

Abstract
Obstructive sleep apnea (OSA) has emerged as a major public health problem and increasing evidence indicates that untreated OSA can lead to the development of various cardiovascular disorders. One important mechanism by which OSA may promote cardiovascular diseases is intermittent hypoxia, in which patients are subjected to repeated episodes of brief oxygen desaturation in the blood, followed by reoxygenation. Such cycles of hypoxia/reoxygenation may result in the generation of reactive oxygen species. Some studies have demonstrated the presence of oxidative stress in OSA patients as well as in animals subjected to intermittent hypoxia. Further, modulations of nitric oxide and biothiol status might also play important roles in the pathogenesis of OSA-associated diseases. Reactive oxygen species and redox events are also involved in the regulation of signal transduction for oxygen-sensing mechanisms. This review summarizes currently available information on the evidence for and against the occurrence of oxidative stress in OSA and the role of reactive oxygen species in cardiovascular changes associated with OSA.
Keywords: Free radical, Intermittent hypoxia, Nitric oxide, Oxidative stress, Reactive oxygen species, Sleep apnea, Superoxide
Introduction
Obstructive sleep apnea (OSA) syndrome is a condition characterized by the occurrence of repetitive episodes of partial or complete obstruction of airflow during sleep. Obstruction usually occurs in the upper airway, and may lead to decreased blood oxygenation and fragmentation of the sleep cycle. Obstructive apnea is defined as complete obstruction of airflow in the upper airway for at least 10 s with persistent effort to breathe [1]. The severity of OSA is measured by the apnea–hypopnea index, obtained by counting the total number of apneas and hypopneas during sleep and dividing that by the hours of sleep. Severity of symptoms increases as the number of respiratory events per hour of sleep increases. An apnea–hypopnea index lower than 5 per h is normal; 5–15 is mild, 15–30 is moderate, and greater than 30 is severe OSA. Common symptoms of OSA include loud snoring, breathing pauses observed by the bed partner, and feelings of nonrefreshing sleep and excessive daytime sleepiness.
OSA is a common disorder in the United States and other Western countries. Young et al. [2] reported, in a study of adults 30–60 years of age, that 24% of North American men and 9% of women had an apnea–hypopnea index greater than 5 events per hour of sleep. When the apnea–hypopnea index greater than 5 events per hour of sleep was combined with daytime symptoms of excessive sleepiness, the prevalence was found to be 4% in males and 2% in females.
Besides the obvious detrimental effect of sleep-disordered breathing, such as daytime sleepiness, deficits in cognitive performance, and mood and behavioral effects, OSA is also associated with an increased risk for cardiovascular diseases. OSA has been implicated in pathogenesis of various cardiovascular diseases including systemic hypertension, pulmonary hypertension, congestive cardiac failure, cardiac arrhythmias, atherosclerosis, ischemic heart disease, and stroke [3-12]. However, the mechanisms by which OSA affects the cardiovascular system are largely unknown.
The currently available treatment for OSA is mostly limited to the application of continuous positive airway pressure (CPAP) [13]. Although it is effective in reducing daytime sleepiness, patients still experience some episodes of apnea while on CPAP treatment [13]. Further, many of the patients prescribed CPAP might choose not to continue their treatment due to inconvenience or intolerance [14]. Because of incomplete effectiveness and inconsistent use of CPAP, it is not yet clear whether this treatment will completely eliminate the long-term consequences of OSA on the cardiovascular system.
Mechanisms of OSA-induced biological changes may involve intermittent hypoxia, intermittent hypercapnea, intrathoracic pressure changes, sympathetic activation, and sleep fragmentation [15]. Among these, intermittent hypoxia is thought to be a major cause of cardiovascular alterations, which may occur in patients with OSA. OSA patients undergo repeated episodes of hypoxia (which can last for 10 s to as long as 2 min) and normoxia (2–3 min), which resemble ischemia/reperfusion events. During the hypoxic/ischemic phase, cells adapt to a low oxygen environment; however, the reoxygenation/reperfusion phase causes a sudden increase of oxygen in the cells. This reoxygenation/reperfusion phase is thought to result in the production of reactive oxygen species (ROS) and the promotion of oxidative stress [15-17]. ROS produced via the electron reduction of molecular oxygen (O2) include superoxide anion radical , hydrogen peroxide (H2O2), and hydroxyl radical (•OH). These molecules can oxidize a variety of biological molecules including lipids, proteins, and DNA, and can alter biological functions. ROS can also serve as signal transduction mediators to elicit oxygen-sensing mechanisms as well as cell growth events, which play critical roles in cardiovascular diseases [18]. ROS production in OSA patients could also occur via inflammatory responses [19], as well as increased sympathetic tone and elevated catecholamine-induced ROS production [20].
Studies in OSA patients have been performed to test the hypothesis that oxidative stress occurs in association with intermittent hypoxia. Although earlier studies failed to validate this hypothesis, some recent experiments using higher numbers of OSA patients and improved methodologies have provided evidence for the induction of oxidative stress. Further, other redox regulating molecules such as nitric oxide and homo-cysteine are also altered in OSA patients. In this article, we review available literature on the studies of the levels of oxidative stress, nitric oxide, and biothiols in OSA patients as well as animal studies for the mechanisms of oxidative stress and oxygen sensing to provide information on the roles of redox biology in the development of cardiovascular diseases associated with OSA.
Evidence for oxidative stress in OSA patients
Oxidative stress markers in OSA
ROS production results in oxidation of biological molecules, which in turn form oxidized products that have been used for assessing the occurrence of oxidative stress [21,22]. Widely used markers include lipid peroxidation products, and oxidized protein and DNA. Susceptibility to in vitro addition of oxidants to biological samples has also been used to estimate the occurrence of oxidative stress. Since environmental, dietary, and genetic factors can influence the levels of oxidative stress in humans, large sample sizes, appropriate storage of samples (to avoid further oxidation), and the use of sensitive and reliable techniques are needed to accurately assess oxidative stress levels in a given condition. Earlier studies of OSA patients used small numbers of patients and control subjects, and used rather crude methods for the measurements of oxidative stress. Studies by Wali et al. [23] with small numbers of patients (6 patients who experienced severe OSA-related hypoxia, 9 OSA patients with low to moderate hypoxia, and 6 healthy subjects) detected no difference in susceptibility of isolated LDL to free radical-induced conjugated diene formation. They also measured glutathione peroxidase and catalase activities in red blood cells, and failed to detect any differences among the groups [23]. Ozturk et al. [24], using red blood cells also from a small number of OSA patients (n = 6), failed to demonstrate changes in hydrogen peroxide-induced lipid peroxidation (as monitored by measuring malondialdehyde), red blood cell fragility (as an indicator of cell membrane oxidative stress), or total cellular thiol levels (as measured by Ellman's reagent in a colorimetric assay).
More recently, increased sample sizes and more sophisticated experimental designs allowed for observing differences in oxidative stress levels in OSA patients and control subjects. In contrast to the above-noted studies, reporting no differences in the susceptibility to lipid peroxidation in samples from OSA patients and control subjects, Christou et al. [25] assessed levels of diacron-reactive oxygen metabolites (D-ROM) in the blood of 21 OSA patients with apnea–hypopnea index greater than 5, compared with 5 controls. Diacron indicates the ability of transition metals to catalyze the formation of free radicals in the presence of peroxide, as monitored by oxidized alchilamine products. This study found that OSA patients had higher diacron-reactive oxygen metabolites.
Barcelo et al. [26] also reported that the lipid peroxidation profile is abnormal in OSA patients. Using LDL particles isolated from 14 patients with severe OSA (59 apnea/h) and 13 healthy subjects, they found that thiobarbituric acid-reactive substance (TBARS) formation was higher in OSA patients. CPAP treatment improved the abnormal lipid peroxidation events in OSA patients. In a study of 114 OSA patients, Lavie et al. [27] reported that morning levels of TBARS and peroxides were significantly higher in OSA patients, with or without cardiovascular disease, than in controls; and the CPAP treatment decreased nocturnal levels of TBARS and peroxides in OSA patients. Increased oxidized LDL levels are also detected in OSA patients [28].
