

Abstract
Cardiac apoptosis diminishes the contractile mass, which leads to heart failure. Apoptosis of cardiac non-myocytes also contributes to maladaptive remodeling and the transition to decompensated congestive heart failure. New antiapoptotic interventions and medications will be available within the next decade. The aim of this study is to provide a critical synopsis of research projects on cardiocyte apoptosis that have implications for current and future practice and to identify methods to prevent or attenuate apoptosis in patients who have poor ventricular function.
A retrospective literature review reveals a great many important publications. However, very few investigators discuss the clinical ramifications of cardiocyte apoptosis, nor do they address the clinician who sees poor ventricular contractility daily.
Most studies are still investigational and involve antiapoptotic agents such as broad-spectrum caspase inhibitors, antioxidants, calcium channel blockers, insulin-like growth-factor 1, and poly(adenosine diphosphate ribose) synthetase inhibitors.
Some options have already been incorporated into the clinical practices of cardiologists and cardiac surgeons: repairing or replacing diseased or damaged valves before ventricular function deteriorates; reducing afterload with medication or intra-aortic balloon pulsation in patients who display acute increases in afterload; decreasing catecholamine-induced cardiotoxicity in hemodynamically compromised patients, by using β-blockers and phosphodiesterase inhibitors; and inserting intra-aortic balloon pumps or ventricular assist devices early in cases of failing myocardium. Coronary revascularization early in myocardial infarction is effective antiapoptotic therapy. Other therapeutic targets are cardiopulmonary bypass and aortic cross-clamping, both of which require reductions in associated myocardial apoptosis.
Key words: Apoptosis; cardiomegaly/pathology; heart failure, congestive; myocardial infarction/complications; myocardial ischemia/complications; therapies, investigational; ventricular remodeling
Cardiac failure is the end stage of all heart disease and is a major cause of morbidity and death. National hospital discharge surveys indicate that approximately 4.8 million Americans have heart failure.1 Despite significant advances in the medical and surgical treatment of heart failure, this important challenge remains: during the last 2 decades, congestive heart failure has become an increasingly frequent reason for hospital admission. Clearly, it is a major health problem.
Within the past decade, there has been increasing evidence that apoptosis contributes substantially to the pathogenesis of heart failure. Cardiocyte apoptosis, a morphologically different mode of cell death from necrosis, is an important component of the remodeling process and of the transition from an adaptive myocardial condition to end-stage cardiac failure (Figs. 1 and 2). Cardiac apoptosis is a genetically programmed and energy-requiring process that is executed by a family of ubiquitously expressed cysteine proteases that are termed caspases. Caspases are present in the cell as inactive pro-caspases, which are activated in response to apoptotic stimuli (Fig. 2). An understanding of the physiology of apoptosis and its clinical implications is important for the physician, because the therapeutic options may improve the outcome of patients who have heart failure. In fact, the attenuation and prevention of apoptotic pathways are new modes of therapy for congestive heart failure that will be applied in clinical practice within the next decade.
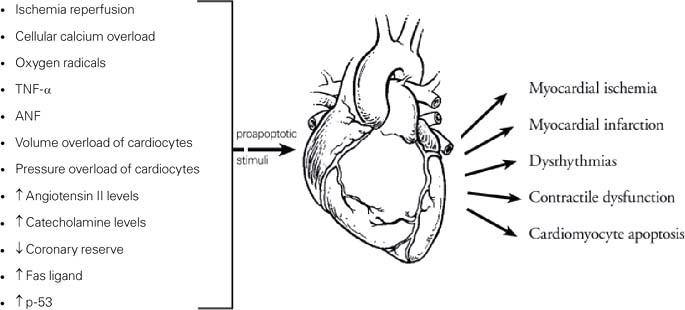
Fig. 1 The most relevant myocardial conditions and agents that induce cardiomyocyte apoptosis are listed (left). These can lead to other clinical entities (right).
ANF = antinuclear factor; TNF-α = tumor necrosis factor-α; p-53 = protein 53
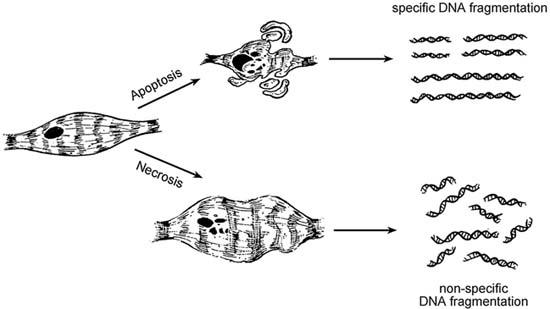
Fig. 2 Early stages of apoptosis are characterized by cell shrinkage and aggregation of chromosomal DNA into small masses and preparation for exocytosis. These apoptotic bodies are membrane bound and are subsequently phagocytosed by macro-phages and neutrophils. Necrosis, on the other hand, is asso-ciated with loss of transmembrane ion gradient and with membrane disruption secondary to depletion of intracellular adenosine tri-phosphate, which causes an inflammatory reaction in surrounding tissue. Specific DNA fragmentation is a hallmark of apoptosis, and it is used as a detection method in “DNA-laddering.”
