was observed when N-methylformamide was replaced with formamide (Figure 2C). During the HI-LIC experiment, some dehydration of acrylamide chains may apparently take place. Therefore, it is advisable not to use more than 95 % ACN in the mobile phase for a longer time. Under these conditions, we observed the loss of efficiency around 10-15 % after just 15 runs, in contrast to the reversed-phase experiments, where majority of aqueous phase is needed [39]. In order to enhance the ruggedness of acrylamide rod, we tested another crosslinker, N,N’-diacryloylpiperazine (PDA). This compound has displayed better mechanical and chemical properties due to the more rigid piperazine ring, while still maintaining good copolymerization parameters with acrylamide [57]. It is observed that this crosslinker offers notably better endurance (Figure 3). Moreover, efficiencies as high as 350,000 TP/m were sustained for PNP-o ligosaccharides under standard hydrophilic interaction conditions at 60 % ACN (Figure 2). Accordingly, PDA crosslinker and N-methylformamide were utilized for the preparation of additional monolithic columns.
Figure 2.
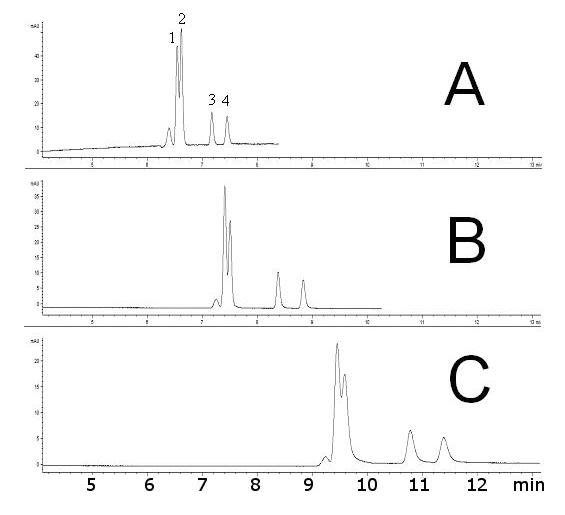
Effect of polymerization crosslinker and organic porogenic solvent on separations of labeled linear oligosaccharides - 4-nitrophenyl (1→4)- α-D-glucopyranan oligomers. (A) crosslinker PDA (column: T 5%, C 66%), solvent: 95% N-methylformamide, resulting efficiency for PNP-Glc5 ∼ 353 000 TP/m (B) crosslinker Bis (column: T 5%, C 60%), solvent: 95% N-methylformamide, resulting efficiency for PNP-Glc5 ∼ 308 000 TP/m (C) crosslinker Bis (column: T 5%, C 60%), solvent: 50% formamide, resulting efficiency for PNP-Glc5 ∼ 114 000 TP/m. Sample: in elution order = 5 % acetone, (1) PNP-Glc1, (2) PNP-Glc2, (3) PNP-Glc5, (4) PNP-Glc6, each oligosaccharide 1 mg/ml, inj. 4 kV/4 s, Agilent CE3D, UV 280 nm, ∼ 600 V/cm, 2.8 μA; Mobile phase: ACN / 240 mM NH +4 formate/H2O (60:1:39); Blank acryl amide columns (ltot = 35.5 cm, ldet = 27.0 cm)
Figure 3.
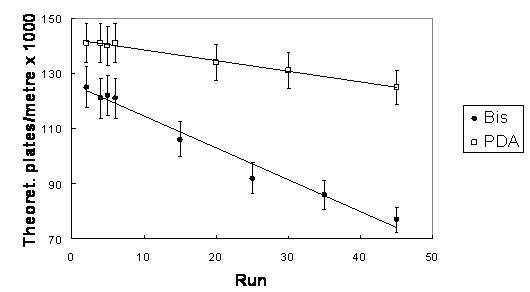
Longevity of columns in 90 % ACN for various crosslinkers. The column-to-column reproducibility is reflected in the listed uncertainty values (n=3). Sample: acetone 10 %, inj. 1 kV/10 s; Mobile phase: ACN/ 240 mM NH+4 formate/H2O (90:1:9), UV 280nm; Blank AAm/Bis (T 5%, C 60%) or AAm/PDA (T 5%, C 66%) columns (ldet = 27.0 cm, ltot = 35.5 cm)
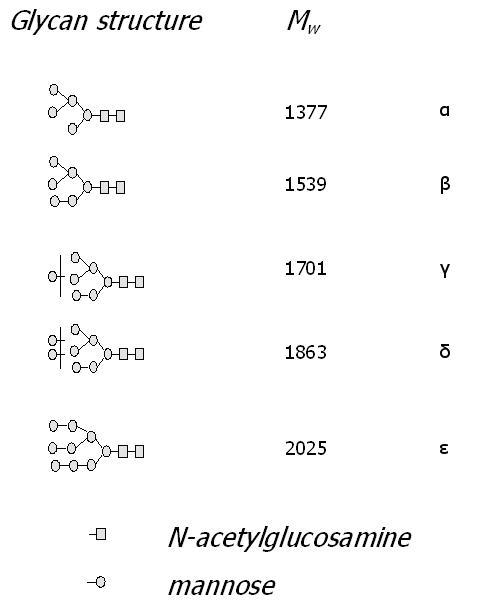
The separation performance of the acrylamide monolith was tested using the glycan residues enzymatically cleaved from ribonuclease B (RiB), a model glycoprotein. Each bovine pancreatic RiB molecule (Mw ≈ 15.5 kDa) contains only one glycosylation site (at asparagine 34) to which one of several oligosaccharide structures could be linked [58]. These glycan structures include exclusively mannose-type glycans diverging in a linkage type and a number of sugar moieties (Figure 4A).