The cellular antioxidant defense mechanisms can be altered in response to oxidation and may serve as oxidative stress markers. Levels of small molecule antioxidants such as ascorbic acid, tocopherols, carotenoids, and flavonoids are often lower in diseases associated with oxidative stress. While comprehensive examinations of antioxidant levels in OSA have not been rigorously performed, one study by Christou et al. [29] assessed the antioxidant capacity in the blood of 14 patients with severe OSA (apnea/hypopnea index of N20) compared with controls using the Trolox Equivalent Antioxidant Capacity assay. This colorimetric assay is based on the scavenging of the 2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid) radical converting it into a colorless product. Similarly, patients with end-stage renal disease and OSA were found to have decreased total antioxidant status and increased coronary artery calcification compared to patients without OSA syndrome [30]. The results showed that patients with severe OSA had reduced antioxidant capacity. Taken together, results from studies described above indicate the occurrence of oxidative stress in systemic circulation of OSA patients.
A recent study by Yamauchi et al. [31] demonstrated that urinary 8-hydroxy-2′-deoxyguanosine excretion was significantly higher in patients with severe OSA (n = 58) compared with control subjects (n = 70), confirming the above studies providing evidence for the occurrence of oxidative stress in OSA patients.
It should be noted that whether oxidative stress occurs in OSA patients is still controversial since some of more recent studies failed to demonstrate increased lipid peroxidation in OSA patients [32,33]. Svatikova et al. [32] suggested that conflicting results may be explained by the presence of comorbidities and/or medications in OSA patients, the absence of control subjects matched closely for body mass index (BMI) and obesity, the presence of undiagnosed OSA in control populations, and the timing of oxidative stress measurements.
Inflammation as a mechanism of ROS production in OSA
Studies described above suggest the occurrence of oxidative stress in systemic circulation and airways of OSA patients. While the mechanisms of ROS production, such as the role of hypoxia/reoxygenation, still need to be defined, evidence suggests the role of inflammatory responses in ROS production in OSA patients. Immune cells are primary sources of ROS generation in inflammation. Therefore, measurements of the levels of ROS-generating immune cells in plasma and in the airways, and their oxidative activities using in vitro assays, can be an estimate of immune cell-derived ROS generation. OSA is associated with increased inflammation; thus immune cells may be a source of ROS in OSA [7].
In 1995, Muns et al. [34] examined the numbers of neutrophils and oxidative burst of granulocytes recovered from nasal lavage of 24 OSA patients compared with controls. These studies measured the conversion rate of radiolabeled dihydrorhodamine elicited by treatment of neutrophils with Escherichia coli. Their results detected no difference in numbers of neutrophils or in the oxidative burst between OSA patients and control subjects.
In contrast, a study examining systemic inflammatory responses by Schulz et al. [35] reported that, compared with controls, patients with OSA had markedly enhanced release of superoxide from stimulated polymorphonuclear neutrophils (PMNs) as measured by superoxide dismutase-inhibitable reduction of cytochrome c. In their study of 18 OSA patients, PMNs were stimulated with the bacterial tripeptide formyl-methionylleucyl-phenylalanine as well as with a calcium ionophore. Both of these stimulants produced more superoxide in PMNs from OSA patients. CPAP therapy resulted in decreased superoxide release in response to these agents.
Dyugovskaya et al. [36] examined the expression of adhesion molecules on the monocytes as another indication of the activation of systemic inflammation in OSA. Using 18 moderate to severe OSA patients and 26 healthy controls, these studies found that OSA was associated with increased expression of adhesion molecules CD15 and CD11c and increased adherence of monocytes in culture to human endothelial cells. Examination of the activation state of the immune cells showed increased intracellular ROS production in some monocyte and granulocyte subpopulations and upregulation of CD15 expression due to hypoxia in vitro. Flow cytometric analysis using dihydroethidium showed that ROS production with phorbol ester stimulation of CD11c+ and CD64+ monocytes and granulocytes was higher in OSA patients. Moreover, ROS production in nonprimed CD11c+ monocytes was also higher in OSA patients. Nasal CPAP (nCPAP) treatment downregulated CD15 and CD11c expression and decreased basal ROS production in CD11c+ monocytes. These results indicate that CPAP treatment is effective in reducing systemic inflammatory responses including those associated with ROS generation.
Evidence for altered nitric oxide production in OSA patients
Nitric oxide is an endothelium-derived vasorelaxing factor for both systemic and pulmonary vasculature (reviewed in [37]). Besides its generation in endothelial cells, nitric oxide can also be generated by various other tissues via the actions of nitric oxide synthetases (NOS). Studies indicate that a primary mechanism of nitric oxide actions is the inhibition of smooth muscle cell contraction through its effect on guanylate cyclase and the activation of protein kinase G, thus inducing vasorelaxation and attenuating the degrees of hypertension [38]. Nitric oxide also suppresses the smooth muscle proliferation and induces apoptosis of these cells, providing beneficial effects on vascular remodeling [39]. Thus, long-term complications of OSA including systemic hypertension, pulmonary hypertension, myocardial infarction, and stroke might be influenced by dysregulated nitric oxide [40]. Experiments by Kato et al. [41] showed that patients with OSA had a blunted vasodilation in response to acetylcholine, but not in response to sodium nitroprusside or verapamil, indicating that OSA patients have impaired resistance-vessel endothelium-dependent vasodilation, but the smooth muscle cell response itself is normal. Providing that there is an increase in oxidative stress in OSA patients as described above, it is expected that these patients might have lower nitric oxide levels as can quench nitric oxide [42].
Human studies have been performed to assess the levels of nitric oxide and its metabolites in OSA patients. Earlier studies by Olopade et al. [43] monitored nitric oxide by using a chemiluminescence analyzer. This study showed that OSA patients (n = 20), but not control subjects, had significantly higher exhaled nasal nitric oxide levels (39.7 ± 3.8 ppb vs 28.4 ± 2.9 ppb) after sleep compared to before sleep, while exhaled oral nitric oxide levels increased in both groups. Basal exhaled nitric oxide levels were not different between OSA and control groups. Agusti et al. [44] measured basal exhaled nitric oxide in 24 OSA patients and 7 controls and found that exhaled nitric oxide levels were not significantly different between the two groups. These results indicate that apneic episodes in OSA patients might promote nasal inflammation and the production of nitric oxide.
Studies on circulating levels of nitric oxide demonstrated that nitric oxide is lower in OSA patients. Ip et al. [45] reported that serum nitrite/nitrate levels were lower in OSA subjects (38.9 ± 22.9 μM; n = 30), compared to controls (63.1 ± 47.5 μM; n = 40). nCPAP significantly increased the serum nitrite/nitrate levels (30.5 ± 14.4 μM without nCPAP vs 81.0 ± 82.1 μM with nCPAP). In 21 OSA patients, Schulz et al. [46] observed that blood levels of nitric oxide measured by chemiluminescence were significantly lower compared to control subjects. Two nights of CPAP treatment increased the nitric oxide levels. Lavie et al. [47] more recently reported that plasma nitric oxide levels measured hourly during sleep were found to be significantly lower in 8 OSA patients compared to 6 control subjects. Nine months treatment with nCPAP increased nitric oxide levels as well as the levels of L-arginine (precursor for nitric oxide) in OSA patients. In the studies by Teramoto et al. [48], the serum level of nitric oxide production was monitored by measuring nitrite/nitrates levels in 24 patients with OSA and 24 controls. Nitric oxide levels were found to be lower in patients with OSA than in controls. They also found that administration of 1–2 L/min of oxygen during night increased the nitric oxide level.