Cardiocyte apoptosis in heart failure has been the topic of research in many recent studies. However, very few of these articles are of clinical use to the physician who treats patients with congestive heart failure. The aims of this paper are to present a critical review of the studies on antiapoptotic therapy for conditions associated with congestive heart failure, to provide ramifications for physicians who treat patients who have congestive heart failure, and to translate the current knowledge into modern clinical practice, both in surgery and in medicine.
Methods
A literature review that used the Ovid search engine to find “heart failure and apoptosis” and its subheadings yielded 62,008 publications. In order to keep our study concise, we subsequently limited the search to articles in English and focused on studies that involved human beings. The articles were evaluated for their validity, their importance, and their applicability to clinical practice. The detection methods were critically reviewed for methodological accuracy, because detection methods with poor sensitivity can lead to skewed results and incorrect conclusions. The articles were reviewed, and clinically important data were collected and incorporated.
Results
Cardiocyte apoptosis, a part of the phenotype of the failing heart, has been detected in studies of both animals and human beings.2–5 Compensatory mechanisms that are closely associated with failing myocardium are left ventricular (LV) hypertrophy and dilation, enhanced and sustained activity of the renin-angiotensin-aldoster-one system, chronically increased intracellular calcium, and increased sympathetic response. These same mechanisms—considered to be potent inducers of apoptosis—are responsible for cardiac remodeling and for the progression of heart failure.6–8 It is well known, of course, that heart failure can result from the acute loss of cardiocytes in an infarcted area; but not infrequently, heart failure is precipitated by delayed remodeling of the myocardium.
Valvular regurgitation is the leading cause of LV dilatation. The volume overload associated with aortic and mitral regurgitation increases LV end-diastolic pressure and isometric stretching of the myocardium. In 1 prominent study that used a rodent model, the papillary muscle was subjected to 50 mN/mm isometric tension.9 The authors found a 21-fold higher apoptotic index than was associated with baseline tension (7–8 mN/mm).9 This evidence supports and explains the occurrence of cardio-cyte apoptosis in volume-overloaded ventricles. Abbate and coworkers10 studied the role of cardiocyte apoptosis in the genesis of LV dilatation and thinning of non-infarcted myocardium after large transmural myocardial infarction. In this postmortem analysis, the apoptotic index in remote non-infarcted regions correlated strongly with dilatation and thinning of the myocardium.10
Ventricular hypertrophy is another compensatory mechanism associated with heart failure; it can be the result of increased afterload, long-lasting hypertension, and aortic or pulmonic stenosis. Cardiocyte apoptosis has been detected in hypertrophied hearts regardless of the cause of the hypertrophy.11,12 In a rodent model of LV hypertrophy, banded rats convincingly showed a substantially higher apoptotic index than did rats in a control group.12 Using isolated adult human cardiocytes, Kajstura and colleagues7 showed that the apoptotic index rose in cardiocytes treated with angiotensin II (AT-II). In the same study, apoptotic cell death was prevented with angiotensin-converting enzyme (ACE) inhibitors. Angiotensin II activates proapoptotic p-53 transcription,13 induces apoptogenic genes (such as cMyc, c-Fos, and c-Jun), and up-regulates genes that encode for proapoptotic atrial natriuretic factor.14,15 Both ACE inhibitors and AT-II blockers can inhibit this effect and reduce cardiocyte death due to apoptosis. Both agents counteract the effects of AT-II, including increase of afterload, ventricular remodeling, and cardiocytic and endothelial necrosis and apoptosis; they also counteract diastolic heart failure due to fibroblast proliferation and collagen deposition in the LV wall.7,16 There is level-1 evidence supporting the use of ACE inhibitors and AT-II blockers,17,18 especially after surgery on patients who have poorly functioning or dilated ventricles.