Figure 4.
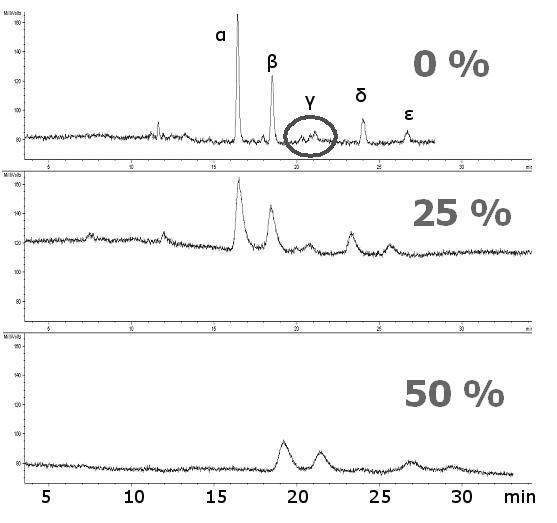
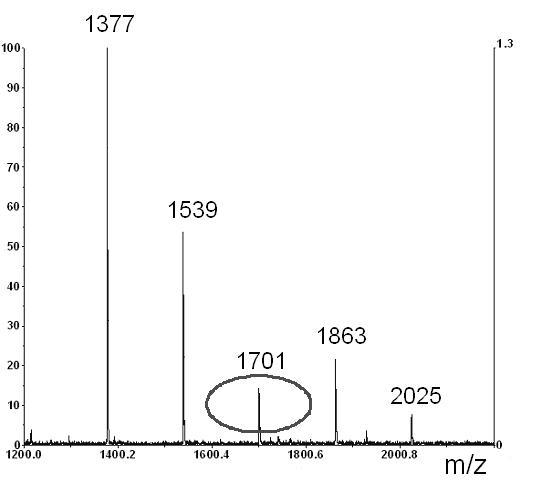
Effect of the amount of copolymerization ligand (x %) in a polymerization feed on separations of glycans cleaved from ribonuclease B. Structures of glycans and their corresponding MALDI-TOF-MS. Sample: Ribonuclease B glycans tagged with 2-AB, ca. 10 μg/20 μl, inj. 8 kV/10 s; Mobile phase: ACN/ 240 mM NH +4 formate/H2O (60:1:39), CEC-LIF, ∼ 600 V/cm, ∼ 3.5 μA; Columns AAm/Bis (T 5%, C 60%, TRIS-AAm x%), (ltot = 40.0 cm, ldet = 33.0 cm); (A) 0 % (blank monolith), (B) 25 % TRIS-AAm, (C) 50 % TRIS-AAm
A separation of labeled glycans in the hydrophilic interaction mode is shown on Figure 4B. Although it is possible to separate fairly polar compounds on various types of ligands [59], the acrylamide backbone (“blank” acrylamide matrix) alone is capable of resolving the carbohydrate moieties even without any polar ligand embodied. The exchange of acrylamide for a hydrophilic ligand did not cause a significant shift in capacity factors of glycans for a very broad range of the ligand content (Table 1). Nevertheless, the most efficient separation was attained in the absence of any copolymerizing ligand (Figure 4B upper trace). The separation efficiency decreased as a function of the amount of ligand added (Figure 4B, middle trace vs. lower trace). The separation efficiencies calculated for Glc7 at different ligand content are 152,000, 68,000, 26,000 for 0%, 25% and 50% ligand content, respectively (Table 1). This is believed to be due to the masking of the backbone by the added ligand. The high separation efficiency furnished by the “blank” or “pure” acrylamide porous media allowed isomeric separation of the branched oligosaccharide chains (Figure 4B). These structures are not commonly distinguishable in mass spectrometry since they feature the same molecular weight (MALDI-TOF mass spectrum of RiB glycans is depicted in Figure 4C). Note that the only peak, marked as γ, at 1701 m/z (Figure 4A) has three isomers which are resolved from each other by CEC, as illustrated in Figure 4C (upper trace).
Table 1.
Separation capacities and efficiencies for the different monoliths. AAm/Bis (T 5%, C 60%, TRIS-AAm x%), conditions as in Figure 5, for mobile-phase consisting of 60% ACN. The column-to-column reproducibility is reflected phase in the uncertainty values (confidence interval, α = 0.05, n = 3).
| Parameter | Ligand content (x) | |||
|---|---|---|---|---|
| 0 % | 25 % | 50 % | ||
| Capacity factor for G7 | 0.74 ± 0.09 | 0.75 ± 0.10 | 0.79 ± 0.08 | |
| Separation efficiency for G7 | [theoretical plates / m] | 152 000 ± 13 600 | 68 000 ± 8 600 | 26 000 ± 5 100 |
The main site of interaction between a sugar analyte and the stationary phase is located on amide groups of the acrylamide backbone, while the separation chemistry is based on the combined effects of hydrogen bonding and hydrophilicity. This is shown in Figure 5, where a “blank” acrylamide monolith allowed the separation of 2AB-labeled oligosaccharide mixture. This maltooligosaccharide ladder contains linear sugar chains with an increasing number of glucose units ranging from 4-10. In order to obtain very high detection sensitivity (down to ca. 200 fmol), the LIF detection was utilized through labeling of the analytes with 2-AB fluorophore.
Figure 5.