Possible mechanisms of OSA-induced reduction of nitric oxide include: (1) superoxide-mediated scavenging of nitric oxide, generating products such as peroxynitrite which can nitrosylate membrane proteins and oxidize lipids [49]; (2) reduction of molecular oxygen, which serves as a cosubstrate for nitric oxide synthesis; (3) increased NOS inhibitors; and (4) suppression of endothelial NOS expression [50]. Two of these hypotheses have been directly addressed in studies of human OSA patients. Studies by Svatikova et al. [51] measured circulating nitrotyrosine, as an indicator of peroxynitrite formation via nitric interactions, in 10 severe OSA patients and 10 controls using liquid chromatography–tandem mass spectrometry. Results showed that nitrotyrosine levels were similar in control and OSA groups before or after sleep. The absence of alterations in nitrotyrosine formation suggests that peroxynitrite formation does not occur in OSA. An endogenous competitive inhibitor of NOS, asymetrical dimethylarginine (ADMA), has been shown to be increased in the plasma of OSA patients [52], indicating the potential importance of NOS inhibition. Further work is needed to determine the mechanism of nitric oxide reduction in OSA patients.
Evidence for altered homocysteine levels in OSA patients
Homocysteine is a thiol-containing compound generated during the metabolism of methionine. In plasma, homocysteine is largely oxidized, existing as homocysteine thiolactone or homocystine. Moderate hyperhomocysteinemia (>14 μM) has been shown to be an independent risk factor for vascular disease [53-56]. Plasma total homocysteine increases with age [57] and increased plasma homocysteine may be responsible for the development of cardiovascular diseases [58]. The mechanism of homocysteine actions has been postulated to involve the production of ROS [59,60]. Further, homocysteine has been shown to influence oxidative stress responsive signal transduction pathways [61,62].
Because OSA is associated with increased risk for various cardiovascular diseases, Lavie et al. [63] measured plasma levels of homocysteine in control healthy subjects (n = 73), OSA patients without any cardiovascular disease (n = 127), OSA patients with hypertension (n = 61), OSA patients with ischemic heart disease (n = 49), and ischemic heart disease patients without OSA (n = 35). Results showed that the mean ± SD value for homocysteine levels from OSA patients with ischemic heart disease was 14.6 ± 6.8 μM, while that of control healthy subjects was 9.8 ± 3.5 μM (P < 0.0001). Homocysteine levels from OSA patients without ischemic heart disease and ischemic heart disease patients without OSA were 9.9 ± 3.0 and 11.8 ± 5.3 μM, respectively. Together these results suggest that increased homocysteine levels are associated with the occurrence of ischemic heart disease in OSA patients, but OSA alone does not appear to be an inducer of increased plasma homocysteine. More rigorous time course studies by Svatikova et al. [64] also showed that having OSA per se did not change homocysteine levels, and CPAP treatment was not effective in lowering homocysteine in patients with OSA [65]. In contrast, Jordan et al. [66] reported that 7 of 16 untreated OSA patients showed homocysteine levels > 11.7 μM, and that the level of homocysteine was effectively lowered by CPAP treatment. Thus, the role of homocysteine in OSA-induced cardiovascular complications is not yet clear, and further investigations are needed.
Oxidative stress in animal models of intermittent hypoxia
While the mechanisms of OSA-induced biologic changes are complex, it has been postulated that chronic intermittent hypoxia is a critical component. Experimental animal models can be useful as they give the opportunity to modulate biological systems to elucidate detailed mechanisms of pathophysiology, which cannot be performed in humans.
While information on the cardiac effects of intermittent hypoxia-produced ROS has recently started to emerge, results generated from rodent models of intermittent hypoxia for understanding the involvement of oxidative stress in neuronal damage and learning impairment should be helpful for future studies on the cardiovascular system.
Row et al. [67] reported that intermittent hypoxia-induced lipid peroxidation and increased isoprostane concentrations in the brain accompanied impairment of spatial learning in rats. Further, learning deficits induced by intermittent hypoxia were attenuated by the antioxidant PNU-101033E. Xu et al. [68] used a mouse model of intermittent hypoxia and demonstrated cortical production of ROS and neuronal cell death. Chronic intermittent hypoxia increased the levels of protein oxidation, lipid peroxidation, and nucleic acid oxidation in mouse brain cortex. Oxidative stress-responsive transcription factors such as NF-κB, c-Fos, and c-Jun were activated in response to intermittent hypoxia. In transgenic mice overexpressing Cu, Zn-SOD, ROS production and neuronal cell apoptosis induced by intermittent hypoxia are attenuated. Similarly, Veasey et al. [69] found, in a mouse model of long-term intermittent hypoxia, oxidative injury in the wake-promoting regions of the basal forebrain and brainstem after a 2-week exposure to intermittent hypoxia as evident by elevated isoprostane, protein carbonyl, nitration, and induction of glutathione reductase and methionine sulfoxide reductase. These studies suggest a link among OSA, ROS generation, and alterations in neurological function.
ROS can also function as regulators of physiologic functions [70]. In response to intermittent hypoxia, ROS might contribute to changes in the carotid body function [71]. To support this hypothesis, a study by Peng et al. [72] demonstrated that chronic intermittent hypoxia induced prolonged augmentation of sensory discharge, called long-term facilitation, of the carotid body hypoxic sensory response; the potentiation of carotid body activity was prevented by a superoxide scavenger in rats. Chronic intermittent hypoxia inhibited complex I activity of the mitochondrial electron transport, suggesting that superoxide might be produced by the mitochondria in response to intermittent hypoxia. A second study of the hypoxic sensory response of the carotid bodies showed that long-term facilitation was induced by short durations of intermittent hypoxia (repeated episodes of 15 s hypoxia/5 min normoxia), but not by long durations of intermittent hypoxia (4 h of hypobaric hypoxia + 20 h of normoxia). Again, the augmentation was prevented by superoxide scavenger, indicating a role for ROS [73]. Similarly, long-term facilitation of breathing elicited by intermittent hypoxia is prevented by a superoxide scavenger [74]. Therefore, ROS, specifically, superoxide anion radicals, are produced from mitochondria in response to intermittent hypoxia and may serve as signaling molecules for neuronal responses.
In contrast to the observations that intermittent hypoxia produces ROS and induces oxidative stress, sleep deprivation alone does not have marked effects on ROS formation. In a rat model of sleep deprivation, Gopalakrishnan et al. [75] measured oxidant production, antioxidant enzyme activities, lipid peroxidation, and protein oxidation. No evidence of oxidative stress was found in brain, liver, and skeletal muscle. Thus, intermittent hypoxia, rather than sleep fragmentation, might play a major role in oxidative stress associated with human OSA.
In 2005, several studies were published to demonstrate the effects of intermittent hypoxia on the heart. Campen et al. [76] compared the effects of acute hypoxia (4 min of 10% O2) and chronic intermittent hypoxia (60 s cycles, 12 h/day for 5 weeks) in mice and found that acute hypoxia induced pulmonary hypertension and increased the load to the right heart, while chronic intermittent hypoxia resulted in mild to moderate systemic and pulmonary hypertension, leading to left and right ventricular hypertrophy. While the reports of the effects of intermittent hypoxia on myocardial oxidative status are limited, Chen et al. [77] reported increased lipid peroxidation in left ventricles of rats subjected to chronic intermittent hypoxia. Oxidative stress induced by chronic intermittent hypoxia might be responsible for increased myocardial damage [78]. Results from our laboratory [79] demonstrated that chronic intermittent hypoxia increased ischemia–reperfusion-induced lipid peroxidation in mouse hearts (Fig. 1A), indicating that myocardial oxidative stress can be enhanced by chronic intermittent hypoxia. This might be responsible for increased susceptibility to ischemia/reperfusion-induced cardiac myocyte apoptosis in mice treated with chronic intermittent hypoxia (Fig. 1B). These results from animal models suggest that chronic intermittent hypoxia can induce oxidative stress in the heart. Myocardial mechanisms of intermittent hypoxia-induced oxidative stress need further investigation. Available information on the brain should help in the design of experiments concerning the effects of intermittent hypoxia on the heart.
Fig. 1.