Congestive heart failure patients have increased baseline intracellular calcium due to high levels of AT-II and catecholamines.7,14,19 An increase in cellular calcium ions is a proapoptotic stimulus.6 Because calcium channel blockers and sodium–hydrogen exchanger inhibitors reduce cellular calcium (the latter by decreasing cellular sodium), they have the potential to decrease the apoptotic index in cardiocytes at risk. Oral administration of sodium–hydrogen exchanger inhibitors has reduced death, arrhythmias, infarct size, apoptotic index, and Bcl-2/Bax ratio in an ischemic rodent model.20 Although this experiment in rodents is promising, the literature still lacks a convincing study in a human model of heart failure. Calcium channel blockers have reduced the direct toxic effects of norepinephrine by decreasing intracellular calcium,19 but no studies that might show decreased apoptosis in heart failure models have been undertaken. In randomized controlled studies of human heart failure, there is no level-1 evidence that calcium channel blockers are of benefit.21 Because calcium channel blockers do not influence the structural cardiac remodeling process, they fail to improve outcomes of patients with congestive heart failure.22
The level of catecholamine activity, which is elevated in failing myocardium, is associated with direct toxicity and with apoptosis of cardiocytes.8,19,23 In 2 independent studies,5,8 the direct toxic and apoptogenic effects of norepinephrine were abolished with β-adrenergic receptor antagonists, but not with an α-receptor antagonist. The antiapoptotic effects of β-adrenergic receptor antagonists on norepinephrine-induced apoptosis have been studied and have proved true for atenolol and car-vedilol.24,25
In clinical application, β-adrenergic receptor antag-o-nists reduce overall death and hospitalization in congestive heart failure patients.26,27 In patients with low-output syndrome after open-heart surgery, acute myocardial infarction, or other conditions that are associated with increased endogenous or exogenous catecholamine, ino-tropic support of the heart can be accomplished with alternative medications, such as milrinone. Physicians with experience in the insertion of intra-aortic balloon pumps or ventricular assist devices might want to use them in patients who display clinically low output, as a bridge to recovery or as destination therapy. The ReMATCH trial28 has provided us with level-1 evidence for the survival benefits associated with ventricular assist devices. Moreover, the literature29 reveals decreased apoptotic DNA laddering and apoptotic DNA fragmentation after the insertion of a ventricular assist device. de Jonge and associates30 have shown a decreased apoptotic index after unloading the heart with a LV assist device, while Rivello and colleagues31 have documented (by nuclear staining) decreased cardiomyocyte nuclear size and ploidy status. Therefore, timely placement of a ventricular assist device is likely to improve the prospect of myocardial recovery by reducing the apoptotic index.
Myocardial infarction is a prominent inducer of cardiocyte apoptosis and is the leading cause of heart failure. Myocytic loss due to apoptotic cell death during the acute stage of myocardial infarction has been well established in both animal and human studies. In rat hearts, an increase in the apoptotic index was present as early as 3 hours and as late as 1 month after coronary occlusion.32 Apoptotic human cardiocytes have been found chiefly in the hypoperfused border zone between the central infarct area and non-ischemic cardiac tissue.33
Broad-spectrum caspase inhibitors can control the signal pathway of cardiocyte apoptosis. Such antiapoptotic agents have been used successfully in a rodent model of acute myocardial infarction.34,35 Armstrong and co-workers35 found that IDN-6734 reduces the size of myocardial infarction in rats if the poly-caspase inhibitor is given before reperfusion of the left anterior descending artery (47% reduction) or 1 hour after reperfusion (45% reduction). In another rodent study, zVAD.fmk reduced infarction size, improved hemodynamics, and attenuated apoptosis, in comparison with a control group.34
Discussion
Cardiocyte apoptosis is a precisely orchestrated process that is hard-wired into all metazoan cells. Apoptosis contributes to the development of congestive heart failure in at least 2 distinct ways. First, it reduces the number of contractile cardiocytes through programmed cell death. This low, but abnormal, rate of cardiocyte apoptosis amounts to a considerable loss for adult human myocardium, because adult human cardiocytes have lost the replicative potential of neonatal cardiocytes. Second, apoptosis of cardiac non-myocytes can contribute substantially to the progressive nature of failing myocardium: Fujiwara and coworkers36,37 have supported the hypothesis that apoptosis of granulation cells is related to cardiac remodeling after myocardial infarction.
For the reasons mentioned above, the apoptotic sig-naling pathway in cardiocytes and non-cardiocytes is an important factor in the transition from compensated to decompensated heart failure. Antiapoptotic therapeutic interventions offer an appealing platform for devising ways in which to retard the maladaptative growth associated with congestive heart failure.
Numerous myocardial conditions and agents have been identified as inducers of cardiocyte apoptosis (Fig. 1). Table I presents a summary of the most prominent antiapoptotic therapies, which are primarily investigational and without direct application to clinical practice. The use of β-adrenergic receptor antagonists, AT-II blockers, and ACE inhibitors has been associated with decreased apoptotic activity. These medications have proved in randomized controlled studies to be of substantial clinical benefit to heart failure patients. What remains unclear is the specific survival benefit that might be attached to the antiapoptotic characteristics of these medications.