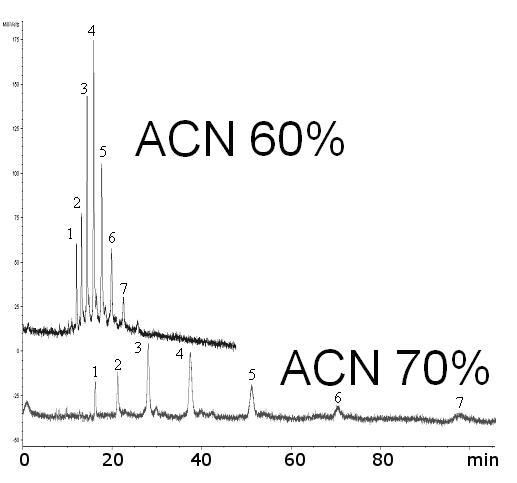
Effect of ACN% in the mobile phase on separation of linear oligosaccharides. Sample: maltooligosaccharides (Glc4-Glc10) tagged with 2-AB, ca. 0.3 mg/ml, inj. 2 kV/10 s; Mobile phase: ACN/ 240 mM NH4 formate/H2O (x: 1: 99-x), CEC-LIF ∼ 600 V/cm, ∼ 3.5 μA; Blank AAm/Bis column (T 5%, C 60%), (ldet = 27.0 cm, ltot = 35.5 cm)
Indeed, retention times increased as the number of glucose units increased (Figure 5). Also, the retention times increased with an increasing amount of organic solvent in the mobile phase (Figure 5), suggesting a hydrophilic interaction mode [60]. The capacity factor k increased from 0.5 to 1.3 for maltotetraose (Glc4) and from 0.5 to 10 for maltodecaose (Glc10) as the ratio of ACN in the running buffer increased from 40 % to 70 % (Figure 6).
Figure 6.
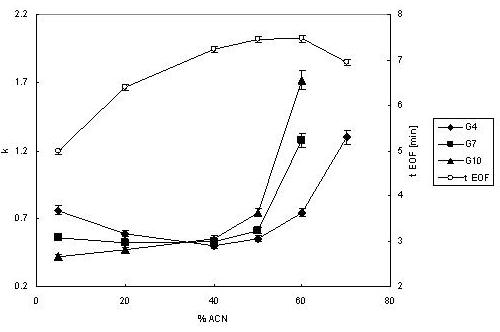
Effect of ACN percentage in the mobile phase on capacity factor k of oligosaccharides and the EOF marker time. The run-to-run reproducibility is reflected by the uncertainty values (n=3). Conditions same as in Figure 4.
It is noticeable that at a low organic content of the mobile phase (<20%), the retention order is switched and the tagged oligosaccharides are eluted in the order of decreasing molecular size. This fact indicates the reversed-phase mode of chromatography, forced by a high polarity of the mobile phase. There is definitely no other apparent reason why polar analytes should become retained on a hydrophilic stationary phase in a reversed order, but an interaction between a hydrophobic tag and the acrylamide phase. While very hydrophilic (and thus perfectly suited for hydrophilic interaction chromatography, as demonstrated thus far), the “blank” acrylamide stationary phase can seriously exhibit a specific interaction of less polar analytes, the so called “aromatic adsorption” [61].
3.3. Structural features of acrylamide monolith
A scrutiny of the network morphology was accomplished through the scanning electron microscopy. Figure 7 reveals the porogenic system furnishing a “cauliflower shape” of the monolith, with aggregates of spherical domains, as is typical for porous polymer monoliths after a phase separation [62,63]. The “cauliflower” shaped-structures are prominent in the “blank” matrix, while less visible at higher ligand content (Figure 7A-C).
Figure 7.



SEM of acrylamide monoliths. Monolith AAm/Bis (T 5%, C 60%, TRIS-AAm x%) with increasing ligand content: A) 0 % (blank), B) 25 %, C) 50 %.
All data pertaining to the morphology of the network as a function of different ligand content are summarized in Table 2. The BET value of specific surface area for “blank” acrylamide network lies around Sg = 74 m2/g, which is comparable with a typical porous monolithic material [64], as is the total cumulative pore volume of about Vg = 1.2 ml/g, determined by HgIP measurements. The measured value of polymer density (d = 1.3 g/ml) is in agreement with those obtained for the less crosslinked acrylamide networks, e.g. d = 1.302 g/ml [65] and d = 1.35 g/ml [66].
Table 2.
Data for porous acrylamide matrixes in dependence on the ligand content.
| Ligand content (x) | 0 % | 25 % | 50 % | |
|---|---|---|---|---|
| Total cumulative volume | [ml/g] | 1.27 | 0.28 | 0.19 |
| Specific surface area | [m2/g] | 73.9 | 35.9 | 0.7 |
| Total porosity | [%] | 62.3 | 26.7 | 19.6 |
| Total porosity (from swelling) | [%] | 68.7 | ||
| Mean pore radius | [nm] | 34.4 | 15.9 | 7.4 |
| Mean particle radius | [nm] | 41 | 84 | 4300 |
| Bulk density (measured with HgIP) | [g/cm3] | 4.8 | 1.8 | 2.1 |
| Apparent density (from HgIP) | [g/cm3] | 0.67 | 1.20 | 1.49 |
| True density (from pycnometry) | [g/cm3] | 1.3 | ||
| Weight swelling ratio | 9.0 | |||
| Volume swelling ratio | 6.9 |
Visually, as the ligand content increases, the globulated pattern disappears (Figure 7A-C) with a consequent deprivation of the monolith opacity in the macroscopic range. This is probably due to a deterioration of phase separation during copolymerization when polar ligands are used, because interaction parameters of solvent and polymer are being approached [67].