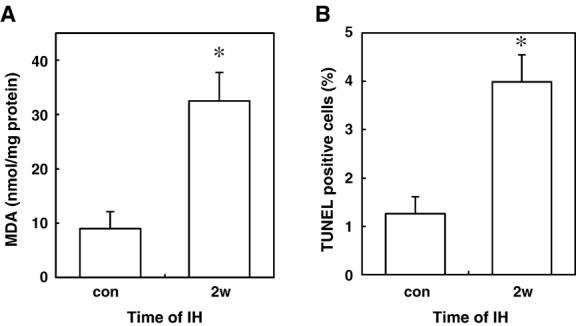
Effects of intermittent hypoxia on ischemia/reperfusion-induced myocardial lipid peroxidation and cardiac muscle cell apoptosis. C57BL/6 mice were subjected to intermittent hypoxia (2 min cycles of 6 and 21% O2) for 8 h/day for 2 weeks (2w). After intermittent hypoxia, isolated hearts were subjected to 30 min ischemia followed by reperfusion. (A) Hearts were homogenized and the levels of malondialdehyde (MDA) were monitored as an indication for lipid peroxidation. (B) Hearts were stained to assess TUNEL-positive cells as an indication for apoptosis. Values represent means ± SE. (*) denotes the values that are significantly different from control (con) at P b 0.05.
Oxygen sensing, redox signaling, and heart failure
Hypoxia results in various biologic events, which might lead to heart failure. For example, under chronic hypoxic conditions such as those that may occur in COPD patients, mean blood pressure of pulmonary circulation rises due to hypoxic vasoconstriction and pulmonary vascular narrowing. Increased pulmonary vascular resistance exerts pressure overload to the right ventricle, resulting in initial concentric hypertrophy followed by dilated cardiomyopathy. During hypoxic vasoconstriction, it is established that pulmonary artery smooth muscle directly serves as an O2-sensing organ and constricts the vessels.
Proposed molecular mechanisms of O2 sensing include mitochondria theory [80] and NAD(P)H oxidase theory [81]. In both mitochondria and NAD(P)H oxidase models of the O2-sensing mechanism, ROS have been proposed to serve to signal the downstream effector molecules including K+ channels which might be important for the acute changes in response to altered oxygen tension. More recently, hemoxygenase-2 has been shown to act as an oxygen sensor, which affects K+ channels [82]. Hypoxia-inducible factor-1 (HIF-1) also plays important roles in more chronic changes in response to altered O2 by regulating gene transcription via the activities of prolyl and asparaginyl hydroxylases [83].
Mechanisms of O2 sensing in carotid body, neuroepithelial body, and pulmonary artery have been well studied. While it is well-established that oxygen sensing which occurs in the carotid body plays a major role in the development of systemic hypertension that is associated with OSA and intermittent hypoxia [84], it is not yet clear how O2 might be sensed to exert other cardiovascular alterations, especially changes in the heart. The direct effects of chronic hypoxia on the heart have been studied in the context of understanding the differences between right and left ventricular hypertrophy. Sharma et al. [85] compared gene expression patterns between right and left ventricles from hypoxia-treated Wistar rats with pulmonary hypertension. They found that, while large changes in gene expression occur in the right ventricle that is subjected to pressure overload as well as hypoxia, altered expressions of genes such as myosin isoforms and pyruvate dehydrogenase kinase-4 were also detected in the left ventricle. Since this study only examined the expression of a selected 9 genes, it is likely that more genes are altered in the heart through hypoxic treatment without exerting pressure overload. Razeghi et al. [86] also reported isoform switches of myosin heavy chain and an increased expression of Nkx2.5 mRNA in left ventricles from hypoxia-treated rats, supporting the notion that the heart can directly sense changes in O2 tension and modulate its gene expression. This is also supported in a study showing distinct biologic events occurring in right and left ventricular hypertrophy induced by chronic hypoxia and norepinephrine infusion [87].
Do cardiac myocytes have O2-sensing mechanisms that might influence the course of the development of cardiac hypertrophy and transition to heart failure? Schumacker and coworkers have shown that cardiac myocytes do sense low oxygen tension via activating the production of ROS from mitochondria [88-90]. Cataldi et al. [91] reported that protein kinase C alpha is involved in sensing intermittent hypoxia in the rat heart. Wright et al. [92] showed that prolyl hydroxylation is the oxygen-sensing mechanism in cardiac myocytes, which in turn regulates HIF-1, heme oxygenase-1, and GLUT1. While the role of NADPH oxidase in O2 sensing in cardiac myocytes has not been reported, this enzyme has been shown to be expressed and play a role in the development of cardiac hypertrophy and in the transition to heart failure [93-100]. The concept that the heart directly senses changes in oxygen tension has recently been supported by results from our laboratory that 20 min of intermittent hypoxia can elicit significant changes in gene expression and protein modifications in mouse hearts [101].
It is not yet clear exactly what the differences might be between cardiac effects of chronic hypoxia and chronic exposures to repeated episodes of hypoxia/reoxygenation, which might occur in OSA patients. A recent study by Fan et al. [102] compared the effects of chronic hypoxic exposure (11% O2 for 24 h/day) and chronic intermittent hypoxia (21% O2,4 min; 11% O2, 4 min for 24 h/day) in neonatal mice. They found a robust right ventricular hypertrophy and cardiac enlargement in mice subjected to chronic hypoxia, but not in chronic intermittent hypoxia. cDNA microarray analyses indicated that the myocardial gene expression patterns are different between chronic hypoxia and chronic intermittent hypoxia, confirming the concept that the reoxygenation phase and, likely, the production of ROS play distinct roles in the cardiac alterations induced by intermittent hypoxia as well as in OSA.
Intermittent hypoxia, characterized by repeated episodes of hypoxia/reoxygenation cycles, likely produces ROS. Substantial evidence now points to the role of oxidant-mediated vascular smooth muscle signal transduction in the promotion of systemic hypertension [70,103-106]. Thus, although one proposed mechanism of OSA-mediated hypertension involves sensing of oxygen by the carotid body [84] and the subsequent activation of the renin–angiotensin II axis [107], ROS produced by hypoxia/reoxygenation cycles might also play a role in the development of systemic hypertension by directly affecting smooth muscle. For example, superoxide can increase calcium signaling in vascular smooth muscle [108] and this ROS has been proposed as an endothelial-derived contracting factor [109]. Further, ROS has been shown to directly promote growth of vascular smooth muscle cells [110]. While the production of ROS during chronic hypoxia has been controversial [111-113], evidence also suggests the roles of ROS in signaling pathways for the development of pulmonary hypertension [114-116].It appears that OSA symptoms are heterogeneous in nature and the resultant consequences differ accordingly. For example, an early study by Bradley et al. [117] identified that 12% of OSA patients had right heart failure and these patients had a substantially lower awake arterial PO2, suggesting that sustained hypoxia activates signals to promote pulmonary hypertension and right ventricular heart failure. In contrast, the majority of the OSA patients do not have daytime hypoxemia, and nighttime intermittent hypoxia appears to elicit systemic hypertension. Fig. 2 depicts two possible mechanisms by which OSA syndrome might result in cardiac hypertrophy and failure through ROS.
Fig. 2.
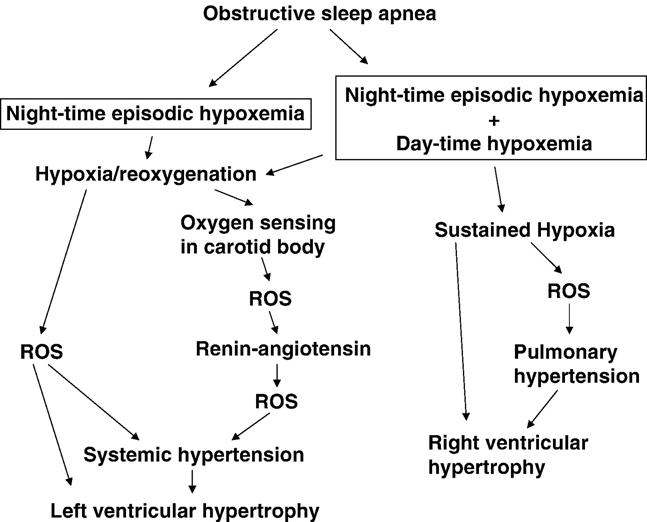
Schematics of the roles of ROS in OSA-mediated cardiac hypertrophy.