TABLE I. Investigational Options for Treatment of the Failing Myocardium
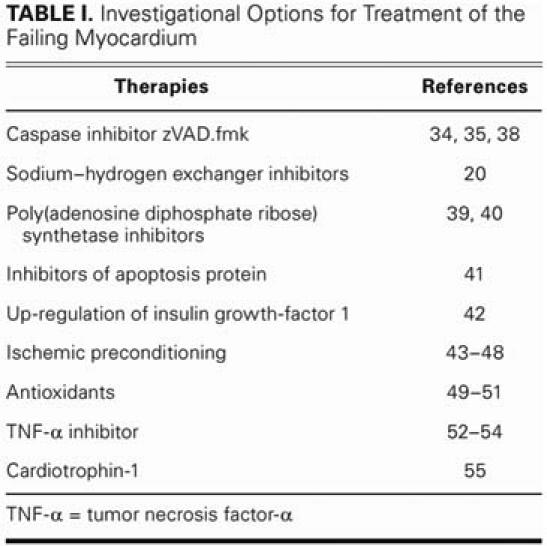
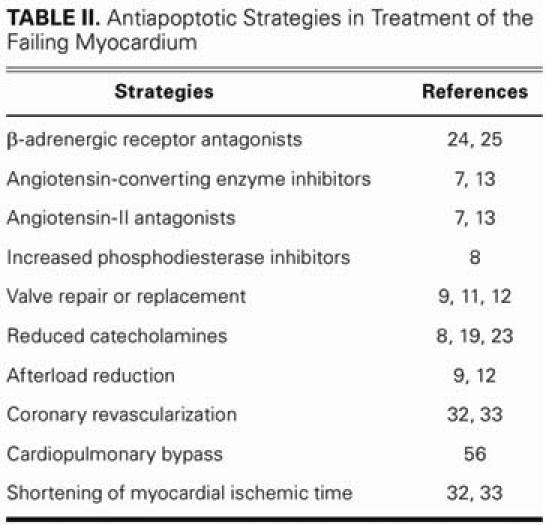
Most antiapoptotic treatment of heart failure is pharmaceutical in nature. The investigations have been led mostly by our nonsurgical colleagues and, therefore, are rarely applicable to the practice of surgeons who treat the heart-failure population. Myocardial infarction, volume and pressure overload, and clinical conditions associated with sustained or acute increases in catecholamine levels are all stimuli of apoptosis that are clinically relevant to the surgeon. The therapeutic ramifications of these events have, in some cases, already been incorporated into the clinical practice of modern heart failure medicine and surgery (Table II). The pertinent preventive measures on the surgical side include reducing myocardial ischemia time with prompt coronary artery revascularization, early valvular repair or replacement (before deterioration of ventricular function), and afterload reduction with intra-aortic balloon pulsation when cardiac function has been compromised after myocardial infarction or ischemic mitral regurgitation.
TABLE II. Antiapoptotic Strategies in Treatment of the Failing Myocardium
The use of cardiopulmonary bypass has been associated with increased apoptotic indices. Blood contact with foreign surfaces raises the serum level of Fas and Fasligand in human beings who undergo cardiopulmonary bypass,56 which in turn increases apoptotic cell death in the myocardium and other organ tissues through the cellular death receptor pathway. Some of the features of post-bypass syndrome, including cerebral dysfunction and renal insufficiency, may be secondary to apoptotic injury to other organs.57,58 Although the apoptosis associated with coronary artery bypass grafting can be avoided by performing the operation without cardio-pulmonary bypass, off-pump processes are not feasible in all currently performed coronary operations or in any other open-heart operation involving valve repair or replacement. However, an important therapeutic goal can still be attained if the pro-inflammatory and proapoptotic state of cardiopulmonary bypass can be ameliorated. Potential therapeutic measures are the reduction of foreign surfaces, the shortening of bypass time, and the addition of antiapoptotic medication to the priming solution in the cardiopulmonary bypass circuit. Myocardial protection itself can be altered to reduce apoptosis. A myocardial protection plan that depends heavily upon hypothermia will result in longer bypass times and increased apoptotic indices. Alternatively, surgeons who use normothermic techniques will achieve faster restoration of cardiac performance, decreased reperfusion time, and less use of catecholamines. Such procedures are beneficial in reducing the apoptotic rate in the failing myocardium.
Reperfusion injury after cardioplegia, another apop-totic state that confronts cardiac surgeons on a daily basis, is another treatment target. Antioxidants, poly(adenosine diphosphate ribose) synthetase inhibitors, and calcium channel blockers may be useful in decreasing reactive oxygen radicals, reducing neutro-phil-mediated inflammatory cascade, and attenuating cellular calcium overload.39,40,49–51 However, all of these agents are investigational for clinical purposes. In order to receive FDA approval for clinical practice, they must be studied and endorsed by a heart failure specialist as a sole treatment, or by a heart failure surgeon as an adjunct to cardioplegic solution or to the cardiopulmonary bypass circuit.