The pore distribution profiles are depicted in Figure 8. Only a unimodal distribution is recorded, with the highest peak interpreted as the mean pore radius. The mean pore radius decreased with an increasing content of ligand from r ≈ 35 nm (Figure 8A), 15 nm (Figure 8B), and down to r ≈ 7 nm (Figure 8C), for the ligand content of 0%, 25% and 50%, respectively. These values exemplify the range for mesopores [68]. Although these mesoporous ranges are not sufficient for the generation of the EOF [69], it is observed with linear velocity flow up to 0.8 mm/s when field strengths of about 800 V/cm were applied (Figure 9). Therefore, we believe that the sought pore sizes must be created during reswelling of the acrylamide network in various solvents as a direct consequence of chain relaxation (and polymer solvation) [70]. The restructuring of polymer networks after swelling in thermodynamically good solvents have been recently discussed by J.L. Cabral and C.D. Skinner [71].
Figure 8.
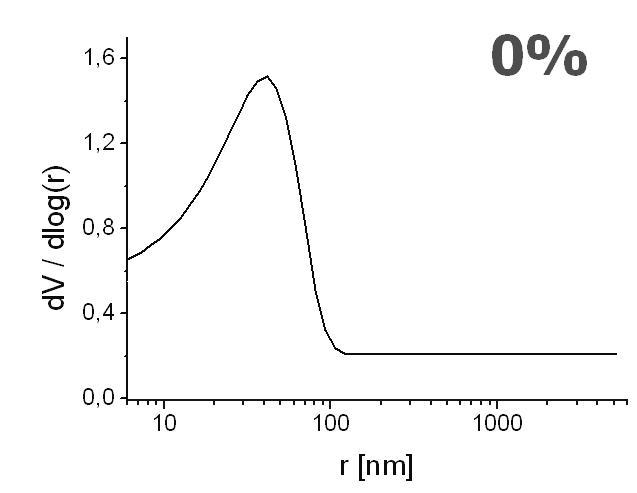
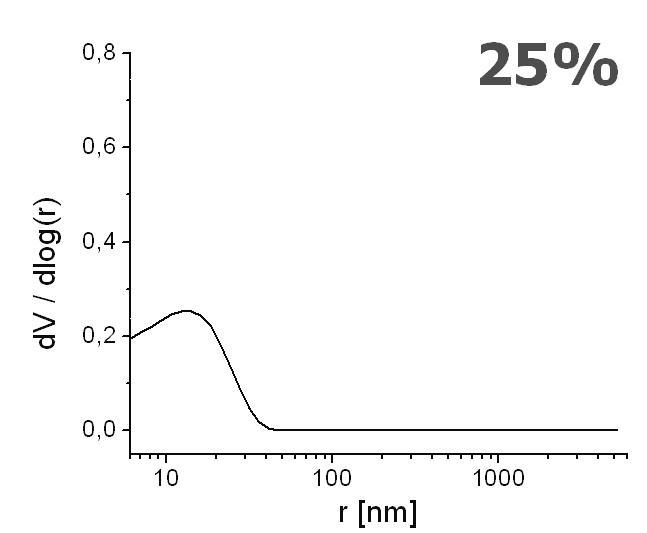
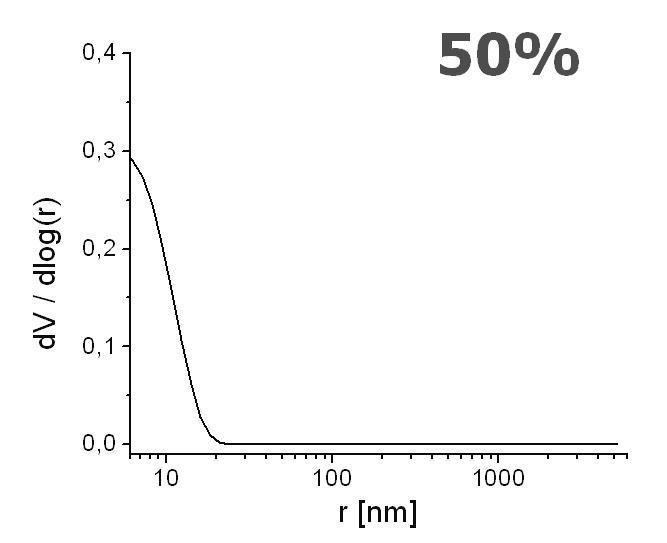
Effect of increasing ligand content on pore size distribution. Monolith AAm/Bis (T 5%, C 60%, TRIS-AAm x%) with increasing ligand content: A) 0 % (blank), B) 25 %, C) 50 % measured with mercury intrusion porosimetry
Figure 9.
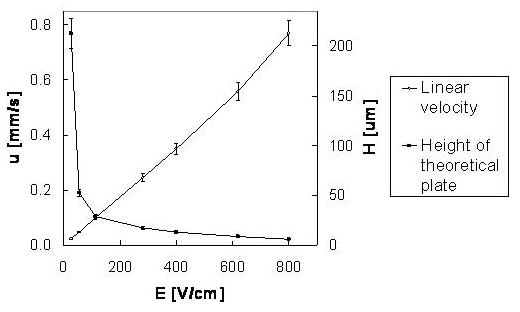
Van Deemter plot and the effect of electric field strength on linear velocity. The run-to-run reproducibility is reflected by the uncertainty values (confidence interval, α = 0.05, n = 3) Sample: acetone 10 %, inj. 1 kV/10 s; Mobile phase: ACN/ 240 mM NH4 formate/H2O (60:1:39), UV 280 nm; Blank AAm/PDA column (T 5%, C 66%), (ldet = 27.0 cm, ltot = 35.5 cm).