Prospective
OSA affects a large number of the adult and pediatric population in the United States and other Western countries. Obesity is a major risk factor for the development of OSA, and the incidence of OSA is expected to increase as the obese population grows. Although CPAP and some forms of surgery can ameliorate some of the causes of upper airway obstruction during sleep, in some cases, apnea episodes are not completely eliminated or patients may choose not to pursue these treatments because of inconvenience or discomfort with CPAP. Moreover, many people, especially in the pediatric population, are unaware of having OSA. Since OSA can cause cardiovascular complications, understanding the mechanisms of OSA actions should help prevent and/or treat detrimental events that are associated with OSA.
One important mechanism by which OSA may cause these long-term pathological effects is intermittent hypoxia, which resembles the event of ischemia/reperfusion injury. Although earlier OSA patient studies failed to provide evidence for oxidative stress, more recent studies demonstrated increased lipid peroxidation and primed ROS generation by blood cells. It should be emphasized that the occurrence of oxidative stress in OSA syndrome is still a controversial subject since some recent studies still failed to demonstrate increased oxidation in OSA patients. Such inconsistencies might be in part due to techniques utilized to measure oxidative stress markers. Techniques used to measure lipid peroxidation so far have been rather crude, and more sophisticated methods should be utilized to accurately monitor lipid peroxidation as well as other forms of oxidative stress markers. Comprehensive studies on antioxidant and oxidative stress status using sensitive biomarker assessment [21,22] should also provide insights to the changes in redox status in OSA patients. Should such measurements indeed demonstrate the occurrence of oxidative stress, whether such changes are due to intermittent hypoxia needs further investigation. Nevertheless, in some studies, CPAP was found to be effective in suppressing OSA-associated changes in redox status (Table 1), providing evidence that apneic events might alter redox biology in OSA patients.
Table 1.
Effects of CPAP treatment on OSA patients
| Decrease lipid peroxidation (Barcelo et al. [26]; Lavie et al. [27]) |
| Decrease superoxide release from stimulated PMNs (Schulz et al. [35]) |
| Decrease ROS production in monocytes (Dyugovskaya et al. [36]) |
| Increase nitric oxide (Ip et al. [45]; Schulz et al. [46]; Lavie et al. [47]) |
| Lower homocysteine (Jordan et al. [66]) |
Animal studies of intermittent hypoxia showed evidence of oxidative stress in brain and more recently in the heart. Further examination of the effects of intermittent hypoxia on oxidative stress status in cardiovascular system such as in the heart and vascular endothelial cells should help determine whether the elimination of oxidative stress should be included to prevent and/or treat OSA-induced cardiovascular complications. In addition to possibly causing oxidative damage, ROS also play important signaling roles for O2 sensing as well as to promote vascular and cardiac hypertrophy. Such oxidant-signaling mechanisms may occur in OSA patients without having obvious changes in measurable oxidative stress status. Future studies may include intervention studies to determine the effects of antioxidants and other redox modulatory agents on OSA-associated complications.
Acknowledgments
This work was supported in part by National Institutes of Health Grants HL67340 (to Y.J.S.), HL72844 (to Y.J.S.), and HL73929 (to R.M.D.). The information in the manuscript is the opinion of the authors and does not reflect the views of the Uniformed Services University of the Health Sciences, the US Department of Defense, or the US Federal Government.
Abbreviations
- CPAP
continuous positive airway pressure
- nCPAP
nasal CPAP
- OSA
obstructive sleep apnea
- ROS
reactive oxygen species
- O2
oxygen
superoxide anion radical
- H2O2
hydrogen peroxide
- • OH
hydroxy radical
- TBARS
thiobarbituric acid-reactive substances
- PMNs
polymorphonuclear neutrophils
- HIF-1
hypoxia-inducible factor-1
References
- 1.Meoli AL, Casey KR, Clark RW, Coleman JA, Jr., Fayle RW, Troell RJ, Iber C. Clinical Practice Review Committee. Hypopnea in sleep-disordered breathing in adults. Sleep. 2001;24:469–470. [PubMed] [Google Scholar]
- 2.Young T, Palta M, Dempsey J, Skatrud J, Weber S, Badr S. The occurrence of sleep-disordered breathing among middle-aged adults. N. Engl. J. Med. 1993;328:1230–1235. doi: 10.1056/NEJM199304293281704. [DOI] [PubMed] [Google Scholar]
- 3.Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S, D'Agostino RB, Newman AB, Lebowitz MD, Pickering TG. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–1836. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- 4.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. New Engl. J. Med. 2000;342:1378–1384. doi: 10.1056/NEJM200005113421901. [DOI] [PubMed] [Google Scholar]
- 5.Shamsuzzaman AS, Gersh BJ, Somers VK. Obstructive sleep apnea: implications for cardiac and vascular disease. JAMA. 2003;290:1906–1914. doi: 10.1001/jama.290.14.1906. [DOI] [PubMed] [Google Scholar]
- 6.Lavie P, Herer P, Hoffstein V. Obstructive sleep apnoea syndrome as a risk factor for hypertension: population study. Br. Med. J. 2000;320:479–482. doi: 10.1136/bmj.320.7233.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qureshi A, Ballard RD. Obstructive sleep apnea. J. Allergy Clin. Immunol. 2003;112:643–651. doi: 10.1016/j.jaci.2003.08.031. [DOI] [PubMed] [Google Scholar]
- 8.Sajkov D, Cowie RJ, Thornton AT, Espinoza HA, McEvoy RD. Pulmonary hypertension and hypoxemia in obstructive sleep apnea syndrome. Am. J. Respir. Crit. Care Med. 1994;149:416–422. doi: 10.1164/ajrccm.149.2.8306039. [DOI] [PubMed] [Google Scholar]
- 9.Peker Y, Kraiczi H, Hedner J, Loth S, Johansson A, Bende M. An independent association between obstructive sleep apnoea and coronary artery disease. Eur. Respir. J. 1999;14:179–184. doi: 10.1034/j.1399-3003.1999.14a30.x. [DOI] [PubMed] [Google Scholar]
- 10.Schafer H, Koehler U, Ewig S, Hasper E, Tasci S, Luderitz B. Obstructive sleep apnea as a risk marker in coronary artery disease. Cardiology. 1999;92:79–84. doi: 10.1159/000006952. [DOI] [PubMed] [Google Scholar]
- 11.Bassetti C, Aldrich MS, Quint D. Sleep-disordered breathing in patients with acute supra- and infratentorial strokes. A prospective study of 39 patients. Stroke. 1997;28:1765–1772. doi: 10.1161/01.str.28.9.1765. [DOI] [PubMed] [Google Scholar]
- 12.Dyken ME, Somers VK, Yamada T, Ren ZY, Zimmerman MB. Investigating the relationship between stroke and obstructive sleep apnea. Stroke. 1996;27:401–407. doi: 10.1161/01.str.27.3.401. [DOI] [PubMed] [Google Scholar]
- 13.Douglas NJ, Engleman HM. CPAP therapy: outcomes and patient use. Thorax. 1998;53:S47–S48. [PMC free article] [PubMed] [Google Scholar]