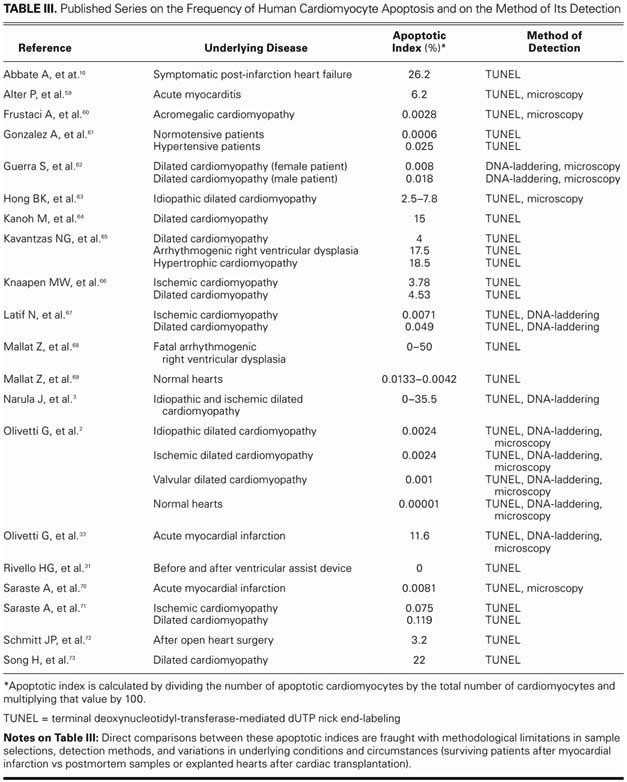
In order to present a critical overview of the current studies on apoptosis in failing myocardium, it is important to understand that our current knowledge about cardiocyte apoptosis has substantial limitations. It is unknown, for example, whether apoptosis is the primary or the secondary event in pathogenic cardiac conditions. Also, further investigation is needed to determine whether inhibition of apoptosis can delay disease progression in animal models and in subsequent clinical applications in human beings. Antiapoptotic pharmaceutical agents have the highest potential to become clinically important as a therapeutic option. These medications could be administered preoperatively as an adjunct to the priming solution of the cardiopulmonary circuit or intraoperatively as an adjunct to the cardioplegic solution. However, the safety and long-term consequences of these therapies have not been adequately investigated. Such therapies could possibly lead to alternative modes of cellular death, such as necrosis, or could increase autoimmune and lymphoproliferative disorders. Also of concern is the imprecision of the techniques used to detect cardiocyte apoptosis. The positive predictive value of terminal deoxynucleotidyl-transferase-mediated dUTP nick end-labeling (TUNEL), which is the method most commonly used to detect apoptosis, is less than satisfactory. The heterogeneity of examined tissue samples can reduce the accuracy of DNA-laddering, because it cannot specify the cell type that is undergoing apoptosis in a tissue sample comprising multiple types of cells. The brevity of apoptosis is another reason for the low sensitivity rates of current diagnostic methods. Indeed, these factors could explain inconsistencies in the reported frequency of human cardiocyte apoptosis (Table III). More accurate detection tools are now under development.
TABLE III. Published Series on the Frequency of Human Cardiomyocyte Apoptosis and on the Method of Its Detection
The prevention or attenuation of cardiocytic apoptosis is a very appealing therapeutic goal in the treatment of congestive heart failure, and as we accumulate more data, the spectrum of therapeutic methods will widen. It is vital for physicians to understand the treatment options and to incorporate them to the extent possible into the practice of modern heart failure medicine and surgery.
Acknowledgment
The authors wish to acknowledge Rita Laire for her help in preparing the manuscript.
Footnotes
Address for reprints: Ali Khoynezhad, MD, PhD, Section of Cardiothoracic Surgery, University of Nebraska Medical Center, 982315 Nebraska Medical Center, Omaha, NE 68198–2315. E-mail: akhoynezhad@unmc.edu
References
- 1.Heart Disease and Stroke Statistics 2004 Update [Internet]. American Heart Association. Dallas: American Heart Asso-ciation; 2003. Available from: http://www.americanheart.org/downloadable/heart/1079736729696HDSStats2004UpdateREV3-19-04.pdf System Requirements: Adobe Acrobat.