The relative values of the weight and the volume ratios of networks can provide information about the internal structure of porous networks in their swollen state. Roughly, the higher difference between qw and qv, the greater is the volume of pores [72]. The water uptake can be very large. The acrylamide matrix is able to gain 9-times its initial weight (qw = 9.0) and nearly 7-times its dry volume (qv = 6.9), which means that the matrix expands freely almost twice in each of its three dimensions. Numerically, the porosity in swollen state averages Ps = 69 %, however, the actual value can be different since the theory is partly based on isotropic swelling [53]. Becoming swollen in good solvents, our acrylamide monolith appears to take over a sponge-like shape (Figure 7A), which is characteristic of macroporous hydrogels with communicating pores [73]. These pores, with the estimated sizes of around 0.5 μm, should easily account for the needed EOF, as discussed above.
4. CONCLUSIONS
Highly crosslinked acrylamide hydrogels could be very effectively employed as hydrophilic stationary phases for chromatographic purposes. Here, they have been primarily targeted at the separations of oligosaccharides in analytical glycobiology. Separations can be achieved using various ligands; however, for hydrophilic interaction chromatography, the best results are achieved with just a “blank” acrylamide network. The blank acrylamide backbone provides the best efficiency at sufficient capacity factors that are easily changed with the ACN content. Although this monolith type is very hydrophilic, some aromatic compounds could be separated as well.
Additionally, the “blank” acrylamide matrix exhibits the highest permeability and porosity values. During polymerization, the phase separation and the lateral aggregation appears to take place, thus forming together a typical “cauliflower pattern” with unimodal porosity distribution. Nevertheless, the final shape of this network is highly affected by swelling in good solvents. Although the most remarkable features of this monolith is its very high efficiency, its lower mechanical stability can only permit its use CEC or in low-pressure chromatography [74].
Acknowledgements
We are indebted to the following organizations for their financial support: the Hlavka Foundation, Czech Republic; the Academy of Sciences of the Czech Republic (the project AVOZ 405000505); the Ministry of Education, Youth and Sports (the project 1M0538) and the National Institute of General Medical Sciences, U.S. Department of Health and Human Services (Grant No. GM24349). This work was also supported by the Indiana Genomics Initiative (INGEN), which is funded in part by the Lilly Endowment, Inc. (USA).
List of abbreviations
- 2-AB
2-aminobenzamide
- AFM
atomic force microscopy
- APS
ammonium persulfate
- BET
Brunauer-Emmett-Teller method for pore size estimation (thermal nitrogen desorption)
- HgIP
mercury intrusion porosimetry
- HI-LIC
hydrophilic interaction liquid chromatography
- k
capacity factor
- PNP
4-nitrophenol
- RiB
ribonuclease B
- SEM
scanning electron microscopy
- TP
theoretical plates
- χ
polymer-liquid interaction parameter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Mechref Y, Novotny MV. Structural investigations of glycoconjugates at high sensitivity. Chem. Rev. 2002;102:321–369. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- [2].El Rassi Z. Carbohydrate Analysis by Modern Chromatography and Electrophoresis. Elsevier Science; Amsterodam: 2002. [Google Scholar]
- [3].Deyl Z, Svec F. Capillary electrochromatography. Elsevier Science; Amsterodam: 2001. [Google Scholar]
- [4].Eeltink S, Rozing GR, Kok WT. Recent applications in capillary electrochromatography. Electrophoresis. 2003;24:3935–3961. doi: 10.1002/elps.200305638. [DOI] [PubMed] [Google Scholar]
- [5].Svec F. Monolithic Stationary Phases. Place of Birth: Prague. Chem. Listy. 2004;98:232–238. [Google Scholar]
- [6].Eeltink S, Decrop WMC, Rozing GP, Schoenmakers PJ, Kok WT. Comparison of the efficiency of microparticulate and monolithic capillary columns. J. Sep. Sci. 2004;27:1431–1440. doi: 10.1002/jssc.200401826. [DOI] [PubMed] [Google Scholar]
- [7].Bedair M, El Rassi Z. Recent advances in polymeric monolithic stationary phases for electrochromatography in capillaries and chips. Electrophoresis. 2004;25:4110–4119. doi: 10.1002/elps.200406136. [DOI] [PubMed] [Google Scholar]
- [8].Legido-Quigley C, Marlin ND, Melin V, Manz A, Smith NW. Advances in capillary electrochromatography and micro-high performance liquid chromatography monolithic columns for separation science. Electrophoresis. 2003;24:917–944. doi: 10.1002/elps.200390136. [DOI] [PubMed] [Google Scholar]
- [9].Jungbauer A, Hahn R. Monoliths for fast bioseparation and bioconversion and their applications in biotechnology. J. Sep. Sci. 2004;27:767–778. doi: 10.1002/jssc.200401812. [DOI] [PubMed] [Google Scholar]