- 14.Engleman HM, Wild MR. Improving CPAP use by patients with the sleep apnoea/hypopnoea syndrome (SAHS) Sleep Med. Rev. 2003;7:81–99. doi: 10.1053/smrv.2001.0197. [DOI] [PubMed] [Google Scholar]
- 15.Lavie L. Obstructive sleep apnoea syndrome—An oxidative stress disorder. Sleep Med. Rev. 2003;7:35–51. doi: 10.1053/smrv.2002.0261. [DOI] [PubMed] [Google Scholar]
- 16.Dean RT, Wilcox I. Possible atherogenic effects of hypoxia during obstructive sleep apnea. Sleep. 1993;16:S15–S21. doi: 10.1093/sleep/16.suppl_8.s15. [DOI] [PubMed] [Google Scholar]
- 17.Prabhakar NR. Sleep apneas: an oxidative stress? Am. J. Respir. Crit. Care Med. 2002;165:859–860. doi: 10.1164/ajrccm.165.7.2202030c. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki YJ, Ford GD. Redox regulation of signal transduction in cardiac and smooth muscle. J. Mol. Cell. Cardiol. 1999;31:345–353. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 19.Conner EM, Grisham MB. Inflammation, free radicals, and antioxidants. Nutrition. 1996;12:274–277. doi: 10.1016/s0899-9007(96)00000-8. [DOI] [PubMed] [Google Scholar]
- 20.Singal PK, Kapur N, Dhillon KS, Beamish RE, Dhalla NS. Role of free radicals in catecholamine-induced cardiomyopathy. Can. J. Physiol. Pharmacol. 1982;60:1390–1397. doi: 10.1139/y82-207. [DOI] [PubMed] [Google Scholar]
- 21.Griffiths HR, Moller L, Bartosz G, Bast A, Bertoni-Freddari C, Collins A, Cooke M, Coolen S, Haenen G, Hoberg AM, Loft S, Lunec J, Olinski R, Parry J, Pompella A, Poulsen H, Verhagen H, Astley SB. Biomarkers. Mol. Aspects Med. 2002;23:101–208. doi: 10.1016/s0098-2997(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 22.Therond P, Bonnefont-Rousselot D, Davit-Spraul A, Conti M, Legrand A. Biomarkers of oxidative stress: an analytical approach. Curr. Opin. Clin. Nutr. Metab. Care. 2000;3:373–384. doi: 10.1097/00075197-200009000-00009. [DOI] [PubMed] [Google Scholar]
- 23.Wali SO, Bahammam AS, Massaeli H, Pierce GN, Iliskovic N, Singal PK, Kryger MH. Susceptibility of LDL to oxidative stress in obstructive sleep apnea. Sleep. 1998;21:290–296. [PubMed] [Google Scholar]
- 24.Ozturk L, Mansour B, Yuksel M, Yalcin AS, Celikoglu F, Gokhan N. Lipid peroxidation and osmotic fragility of red blood cells in sleep-apnea patients. Clin. Chim. Acta. 2003;332:83–88. doi: 10.1016/s0009-8981(03)00126-8. [DOI] [PubMed] [Google Scholar]
- 25.Christou K, Markoulis N, Moulas AN, Pastaka C, Gourgoulianis KI. Reactive oxygen metabolites (ROMs) as an index of oxidative stress in obstructive sleep apnea patients. Sleep Breath. 2003;7:105–110. doi: 10.1007/s11325-003-0105-9. [DOI] [PubMed] [Google Scholar]
- 26.Barcelo A, Miralles C, Barbe F, Vila M, Pons S, Agusti AG. Abnormal lipid peroxidation in patients with sleep apnea. Eur. Respir. J. 2000;16:644–647. doi: 10.1034/j.1399-3003.2000.16d13.x. [DOI] [PubMed] [Google Scholar]
- 27.Lavie L, Vishnevsky A, Lavie P. Evidence for lipid peroxidation in obstructive sleep apnea. Sleep. 2004;27:123–128. [PubMed] [Google Scholar]
- 28.Tan KC, Chow WS, Lam JC, Lam B, Wong WK, Tam S, Ip MS. HDL dysfunction in obstructive sleep apnea. Atherosclerosis. 2006;184:377–382. doi: 10.1016/j.atherosclerosis.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 29.Christou K, Moulas AN, Pastaka C, Gourgoulianis KI. Antioxidant capacity in obstructive sleep apnea patients. Sleep Med. 2003;4:225–228. doi: 10.1016/s1389-9457(02)00253-8. [DOI] [PubMed] [Google Scholar]
- 30.Jung HH, Han H, Lee JH. Sleep apnea, coronary artery disease, and antioxidant status in hemodialysis patients. Am. J. Kidney Dis. 2005;45:875–882. doi: 10.1053/j.ajkd.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Yamauchi M, Nakano H, Maekawa J, Okamoto Y, Ohnishi Y, Suzuki T, Kimura H. Oxidative stress in obstructive sleep apnea. Chest. 2005;127:1674–1679. doi: 10.1378/chest.127.5.1674. [DOI] [PubMed] [Google Scholar]
- 32.Svatikova A, Wolk R, Lerman LO, Juncos LA, Greene EL, McConnell JP, Somers VK. Oxidative stress in obstructive sleep apnoea. Eur. J. Heart. 2005;26:2435–2439. doi: 10.1093/eurheartj/ehi440. [DOI] [PubMed] [Google Scholar]
- 33.Alzoghaibi MA, Bahammam AS. Lipid peroxides, superoxide dismutase and circulating IL-8 and GCP-2 in patients with severe obstructive sleep apnea: a pilot study. Sleep Breath. 2005;9:119–126. doi: 10.1007/s11325-005-0022-1. [DOI] [PubMed] [Google Scholar]
- 34.Muns G, Rubinstein I, Singer P. Phagocytosis and oxidative burst of granulocytes in the upper respiratory tract in chronic and acute inflammation. J. Otolaryngol. 1995;24:105–110. [PubMed] [Google Scholar]
- 35.Schulz R, Mahmoudi S, Hattar K, Sibelius U, Olschewski H, Mayer K, Seeger W, Grimminger F. Enhanced release of superoxide from polymorphonuclear neutrophils in obstructive sleep apnea. Impact of continuous positive airway pressure therapy. Am. J. Respir. Crit. Care Med. 2000;162:566–570. doi: 10.1164/ajrccm.162.2.9908091. [DOI] [PubMed] [Google Scholar]
- 36.Dyugovskaya L, Lavie P, Lavie L. Increased adhesion molecules expression and production of reactive oxygen species in leukocytes of sleep apnea patients. Am. J. Respir. Crit. Care Med. 2002;165:934–939. doi: 10.1164/ajrccm.165.7.2104126. [DOI] [PubMed] [Google Scholar]
- 37.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic. Biol. Med. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 38.Huang QQ, Fisher SA, Brozovich FV. Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J. Biol. Chem. 2004;279:597–603. doi: 10.1074/jbc.M308496200. [DOI] [PubMed] [Google Scholar]
- 39.Pilz RB, Casteel DE. Regulation of gene expression by cyclic GMP. Circ. Res. 2003;93:1034–1046. doi: 10.1161/01.RES.0000103311.52853.48. [DOI] [PubMed] [Google Scholar]
- 40.Haight JS, Djupesland PG. Nitric oxide (NO) and obstructive sleep apnea (OSA) Sleep Breath. 2003;7:53–62. doi: 10.1007/s11325-003-0053-4. [DOI] [PubMed] [Google Scholar]
- 41.Kato M, Roberts-Thomson P, Phillips BG, Haynes WG, Winnicki M, Accurso V, Somers VK. Impairment of endothelium-dependent vasodilation of resistance vessels in patients with obstructive sleep apnea. Circulation. 2000;102:2607–2610. doi: 10.1161/01.cir.102.21.2607. [DOI] [PubMed] [Google Scholar]
- 42.Saran M, Michel C, Bors W. Reaction of NO with implications for the action of endothelium-derived relaxing factor (EDRF) Free Radic. Res. Commun. 1990;10:221–226. doi: 10.3109/10715769009149890. [DOI] [PubMed] [Google Scholar]
- 43.Olopade CO, Christon JA, Zakkar M, Hua C, Swedler WI, Scheff PA, Rubinstein I. Exhaled pentane and nitric oxide levels in patients with obstructive sleep apnea. Chest. 1997;111:1500–1504. doi: 10.1378/chest.111.6.1500. [DOI] [PubMed] [Google Scholar]
- 44.Agusti AG, Barbe F, Togores B. Exhaled nitric oxide in patients with sleep apnea. Sleep. 1999;22:231–235. [PubMed] [Google Scholar]
- 45.Ip MS, Lam B, Chan LY, Zheng L, Tsang KW, Fung PC, Lam WK. Circulating nitric oxide is suppressed in obstructive sleep apnea and is reversed by nasal continuous positive airway pressure. Am. J. Respir. Crit. Care Med. 2000;162:2166–2171. doi: 10.1164/ajrccm.162.6.2002126. [DOI] [PubMed] [Google Scholar]