- 2.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med 1997;336:1131–41. [DOI] [PubMed]
- 3.Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996;335:1182–9. [DOI] [PubMed]
- 4.Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M, Goldstein S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am J Pathol 1996; 148:141–9. [PMC free article] [PubMed]
- 5.Yao M, Keogh A, Spratt P, dos Remedios CG, Kiessling PC. Elevated DNase I levels in human idiopathic dilated cardiomyopathy: an indicator of apoptosis? J Mol Cell Cardiol 1996; 28:95–101. [DOI] [PubMed]
- 6.Orrenius S, McConkey DJ, Bellomo G, Nicotera P. Role of Ca2+ in toxic cell killing. Trends Pharmacol Sci 1989;10:281–5. [DOI] [PubMed]
- 7.Kajstura J, Cigola E, Malhotra A, Li P, Cheng W, Meggs LG, Anversa P. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J Mol Cell Cardiol 1997;29:859–70. [DOI] [PubMed]
- 8.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta-adrenergic pathway. Circulation 1998;98:1329–34. [DOI] [PubMed]
- 9.Cheng W, Li B, Kajstura J, Li P, Wolin MS, Sonnenblick EH, et al. Stretch-induced programmed myocyte cell death. J Clin Invest 1995;96:2247–59. [DOI] [PMC free article] [PubMed]
- 10.Abbate A, Biondi-Zoccai GG, Bussani R, Dobrina A, Camilot D, Feroce F, et al. Increased myocardial apoptosis in patients with unfavorable left ventricular remodeling and early symptomatic post-infarction heart failure. J Am Coll Cardiol 2003;41:753–60. [DOI] [PubMed]
- 11.Li Z, Bing OH, Long X, Robinson KG, Lakatta EG. Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. Am J Physiol 1997;272 (5 Pt 2):H2313–9. [DOI] [PubMed]
- 12.Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, et al. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest 1996;97:2891–7. [DOI] [PMC free article] [PubMed]
- 13.Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, et al. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest 1998;101:1326–42. [DOI] [PMC free article] [PubMed]
- 14.Koglin J, Granville DJ, Glysing-Jensen T, Mudgett JS, Carthy CM, McManus BM, Russell ME. Attenuated acute cardiac rejection in NOS2 -/- recipients correlates with reduced apoptosis. Circulation 1999;99:836–42. [DOI] [PubMed]
- 15.Jiang L, Huang Y, Yuasa T, Hunyor S, dos Remedios CG. Elevated DNase activity and caspase expression in association with apoptosis in failing ischemic sheep left ventricles. Electrophoresis 1999;20:2046–52. [DOI] [PubMed]
- 16.Weber KT. Extracellular matrix remodeling in heart failure: a role for de novo angiotensin II generation. Circulation 1997;96:4065–82. [DOI] [PubMed]
- 17.Garg R, Yusuf S. Overview of randomized trials of angio-tensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials [published erratum appears in JAMA 1995;274:462]. JAMA 1995;273:1450–6. [PubMed]
- 18.Sharma D, Buyse M, Pitt B, Rucinska EJ. Meta-analysis of observed mortality data from all-controlled, double-blind, multiple-dose studies of losartan in heart failure. Losartan Heart Failure Mortality Meta-analysis Study Group. Am J Cardiol 2000;85:187–92. [DOI] [PubMed]
- 19.Mann DL, Kent RL, Parsons B, Cooper G 4th. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation 1992;85:790–804. [DOI] [PubMed]
- 20.Humphreys RA, Haist JV, Chakrabarti S, Feng Q, Arnold JM, Karmazyn M. Orally administered NHE1 inhibitor cariporide reduces acute responses to coronary occlusion and reperfusion. Am J Physiol 1999;276(2 Pt 2):H749–57. [DOI] [PubMed]
- 21.Packer M, O'Connor CM, Ghali JK, Pressler ML, Carson PE, Belkin RN, et al. Effect of amlodipine on morbidity and mortality in severe chronic heart failure. Prospective Randomized Amlodipine Survival Evaluation Study Group. N Engl J Med 1996;335:1107–14. [DOI] [PubMed]
- 22.Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. In behalf of an In-ternational Forum on Cardiac Remodeling. J Am Coll Car-diol 2000;35:569–82. [DOI] [PubMed]
- 23.Colucci WS, Sawyer DB, Singh K, Communal C. Adrenergic overload and apoptosis in heart failure: implications for therapy. J Card Fail 2000;6(2 Suppl 1):1–7. [PubMed]
- 24.Rossig L, Haendeler J, Mallat Z, Hugel B, Freyssinet JM, Tedgui A, et al. Congestive heart failure induces endothelial cell apoptosis: protective role of carvedilol. J Am Coll Cardiol 2000;36:2081–9. [DOI] [PubMed]
- 25.Romeo F, Li D, Shi M, Mehta JL. Carvedilol prevents epi-nephrine-induced apoptosis in human coronary artery endo-thelial cells: modulation of Fas/Fas ligand and caspase-3 pathway. Cardiovasc Res 2000;45:788–94. [DOI] [PubMed]