- [10].Hilder EF, Svec F, Frechet JM. Development and application of polymeric monolithic stationary phases for capillary electrochromatography. J. Chromatogr. A. 2004;1044:3–22. doi: 10.1016/j.chroma.2004.04.057. [DOI] [PubMed] [Google Scholar]
- [11].Hilder EF, Svec F, Frechet JMJ. Polymeric monolithic stationary phases for capillary electrochromatography. Electrophoresis. 2002;23:3934–3953. doi: 10.1002/elps.200290011. [DOI] [PubMed] [Google Scholar]
- [12].Svec F, Peters EC, Sykora D, Frechet JMJ. Design of the monolithic polymers used in capillary electrochromatography columns. J. Chromatogr. A. 2000;887:3–29. doi: 10.1016/s0021-9673(99)01232-7. [DOI] [PubMed] [Google Scholar]
- [13].Palmer CP, McCarney JP. Developments in the use of soluble ionic polymers as pseudo-stationary phases for electrokinetic chromatography and stationary phases for electrochromatography. J. Chromatogr. A. 2004;1044:159–176. doi: 10.1016/j.chroma.2004.04.068. [DOI] [PubMed] [Google Scholar]
- [14].Hjerten S, Liao JL, Zhang R. High-Performance Liquid-Chromatography on Continuous Polymer Beds. J. Chromatogr. 1989;473:273–275. [Google Scholar]
- [15].Hjerten S, Li YM, Liao JL, Mohammad J, Nakazato K, Pettersson G. Continuous Beds - High-Resolving, Cost-Effective Chromatographic Matrices. Nature. 1992;356:810–811. [Google Scholar]
- [16].Xie SF, Svec F, Frechet JMJ. Preparation of porous hydrophilic monoliths: Effect of the polymerization conditions on the porous properties of poly(acrylamide-co-N,N’-methylenebisacrylamide) monolithic rods. J. Polym. Sci. Polym. Chem. 1997;35:1013–1021. [Google Scholar]
- [17].Xie SF, Svec F, Frechet JMJ. Rigid porous polyacrylamide-based monolithic columns containing butyl methacrylate as a separation medium for the rapid hydrophobic interaction chromatography of proteins. J. Chromatogr. A. 1997;775:65–72. doi: 10.1016/s0021-9673(97)00254-9. [DOI] [PubMed] [Google Scholar]
- [18].Liao JL, Li YM, Hjerten S. Continuous beds for microchromatography: Reversed-phase chromatography. Anal. Biochem. 1996;234:27–30. doi: 10.1006/abio.1996.0045. [DOI] [PubMed] [Google Scholar]
- [19].Liao JL, Chen N, Ericson C, Hjerten S. Preparation of continuous beds derivatized with one-step alkyl and sulfonate groups for capillary electrochromatography. Anal. Chem. 1996;68:3468–3472. doi: 10.1021/ac960261k. [DOI] [PubMed] [Google Scholar]
- [20].Ericson C, Hjerten S. Reversed-phase electrochromatography of proteins on modified continuous beds using normal-flow and counterflow gradients. Theoretical and practical considerations. Anal. Chem. 1999;71:1621–1627. [Google Scholar]
- [21].Righetti PG. Bioanalysis: Its past, present, and some future. Electrophoresis. 2004;25:2111–2127. doi: 10.1002/elps.200305808. [DOI] [PubMed] [Google Scholar]
- [22].Maruska A, Kornysova O. Continuous beds (monoliths): stationary phases for liquid chromatography formed using the hydrophobic interaction-based phase separation mechanism. J. Biochem. Biophys. Meth. 2004;59:1–48. doi: 10.1016/j.jbbm.2003.12.003. [DOI] [PubMed] [Google Scholar]
- [23].Maruska A. Monolithic materials. Preparation, properties and applications. Elsevier Science; Amsterodam: 2003. Monolithic Continuous Beds Prepared from Water-Soluble Acrylamide-Based Monomers; pp. 143–172. [Google Scholar]
- [24].Sayil C, Okay O. Macroporous poly(N-isopropyl)acrylamide networks: formation conditions. Polymer. 2001;42:7639–7652. [Google Scholar]
- [25].Durmaz S, Okay O. Phase separation during the formation of poly(acrylamide) hydrogels. Polymer. 2000;41:5729–5735. [Google Scholar]
- [26].Okay O. Macroporous copolymer networks. Prog. Polym. Sci. 2000;25:711–779. [Google Scholar]
- [27].Hoegger D, Freitag R. Acrylamide-based monoliths as robust stationary phases for capillary electrochromatography. J. Chromatogr. A. 2001;914:211–222. doi: 10.1016/s0021-9673(00)01119-5. [DOI] [PubMed] [Google Scholar]
- [28].Hoegger D, Freitag R. Investigation of conditions allowing the synthesis of acrylamide-based monolithic microcolumns for capillary electrochromatography and of factors determining the retention of aromatic compounds on these stationary phases. Electrophoresis. 2003;24:2958–2972. doi: 10.1002/elps.200305522. [DOI] [PubMed] [Google Scholar]
- [29].Hoegger D, Freitag R. Investigation of mixed-mode monolithic stationary phases for the analysis of charged amino acids and peptides by capillary electrochromatography. J. Chromatogr. A. 2003;1004:195–208. doi: 10.1016/s0021-9673(03)00563-6. [DOI] [PubMed] [Google Scholar]
- [30].Freitag R. Comparison of the chromatographic behavior of monolithic capillary columns in capillary electrochromatography and nano-high-performance liquid chromatography. J. Chromatogr. A. 2004;1033:267–273. doi: 10.1016/j.chroma.2004.01.053. [DOI] [PubMed] [Google Scholar]
- [31].Maruska A, Ericson C, Vegvari A, Hjerten S. (Normal-phase) capillary chromatography using acrylic polymer-based continuous beds. J. Chromatogr. A. 1999 Apr 2;837:25–33. [Google Scholar]