- 46.Schulz R, Schmidt D, Blum A, Lopes-Ribeiro X, Lucke C, Mayer K, Olschewski H, Seeger W, Grimminger F. Decreased plasma levels of nitric oxide derivatives in obstructive sleep apnoea: response to CPAP therapy. Thorax. 2000;55:1046–1051. doi: 10.1136/thorax.55.12.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lavie L, Hefetz A, Luboshitzky R, Lavie P. Plasma levels of nitric oxide and L-arginine in sleep apnea patients: effects of nCPAP treatment. J. Mol. Neurosci. 2003;21:57–63. doi: 10.1385/JMN:21:1:57. [DOI] [PubMed] [Google Scholar]
- 48.Teramoto S, Kume H, Matsuse T, Ishii T, Miyashita A, Akishita M, Toba K, Ouchi Y. Oxygen administration improves the serum level of nitric oxide metabolites in patients with obstructive sleep apnea syndrome. Sleep Med. 2003;4:403–407. doi: 10.1016/s1389-9457(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 49.Spieker LE, Noll G, Ruschitzka FT, Maier W, Luscher TF. Working under pressure: the vascular endothelium in arterial hypertension. J. Hum. Hyperten. 2000;14:617–630. doi: 10.1038/sj.jhh.1001012. [DOI] [PubMed] [Google Scholar]
- 50.Schulz R, Seeger W, Grimminger F. Serum nitrite/nitrate levels in obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2001;164:1997–1998. doi: 10.1164/ajrccm.164.10.correspondence_b. [DOI] [PubMed] [Google Scholar]
- 51.Svatikova A, Wolk R, Wang HH, Otto ME, Bybee KA, Singh RJ, Somers VK. Circulating free nitrotyrosine in obstructive sleep apnea. Am. J. Physiol. 2004;287:R284–R287. doi: 10.1152/ajpregu.00241.2004. [DOI] [PubMed] [Google Scholar]
- 52.Carlson J, Hedner J, Pettersson A. Increased plasma concentration of ADMA, a naturally occurring nitric oxide synthesis inhibitor, in OSA patients. Am. J. Respir. Crit. Care Med. 1997;155:A869. [Google Scholar]
- 53.Clarke R, Daly L, Robinson K, Naughten E, Cahalane S, Fowler B, Graham I. Hyperhomocysteinemia: an independent risk factor for vascular disease. N. Engl. J. Med. 1991;324:1149–1155. doi: 10.1056/NEJM199104253241701. [DOI] [PubMed] [Google Scholar]
- 54.Boushey CJ, Beresford SAA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. J. Am. Med. Assoc. 1995;274:1049–1057. doi: 10.1001/jama.1995.03530130055028. [DOI] [PubMed] [Google Scholar]
- 55.Perry IJ, Refsum H, Morris RW, Ueland PM, Shaper AG. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet. 1995;346:1395–1398. doi: 10.1016/s0140-6736(95)92407-8. [DOI] [PubMed] [Google Scholar]
- 56.Selhub J, Jacques PF, Bostom AG, D'agostino RB, Wilson PWF, Belanger AJ, O'Leary DH, Wolf PA, Schaefer EJ, Rosenberg IH. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N. Engl. J. Med. 1995;332:286–291. doi: 10.1056/NEJM199502023320502. [DOI] [PubMed] [Google Scholar]
- 57.Nygard O, Vollset SE, Refsum H, Stensvold I, Tverdal A, Nordrehaug JE, Ueland PM, Kvale G. Total plasma homocysteine and cardiovascular risk profile. The Hordaland homocysteine study. JAMA. 1995;274:1526–1533. doi: 10.1001/jama.1995.03530190040032. [DOI] [PubMed] [Google Scholar]
- 58.McCully KS. Homocysteine and vascular disease. Nature Med. 1996;2:386–389. doi: 10.1038/nm0496-386. [DOI] [PubMed] [Google Scholar]
- 59.Olszewski AJ, McCully KS. Homocysteine metabolism and the oxidative modification of proteins and lipids. Free Radic. Biol. Med. 1993;14:683–693. doi: 10.1016/0891-5849(93)90151-j. [DOI] [PubMed] [Google Scholar]
- 60.Loscalzo J. The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Invest. 1996;98:5–7. doi: 10.1172/JCI118776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi SS, Day RM, Halpner AD, Blumberg JB, Suzuki YJ. Homocysteine and α-lipoic acid regulate p44/42 MAP kinase phosphorylation in NIH/3T3 cells. Antiox. Redox. Signal. 1999;1:123–128. doi: 10.1089/ars.1999.1.1-123. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki YJ, Lorenzi MV, Shi SS, Day RM, Blumberg JB. Homocysteine exerts cell type-specific inhibition of AP-1 transcription factor. Free Radic. Biol. Med. 2000;28:39–45. doi: 10.1016/s0891-5849(99)00200-2. [DOI] [PubMed] [Google Scholar]
- 63.Lavie L, Perelman A, Lavie P. Plasma homocysteine levels in obstructive sleep apnea: association with cardiovascular morbidity. Chest. 2001;120:900–908. doi: 10.1378/chest.120.3.900. [DOI] [PubMed] [Google Scholar]
- 64.Svatikova A, Wolk R, Magera MJ, Shamsuzzaman AS, Phillips BG, Somers VK. Plasma homocysteine in obstructive sleep apnoea. Eur. J. Heart. 2004;25:1325–1329. doi: 10.1016/j.ehj.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 65.Robinson GV, Pepperell JC, Segal HC, Davies RJ, Stradling JR. Circulating cardiovascular risk factors in obstructive sleep apnoea: data from randomised controlled trials. Thorax. 2004;59:777–782. doi: 10.1136/thx.2003.018739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jordan W, Berger C, Cohrs S, Rodenbeck A, Mayer G, Niedmann PD, von Ahsen N, Ruther E, Kornhuber J, Bleich S. CPAP-therapy effectively lowers serum homocysteine in obstructive sleep apnea syndrome. J. Neural. Transm. 2004;111:683–689. doi: 10.1007/s00702-004-0130-2. [DOI] [PubMed] [Google Scholar]
- 67.Row BW, Liu R, Xu W, Kheirandish L, Gozal D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am. J. Respir. Crit. Care Med. 2003;167:1548–1553. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- 68.Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D, Liu R. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- 69.Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, Gow A. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep. 2004;27:194–201. doi: 10.1093/sleep/27.2.194. [DOI] [PubMed] [Google Scholar]
- 70.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 71.Prabhakar NR. Oxygen sensing during intermittent hypoxia: cellular and molecular mechanisms. J. Appl. Physiol. 2001;90:1986–1994. doi: 10.1152/jappl.2001.90.5.1986. [DOI] [PubMed] [Google Scholar]
- 72.Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc. Natl. Acad. Sci. USA. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J. Appl. Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- 74.Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J. Appl. Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- 75.Gopalakrishnan A, Ji LL, Cirelli C. Sleep deprivation and cellular responses to oxidative stress. Sleep. 2004;27:27–35. doi: 10.1093/sleep/27.1.27. [DOI] [PubMed] [Google Scholar]
- 76.Campen MJ, Shimoda LA, O'Donnell CP. Acute and chronic cardiovascular effects of intermittent hypoxia in C57BL/6J mice. J. Appl. Physiol. 2005;99:2028–2035. doi: 10.1152/japplphysiol.00411.2005. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Einbinder E, Zhang Q, Hasday J, Balke WC, Scharf SM. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. Am. J. Respir. Crit. Care Med. 2005;172:915–920. doi: 10.1164/rccm.200504-560OC. [DOI] [PubMed] [Google Scholar]
- 78.Joyeux-Faure M, Stanke-Labesque F, Lefebvre B, Beguin P, Godin-Ribuot D, Ribuot C, Launois SH, Bessard G, Levy P. Chronic intermittent hypoxia increases infarction in the isolated rat heart. J. Appl. Physiol. 2005;98:1691–1696. doi: 10.1152/japplphysiol.01146.2004. [DOI] [PubMed] [Google Scholar]
- 79.Park A-M, Suzuki YJ. Adaptation to chronic intermittent hypoxia in the heart. Proc. Am. Thorac. Soc. 2006 (abstract in press) [Google Scholar]