- 26.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. U.S. Carvedilol Heart Failure Study Group. N Engl J Med 1996;334:1349–55. [DOI] [PubMed]
- 27.Lechat P, Packer M, Chalon S, Cucherat M, Arab T, Boissel JP. Clinical effects of beta-adrenergic blockade in chronic heart failure: a meta-analysis of double-blind, placebo-controlled, randomized trials. Circulation 1998;98:1184–91. [DOI] [PubMed]
- 28.Rose EA, Moskowitz AJ, Packer M, Sollano JA, Williams DL, Tierney AR, et al. The REMATCH trial: rationale, design, and end points. Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure. Ann Thorac Surg 1999;67:723–30. [DOI] [PubMed]
- 29.Bartling B, Milting H, Schumann H, Darmer D, Arusoglu L, Koerner MM, et al. Myocardial gene expression of regulators of myocyte apoptosis and myocyte calcium homeostasis during hemodynamic unloading by ventricular assist devices in patients with end-stage heart failure. Circulation 1999;100(19 Suppl):II216–23. [DOI] [PubMed]
- 30.de Jonge N, van Wichen DF, van Kuik J, Kirkels H, Lahpor JR, Gmelig-Meyling FH, et al. Cardiomyocyte death in patients with end-stage heart failure before and after support with a left ventricular assist device: low incidence of apoptosis despite ubiquitous mediators. J Heart Lung Transplant 2003; 22:1028–36. [DOI] [PubMed]
- 31.Rivello HG, Meckert PC, Vigliano C, Favaloro R, Laguens RP. Cardiac myocyte nuclear size and ploidy status decrease after mechanical support. Cardiovasc Pathol 2001;10:53–7. [DOI] [PubMed]
- 32.Cheng W, Kajstura J, Nitahara JA, Li B, Reiss K, Liu Y, et al. Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res 1996;226:316–27. [DOI] [PubMed]
- 33.Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bona-cina E, et al. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 1996;28: 2005–16. [DOI] [PubMed]
- 34.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 1998;97:276–81. [DOI] [PubMed]
- 35.Armstrong RC, Li F, Smiley R, Miao W, Chen W, Hanks M, et al. Caspase inhibitors reduce infarct size when dosed post-reperfusion in a rodent cardiac ischemia/reperfusion model [abstract]. Circulation 2001;104(Suppl):II–12.
- 36.Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, et al. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res 1998;82:1130–8. [DOI] [PubMed]
- 37.Hayakawa K, Takemura G, Kanoh M, Li Y, Koda M, Kawase Y, et al. Inhibition of granulation tissue cell apoptosis during the subacute stage of myocardial infarction improves cardiac remodeling and dysfunction at the chronic stage. Circulation 2003;108:104–9. [DOI] [PubMed]
- 38.Abbate A, Biondi-Zoccai GG, Baldi A. Pathophysiologic role of myocardial apoptosis in post-infarction left ventricular remodeling. J Cell Physiol 2002;193:145–53. [DOI] [PubMed]
- 39.Yaoita H, Ogawa K, Maehara K, Maruyama Y. Apoptosis in relevant clinical situations: contribution of apoptosis in myocardial infarction. Cardiovasc Res 2000;45:630–41. [DOI] [PubMed]
- 40.Thiemermann C, Bowes J, Myint FP, Vane JR. Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia-reperfusion injury in the heart and skeletal muscle. Proc Natl Acad Sci U S A 1997;94:679–83. [DOI] [PMC free article] [PubMed]
- 41.Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev 1999;13:239–52. [DOI] [PubMed]
- 42.Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, et al. Overexpression of insulin-like growth factor-1 in mice protects from myocyte death after infarction, attenuating ventricular dilation, wall stress, and cardiac hypertrophy. J Clin Invest 1997;100:1991–9. [DOI] [PMC free article] [PubMed]
- 43.Yellon DM, Alkhulaifi AM, Pugsley WB. Preconditioning the human myocardium. Lancet 1993;342:276–7. [DOI] [PubMed]
- 44.Przyklenk K, Kloner RA. Ischemic preconditioning: exploring the paradox. Prog Cardiovasc Dis 1998;40:517–47. [DOI] [PubMed]
- 45.Brocheriou V, Hagege AA, Oubenaissa A, Lambert M, Mallet VO, Duriez M, et al. Cardiac functional improvement by a human Bcl-2 transgene in a mouse model of ischemia/reperfusion injury. J Gene Med 2000;2:326–33. [DOI] [PubMed]
- 46.Maulik N, Engelman RM, Rousou JA, Flack JE 3rd, Deaton D, Das DK. Ischemic preconditioning reduces apoptosis by upregulating anti-death gene Bcl-2. Circulation 1999;100(19 Suppl):II369–75. [DOI] [PubMed]
- 47.Jenkins DP, Pugsley WB, Alkhulaifi AM, Kemp M, Hooper J, Yellon DM. Ischaemic preconditioning reduces troponin T release in patients undergoing coronary artery bypass surgery. Heart 1997;77:314–8. [DOI] [PMC free article] [PubMed]