- [32].Kornysova O, Owens PK, Maruska A. Continuous beds with vancomycin as chiral stationary phase for capillary electrochromatography. Electrophoresis. 2001;22:3335–3338. doi: 10.1002/1522-2683(200109)22:15<3335::AID-ELPS3335>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [33].Schmid MG, Grobuschek N, Lecnik O, Gubitz G, Vegvari A, Hjerten S. Enantioseparation of hydroxy acids on easy-to-prepare continuous beds for capillary electrochromatography. Electrophoresis. 2001;22:2616–2619. doi: 10.1002/1522-2683(200107)22:12<2616::AID-ELPS2616>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- [34].Schmid MG, Grobuschek N, Tuscher C, Gubitz G, Vegvari A, Machtejevas E, Maruska A, Hjerten S. Chiral separation of amino acids by ligand-exchange capillary electrochromatography using continuous beds. Electrophoresis. 2000;21:3141–3144. doi: 10.1002/1522-2683(20000901)21:15<3141::AID-ELPS3141>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- [35].Kornysova O, Machtejevas E, Kudirkaite V, Pyell U, Maruska A. Synthesis and characterization of polyrotaxane-based polymeric continuous beds for capillary electrochromatography. J. Biochem. Biophys. Meth. 2002;50:217–232. doi: 10.1016/s0165-022x(01)00233-0. [DOI] [PubMed] [Google Scholar]
- [36].Plieva FM, Andersson J, Galaev IY, Mattiasson B. Characterization of polyacrylamide based monolithic columns. J. Sep. Sci. 2004;27:828–836. doi: 10.1002/jssc.200401836. [DOI] [PubMed] [Google Scholar]
- [37].Plieva FM, Savina IN, Deraz S, Andersson J, Galaev IY, Mattiasson B. Characterization of supermacroporous monolithic polyacrylamide based matrices designed for chromatography of bioparticles. J. Chromatogr. B. 2004;807:129–137. doi: 10.1016/j.jchromb.2004.01.050. [DOI] [PubMed] [Google Scholar]
- [38].Zhang K, Yan C, Yang JJ, Zhang ZC, Wang QS, Gao RY. Preparation and characterization of C-16 monolithic columns for capillary electrochromatography. J. Sep. Sci. 2005;28:217–224. doi: 10.1002/jssc.200401873. [DOI] [PubMed] [Google Scholar]
- [39].Palm A, Novotny MV. Macroporous polyacrylamide poly(ethylene glycol) matrixes as stationary phases in capillary electrochromatography. Anal. Chem. 1997;69:4499–4507. [Google Scholar]
- [40].Zhang MQ, El Rassi Z. Capillary electrochromatography with polyacrylamide monolithic stationary phases having bonded dodecyl ligands and sulfonic acid groups: Evaluation of column performance with alkyl phenyl ketones and neutral moderately polar pesticides. Electrophoresis. 2001;22:2593–2599. doi: 10.1002/1522-2683(200107)22:12<2593::AID-ELPS2593>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- [41].Que AH, Novotny MV. Separation of neutral saccharide mixtures with capillary electrochromatography using hydrophilic monolithic columns. Anal. Chem. 2002;74:5184–5191. doi: 10.1021/ac025781w. [DOI] [PubMed] [Google Scholar]
- [42].Que AH, Novotny MV. Structural characterization of neutral oligosaccharide mixtures through a combination of capillary electrochromatography and ion trap tandem mass spectrometry. Anal. Bioanal. Chem. 2003;375:599–608. doi: 10.1007/s00216-003-1766-8. [DOI] [PubMed] [Google Scholar]
- [43].Tegeler TJ, Mechref Y, Boraas K, Reilly JP, Novotny MV. Microdeposition device interfacing capillary electrochromatography and microcolumn liquid chromatography with matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2004;76:6698–6706. doi: 10.1021/ac049341b. [DOI] [PubMed] [Google Scholar]
- [44].Que AH, Mechref Y, Huang YP, Taraszka JA, Clemmer DE, Novotny MV. Coupling capillary electrochromatography with electrospray Fourier transform mass spectrometry for characterizing complex oligosaccharide pools. Anal. Chem. 2003;75:1684–1690. doi: 10.1021/ac025985c. [DOI] [PubMed] [Google Scholar]
- [45].Starkey JA, Abrantes S, Mechref Y, Novotny MV. Sensitive analyses of agricultural chemicals by capillary electrochromatography. J. Sep. Sci. 2003;26:1635–1642. [Google Scholar]
- [46].Starkey JA, Mechref Y, Byun CK, Steinmetz R, Fuqua JS, Pescovitz OH, Novotny MV. Determination of trace isoflavone phytoestrogens in biological materials by capillary electrochromatography. Anal. Chem. 2002;74:5998–6005. doi: 10.1021/ac025929b. [DOI] [PubMed] [Google Scholar]
- [47].Que AH, Palm A, Baker AG, Novotny MV. Steroid profiles determined by capillary electrochromatography, laser-induced fluorescence detection and electrospraymass spectrometry. J. Chromatogr. A. 2000;887:379–391. doi: 10.1016/s0021-9673(00)00427-1. [DOI] [PubMed] [Google Scholar]
- [48].Hjerten S. Arch. Biochem. Biophys. 1962:147–151. [PubMed] [Google Scholar]
- [49].Mechref Y, Novotny MV. Mass spectrometric mapping and sequencing of Nlinked oligosaccharides derived from submicrogram amounts of glycoproteins. Anal. Chem. 1998;70:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- [50].Bigge JC, Patel TP, Bruce JA, Goulding PN, Charles SM, Parekh RB. Nonselective and Efficient Fluorescent Labeling of Glycans Using 2-Amino Benzamide and Anthranilic Acid. Anal. Biochem. 1995;230:229–238. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- [51].Liu JP, Hsieh YZ, Wiesler D, Novotny M. Design of 3-(4-Carboxybenzoyl)-2-Quinolinecarboxaldehyde As A Reagent for Ultrasensitive Determination of Primary Amines by Capillary Electrophoresis Using Laser Fluorescence Detection. Anal. Chem. 1991;63:408–412. doi: 10.1021/ac00005a004. [DOI] [PubMed] [Google Scholar]