- 80.Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J. Appl. Physiol. 2000;88:1880–1889. doi: 10.1152/jappl.2000.88.5.1880. [DOI] [PubMed] [Google Scholar]
- 81.Jones RD, Hancock JT, Morice AH. NADPH oxidase: a universal oxygen sensor? Free Radic. Biol. Med. 2000;29:416–424. doi: 10.1016/s0891-5849(00)00320-8. [DOI] [PubMed] [Google Scholar]
- 82.Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- 83.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 84.Fletcher EC, Lesske J, Behm R, Miller CC, III, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J. Appl. Physiol. 1992;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- 85.Sharma S, Taegtmeyer H, Adrogue J, Razeghi P, Sen S, Ngumbela K, Essop MF. Dynamic changes of gene expression in hypoxia-induced right ventricular hypertrophy. Am. J. Physiol. 2004;286:H1185–H1192. doi: 10.1152/ajpheart.00916.2003. [DOI] [PubMed] [Google Scholar]
- 86.Razeghi P, Essop MF, Huss JM, Abbasi S, Manga N, Taegtmeyer H. Hypoxia-induced switches of myosin heavy chain iso-gene expression in rat heart. Biochem. Biophys. Res. Commun. 2003;303:1024–1027. doi: 10.1016/s0006-291x(03)00478-9. [DOI] [PubMed] [Google Scholar]
- 87.Leon-Velarde F, Bourin MC, Germack R, Mohammadi K, Crozatier B, Richalet JP. Differential alterations in cardiac adrenergic signaling in chronic hypoxia or norepinephrine infusion. Am. J. Physiol. 2001;280:R274–R281. doi: 10.1152/ajpregu.2001.280.1.R274. [DOI] [PubMed] [Google Scholar]
- 88.Vanden Hoek TL, Becker LB, Shao Z, Li C, Schumacker PT. Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 1998;273:18092–18098. doi: 10.1074/jbc.273.29.18092. [DOI] [PubMed] [Google Scholar]
- 89.Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J. Biol. Chem. 1998;273:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- 90.Budinger GR, Duranteau J, Chandel NS, Schumacker PT. Hibernation during hypoxia in cardiomyocytes. Role of mitochondria as the O2 sensor. J. Biol. Chem. 1998;273:3320–3326. doi: 10.1074/jbc.273.6.3320. [DOI] [PubMed] [Google Scholar]
- 91.Cataldi A, Bianchi G, Rapino C, Sabatini N, Centurione L, Di Giulio C, Bosco D, Antonucci A. Molecular and morphological modifications occurring in rat heart exposed to intermittent hypoxia: role for protein kinase C alpha. Exp. Gerontol. 2004;39:395–405. doi: 10.1016/j.exger.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 92.Wright G, Higgin JJ, Raines RT, Steenbergen C, Murphy E. Activation of the prolyl hydroxylase oxygen-sensor results in induction of GLUT1, heme oxygenase-1, and nitric-oxide synthase proteins and confers protection from metabolic inhibition to cardiomyocytes. J. Biol. Chem. 2003;278:20235–20239. doi: 10.1074/jbc.M301391200. [DOI] [PubMed] [Google Scholar]
- 93.Hool LC, Di Maria CA, Viola HM, Arthur PG. Role of NAD(P) H oxidase in the regulation of cardiac L-type Ca2+ channel function during acute hypoxia. Cardiovasc. Res. 2005;67:624–635. doi: 10.1016/j.cardiores.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 94.Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J. Gen. Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Privratsky JR, Wold LE, Sowers JR, Quinn MT, Ren J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension. 2003;42:206–212. doi: 10.1161/01.HYP.0000082814.62655.85. [DOI] [PubMed] [Google Scholar]
- 96.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2003;35:851–859. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- 97.Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha (1)-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. 2002;282:C926–C934. doi: 10.1152/ajpcell.00254.2001. [DOI] [PubMed] [Google Scholar]
- 98.Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, Shah AM. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003;41:2164–2171. doi: 10.1016/s0735-1097(03)00471-6. [DOI] [PubMed] [Google Scholar]
- 99.Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 100.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91phox-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 101.Park A-M, Suzuki YJ. Effects of intermittent hypoxia on protein acetylation and sumoylation in the heart. (abstract in press) [Google Scholar]
- 102.Fan C, Iacobas DA, Zhou D, Chen Q, Lai JK, Gavrialov O, Haddad GG. Gene expression and phenotypic characterization of mouse heart after chronic constant or intermittent hypoxia. Physiol. Genomics. 2005;22:292–307. doi: 10.1152/physiolgenomics.00217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Suzuki YJ, Ford GD. Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am. J. Physiol. 1992;262:H114–H116. doi: 10.1152/ajpheart.1992.262.1.H114. [DOI] [PubMed] [Google Scholar]
- 104.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 105.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 107.Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J. Appl. Physiol. 2002;92:627–633. doi: 10.1152/japplphysiol.00152.2001. [DOI] [PubMed] [Google Scholar]
- 108.Suzuki YJ, Ford GD. Superoxide stimulates IP3-induced Ca2+ release from vascular smooth muscle sarcoplasmic reticulum. Am. J. Physiol. 1992;262:H114–H116. doi: 10.1152/ajpheart.1992.262.1.H114. [DOI] [PubMed] [Google Scholar]
- 109.Katusic ZS, Vanhoutte PM. Superoxide anion is an endothelium-derived contracting factor. Am. J. Physiol. 1989;257:H33–H37. doi: 10.1152/ajpheart.1989.257.1.H33. [DOI] [PubMed] [Google Scholar]
- 110.Rao GN, Berk BC. Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 1992;70:593–599. doi: 10.1161/01.res.70.3.593. [DOI] [PubMed] [Google Scholar]
- 111.Archer SL, Huang J, Henry T, Peterson D, Weir EK. A redox-based O2 sensor in rat pulmonary vasculature. Circ. Res. 1993;73:1100–1112. doi: 10.1161/01.res.73.6.1100. [DOI] [PubMed] [Google Scholar]
- 112.Michelakis ED, Hampl V, Nsair A, Wu X, Harry G, Haromy A, Gurtu R, Archer SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res. 2002;90:1307–1315. doi: 10.1161/01.res.0000024689.07590.c2. [DOI] [PubMed] [Google Scholar]
- 113.Waypa GB, Marks JD, Mack MM, Boriboun C, Mungai PT, Schumacker PT. Mitochondrial reactive oxygen species trigger calcium increases during hypoxia in pulmonary arterial myocytes. Circ. Res. 2002;91:719–726. doi: 10.1161/01.res.0000036751.04896.f1. [DOI] [PubMed] [Google Scholar]
- 114.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 115.Lee SL, Simon AR, Wang WW, Fanburg BL. H2O2 signals 5-HT-induced ERK MAP kinase activation and mitogenesis of smooth muscle cells. Am. J. Physiol. 2001;281:L646–L652. doi: 10.1152/ajplung.2001.281.3.L646. [DOI] [PubMed] [Google Scholar]
- 116.Wedgwood S, Black SM. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid. Redox. Signal. 2003;5:759–769. doi: 10.1089/152308603770380061. [DOI] [PubMed] [Google Scholar]
- 117.Bradley TD, Rutherford R, Grossman RF, Lue F, Zamel N, Moldofsky H, Phillipson EA. Role of daytime hypoxemia in the pathogenesis of right heart failure in the obstructive sleep apnea syndrome. Am. Rev. Respir. Dis. 1985;131:835–839. doi: 10.1164/arrd.1985.131.6.835. [DOI] [PubMed] [Google Scholar]