- 48.Perrault LP, Menasche P, Bel A, de Chaumaray T, Peynet J, Mondry A, et al. Ischemic preconditioning in cardiac surgery: a word of caution. J Thorac Cardiovasc Surg 1996;112:1378–86. [DOI] [PubMed]
- 49.Oskarsson HJ, Coppey L, Weiss RM, Li WG. Antioxidants attenuate myocyte apoptosis in the remote non-infarcted myocardium following large myocardial infarction. Cardiovasc Res 2000;45:679–87. [DOI] [PubMed]
- 50.Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc Res 2000;47:446–56. [DOI] [PubMed]
- 51.Zhao ZQ, Nakamura M, Wang NP, Wilcox JN, Shearer S, Ronson RS, et al. Reperfusion induces myocardial apoptotic cell death. Cardiovasc Res 2000;45:651–60. [DOI] [PubMed]
- 52.Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenar-do MJ. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995;377:348–51. [DOI] [PubMed]
- 53.Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest 1996; 98: 2854–65. [DOI] [PMC free article] [PubMed]
- 54.Haunstetter A, Izumo S. Future perspectives and potential implications of cardiac myocyte apoptosis. Cardiovasc Res 2000; 45:795–801. [DOI] [PubMed]
- 55.Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR. Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy. J Biol Chem 1997; 272:5783–91. [DOI] [PubMed]
- 56.Kawahito K, Misawa Y, Fuse K. Transient rise in serum soluble Fas (APO-1/CD95) in patients undergoing cardiac surgery. Artif Organs 2000;24:628–31. [DOI] [PubMed]
- 57.Baumgartner WA, Walinsky PL, Salazar JD, Tseng EE, Brock MV, Doty JR, et al. Assessing the impact of cerebral injury after cardiac surgery: will determining the mechanism reduce this injury? Ann Thorac Surg 1999;67:1871–3; discussion 1891–4. [DOI] [PubMed]
- 58.Meldrum DR, Donnahoo KK. Role of TNF in mediating renal insufficiency following cardiac surgery: evidence of a postbypass cardiorenal syndrome. J Surg Res 1999;85:185–99. [DOI] [PubMed]
- 59.Alter P, Jobmann M, Meyer E, Pankuweit S, Maisch B. Apop–tosis in myocarditis and dilated cardiomyopathy: does enterovirus genome persistence protect from apoptosis? An endomyocardial biopsy study. Cardiovasc Pathol 2001;10: 229–34. [DOI] [PubMed]
- 60.Frustaci A, Chimenti C, Setoguchi M, Guerra S, Corsello S, Crea F, et al. Cell death in acromegalic cardiomyopathy. Circulation 1999;99:1426–34. [DOI] [PubMed]
- 61.Gonzalez A, Lopez B, Ravassa S, Querejeta R, Larman M, Diez J, Fortuno MA. Stimulation of cardiac apoptosis in essential hypertension: potential role of angiotensin II. Hypertension 2002;39:75–80. [DOI] [PubMed]
- 62.Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA, et al. Myocyte death in the failing human heart is gender dependent. Circ Res 1999;85:856–66. [DOI] [PubMed]
- 63.Hong BK, Kwon HM, Byun KH, Kim D, Choi EY, Kang TS, et al. Apoptosis in dilated cardiomyopathy. Korean J Intern Med 2000;15:56–64. [DOI] [PMC free article] [PubMed]
- 64.Kanoh M, Takemura G, Misao J, Hayakawa Y, Aoyama T, Nishigaki K, et al. Significance of myocytes with positive DNA in situ nick end-labeling (TUNEL) in hearts with dilated cardiomyopathy: not apoptosis but DNA repair. Circulation 1999;99:2757–64. [DOI] [PubMed]
- 65.Kavantzas NG, Lazaris AC, Agapitos EV, Nanas J, Davaris PS. Histological assessment of apoptotic cell death in cardiomyopathies. Pathology 2000;32:176–80. [PubMed]
- 66.Knaapen MW, Davies MJ, De Bie M, Haven AJ, Martinet W, Kockx MM. Apoptotic versus autophagic cell death in heart failure. Cardiovasc Res 2001;51:304–12. [DOI] [PubMed]
- 67.Latif N, Khan MA, Birks E, O'Farrell A, Westbrook J, Dunn MJ, Yacoub MH. Upregulation of the Bcl-2 family of proteins in end stage heart failure. J Am Coll Cardiol 2000;35:1769–77. [DOI] [PubMed]
- 68.Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 1996;335:1190–6. [DOI] [PubMed]
- 69.Mallat Z, Fornes P, Costagliola R, Esposito B, Belmin J, Lecomte D, Tedgui A. Age and gender effects on cardiomyocyte apoptosis in the normal human heart. J Gerontol A Biol Sci Med Sci 2001;56:M719–23. [DOI] [PubMed]
- 70.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation 1997;95:320–3. [DOI] [PubMed]
- 71.Saraste A, Pulkki K, Kallajoki M, Heikkila P, Laine P, Mattila S, et al. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Invest 1999;29:380–6. [DOI] [PubMed]
- 72.Schmitt JP, Schroder J, Schunkert H, Birnbaum DE, Aebert H. Role of apoptosis in myocardial stunning after open heart surgery. Ann Thorac Surg 2002;73:1229–35. [DOI] [PubMed]
- 73.Song H, Conte JV Jr, Foster AH, McLaughlin JS, Wei C. Increased p53 protein expression in human failing myocardium. J Heart Lung Transplant 1999;18:744–9. [DOI] [PubMed]