- [52].Uzun L, Say R, Denizli A. Porous poly(hydroxyethyl methacrylate) based monolith as a new adsorbent for affinity chromatography. React. Funct. Polym. 2005;64:93–102. [Google Scholar]
- [53].Beranova H, Dusek K. Swelling of homogeneous and macroporous styrenedivinylbenzene copolymers:copolymer-solvent interactions and the structure of heterogeneous networks. Collect. Czech. Chem. Commun. 1969;34:2932–2941. [Google Scholar]
- [54].Baker JP, Hong LH, Blanch HW, Prausnitz JM. Effect of Initial Total Monomer Concentration on the Swelling Behavior of Cationic Acrylamide-Based Hydrogels. Macromolecules. 1994;27:1446–1454. [Google Scholar]
- [55].Righetti PG. Macroporous Gels - Facts and Misfacts. J. Chromatogr. A. 1995;698:3–17. [Google Scholar]
- [56].Miertus S, Righetti PG, Chiari M. Molecular Modeling of Acrylamide Derivatives - the Case of N-Acryloylaminoethoxyethanol Versus Acrylamide and Trisacryl. Electrophoresis. 1994;15:1104–1111. doi: 10.1002/elps.11501501166. [DOI] [PubMed] [Google Scholar]
- [57].Hjerten S, Mohammad J, Nakazato K. Improvement in Flow Properties and Ph Stability of Compressed, Continuous Polymer Beds for High-Performance Liquid-Chromatography. J. Chromatogr. 1993;646:121–128. [Google Scholar]
- [58].Mechref Y, Baker AG, Novotny MV. Matrix-assisted laser desorption/ionization mass spectrometry of neutral and acidic oligosaccharides with collision-induced dissociation. Carbohydr. Res. 1998;313:145–155. doi: 10.1016/s0008-6215(98)00264-x. [DOI] [PubMed] [Google Scholar]
- [59].Que AH, Konse T, Baker AG, Novotny MV. Analysis of bile acids and their conjugates by capillary electrochromatography/electrospray ion trap mass spectrometry. Anal. Chem. 2000;72:2703–2710. doi: 10.1021/ac991059v. [DOI] [PubMed] [Google Scholar]
- [60].Koizumi K, Utamura T, Kubota Y, Hizukuri S. 2 High-Performance Liquid-Chromatographic Columns for Analyses of Maltooligosaccharides. J. Chromatogr. 1987;409:396–403. [Google Scholar]
- [61].Gelotte B. J. Chromatogr. 1960;3:330–335. [Google Scholar]
- [62].Svec F, Frechet JMJ. Monolithic materials. Preparation, properties and applications. Elsevier Science; Amsterdam: 2003. Rigid macroporous organic polymer monoliths prepared by free radical polymerization; pp. 19–50. [Google Scholar]
- [63].Tobita H, Hamielec AE. Cross-Linking Kinetics in Polyacrylamide Networks. Polymer. 1990;31:1546–1552. [Google Scholar]
- [64].Yu C, Xu MC, Svec F, Frechet JMJ. Preparation of monolithic polymers with controlled porous properties for microfluidic chip applications using photoinitiated free-radical polymerization. J. Polym. Sci. Polym. Chem. 2002;40:755–769. [Google Scholar]
- [65].Saraydin D, Karadag E, Isikver Y, Sahiner N, Guven O. The influence of preparation methods on the swelling and network properties of acrylamide hydrogels with crosslinkers. J. Macromol. Chem. - Pure Appl. Chem. 2004;A41:419–431. [Google Scholar]
- [66].Ilavsky M. Effect of Electrostatic Interactions on Phase-Transition in the Swollen Polymeric Network. Polymer. 1981;22:1687–1691. [Google Scholar]
- [67].Ousalem M, Zhu XX, Hradil J. Evaluation of the porous structures of new polymer packing materials by inverse size-exclusion chromatography. J. Chromatogr. A. 2000;903:13–19. doi: 10.1016/s0021-9673(00)00920-1. [DOI] [PubMed] [Google Scholar]
- [68].Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, Siemieniewska T. Reporting Physisorption Data for Gas Solid Systems with Special Reference to the Determination of Surface-Area and Porosity (Recommendations 1984) Pure Appl. Chem. 1985;57:603–619. [Google Scholar]
- [69].Hjerten S, Vegvari A, Srichaiyo T, Zhang HX, Ericson C, Eaker D. An approach to ideal separation media for (electro)chromatography. J. Capil. Electroph. 1998;5:13–26. [PubMed] [Google Scholar]
- [70].Jerabek K. Characterization of Swollen Polymer Gels Using Size Exclusion Chromatography. Anal. Chem. 1985;57:1598–1602. [Google Scholar]
- [71].Cabral JL, Bandilla D, Skinner CD. Pore size characterization of monolith for electrochromatography via atomic force microscopy studies in air and liquid phase. J. Chromatogr. A. 2006;1108:83–89. doi: 10.1016/j.chroma.2005.12.037. [DOI] [PubMed] [Google Scholar]
- [72].Sayil C, Okay O. Macroporous poly(N-isopropylacrylamide) networks. Polym. Bull. 2002;48:499–506. [Google Scholar]
- [73].Pradny M, Lesny P, Fiala J, Vacik J, Slouf M, Michalek J, Sykova E. Macroporous hydrogels based on 2-hydroxyethyl methacrylate. Part 1. Copolymers of 2-hydroxyethyl methacrylate with methacrylic acid. Collect. Czech. Chem. Commun. 2003;68:812–822. [Google Scholar]
- [74].Palm AK, Novotny MV. Analytical characterization of a facile porous polymer monolithic trypsin microreactor enabling peptide mass mapping using mass spectrometry. Rapid Commun. Mass Spectrom. 2004;18:1374–1382. doi: 10.1002/rcm.1500. [DOI] [PubMed] [Google Scholar]