

Abstract
Rheumatoid arthritis and its animal model, collagen-induced arthritis, are known as a T and B cell dependent disease. To analyze the role of B cells in arthritis, we generated B cell deficient (muMT) mice carrying HLA-DQ8 as transgene, Aβo.DQ8.μmt mice. HLA-DQ8 transgenic mice (Aβo.DQ8) are susceptible to collagen induced arthritis, an animal model for inflammatory arthritis. Deletion of IgM gene led to the absence of B cells while T cells were comparable to Aβo.DQ8 mice. Arthritis and autoantibodies was completely abrogated in B cell deficient DQ8 mice. T cell response and proinflammatory cytokine production in response to type II collagen and its derived peptides in vitro was significantly decreased despite an increased number of Mac-1 positive cells in DQ8.μmt mice compared to DQ8 mice suggesting B cells could be important for antigen presentation as well. In vitro substitution of B cells from wild type mice restored the response in DQ8.umt mice. B cells could also present CII-derived peptides to antigen-specific DQ8-restricted hybridomas reinforcing the role of B cells in presentation of antigens to T cells. The data suggest that B cells can be involved in pathogenesis of arthritis by producing autoantibodies and antigen presentation.
Keywords: B cells, transgenic/knockout mice, antigen presentation, rheumatoid arthritis, autoantibodies
Introduction
Rheumatoid arthritis (RA) is an autoimmune disorder characterized by synovial inflammation, erosion of bone and cartilage leading to destruction of joints. Genetics studies have demonstrated a strong correlation of RA with certain human Major Histocompatibility Complex (MHC) class II alleles, coding for molecules which present antigenic peptides to T cells (Goldstein and Arnett, 1987, Nepom, 1989). Published data is consistent with an involvement of T cells; especially CD4+ T cells, in pathogenesis of RA ( Strober and Hplpshitz, 1990, Gonzalez-Quintial et al, 1996). However, in synovial tissue of RA patients, both T and B cell infiltrates are observed (VanVoxel and Paget, 1975, Kim et al, 1999). B cells could contribute towards pathogenesis by 1) producing autoantibodies directed to self-antigen such as rheumatoid factor, anti-CII antibodies and anti-citrullinated antibodies 2) presenting antigen and activation of T cells through expression of costimulatory molecules and 3) by producing inflammatory cytokines like TNF-α (Bazzoni and Beutler, 1996, Lanzavechhia, 1987, Nielen et al, 2004, Panayi 2005, Silverman and Carson, 2003, Vidard et al, 1996). However, the role of B cells in presenting antigenic peptides to naïve CD4+ T cells is controversial (Constant et al, 1995, Kurt Jones et al, 1988, Ron and Sprent, 1987). Recent studies suggest that B cells can act as APCs by internalizing antigen through BCR or by formation of immune complexes and their internalization through FcgR expressed on professional APCs (Hamano et al, 2000, Amigorena et al, 1992, Amigorena et al, 1998).
A critical role of B cells has been demonstrated in several models of autoimmune diseases that include lupus, arthritis and diabetes (Falcone et al,1998, Serreze et al, 1998, Chan et al 1999, Shlomichik et al 1994, Svensson et al, 1998, Matsumoto et al, 2002). In experimental model of arthritis transfer of autoantibodies can induce transient and mild arthritis (Stuart and Dixon, 1983, Wooley et al, 1984). Using B cell deficient mice, it was shown that Balb/c mice were resistant to proteoglycan-induced arthritis (O’Neil et al, 2005) suggesting that B cells are required as antigen presenting cells for inducing arthritis. Similarly in other autoimmune disease models like lupus and diabetes a potential role of B cells as antigen presenting cells for T cell activation has been demonstrated (Serreze et al, 1998, Chan et al, 1999).
Collagen-induced arthritis is an animal model for rheumatoid arthritis and is known to involve both T and B cell dependent mechanism for pathogenesis and is dependent on MHC polymorphism (Trentham et al, 1977, Wooley et al, 1981). The advent of mouse class II knockout mice expressing human HLA-DR and HLA-DQ transgenes has significantly advanced the understanding of the role of individual HLA class II molecules in various clinical conditions including RA.
In this study, we investigated the role of B cells as antigen presenting cells and in initiation of CIA in transgenic mice expressing a RA associated HLA class II molecule, DQ8 (DQA1*0301, DQB1*0302). Previous studies from our laboratory have shown that Aβo.DQ8 mice elicit a vigorous CD4 mediated, DQ8 restricted cellular response following immunization using CII which progresses to a severe form of CIA (Taneja et al, 2002). To investigate the role of B cells in CIA in Aβo.DQ8 mice, we generated Aβo.DQ8 mice deficient in B cells by mating DQ8 mice with mice lacking B cells due to the deletion of IgM heavy chain gene (μmt). In vivo studies showed that DQ8.umt mice are resistant to develop collagen-induced arthritis and do not produce anti-CII antibodies. In vitro, T cell response to CII and its derived peptide was also lower in DQ8.umt mice than DQ8 mice. Reconstitution of B cells from wild type mice could restore the in vitro response of T cells from DQ8.umt mice suggesting B cells can function as antigen presenting cells in this model. The antigen presenting capacity of B cells was compared with professional antigen presenting cells like dendritic cells by in vtro expansion of DQ8-restricted CII-specific hybridomas. This study suggests that both functions of B cells, antigen presentation and antibody production, can contribute to the development of the autoimmune disease.
Material and Methods
Mice
μmt mice, lacking μ chain, (Jackson Laboratories) were mated with Aβo mice.Aβo.DQ8. μmt deficient was generated by mating Aβo.DQ8 mice, described previously (Taneja et al, 2002), with Aβo. μmt mice. For all the groups, parental mice and negative littermates were included as controls. All mice used in this study lack endogenous class II molecules (Aβo). All mice were bred in pathogen-free facility and maintained in a clean conventional colony in Immunogenetics Mouse Colony at the Mayo Clinic (Rochester, MN). All experiments were performed with the approval of Institutional Animal Care and Use Committee. Experimental mice represented both sexes and were 8−12 weeks when immunized with collagen.
Flow cytometry
All mice were typed for phenotypic expression of DQ8 molecules. Expression of HLA -DQ, H2A, and T-cell receptor Vβ chains molecules on peripheral blood lymphocytes (PBLs) was analyzed by flow cytometry using FACS IV (Beckton Dickinson) as described earlier (30). Antibodies used for staining are IVD12 (anti DQB1), HB163 (anti-Ab), GK1.5 (anti-CD4), 53.6.8 (anti CD8), MR9−4(anti Vβ5.1), MR9−8 (anti Vβ5.1.2), 44−22−1(anti Vβ6), F23.1 (anti Vβ8.1.2.3), KJ-16 (anti Vβ8.1.2), F23.2 (anti Vβ8.2), KT11 (antiVβ11). Splenic or Lymph node cells were stained for CD3, CD4, CD8, Mac-1, B220, CD95 and costimulatory molecules CD28, CD40 using specific antibodies and analyzing by Flow cytometry.
Induction and evaluation of CIA
Pure native bovine type II collagen (BII) was obtained by multiple step purification described previously (Griffiths et al, 1981). To induce CIA, 8−12 weeks old transgene positive mice and negative littermates were immunized with 100 μg of CII emulsified 1:1 with complete Freunds’ adjuvant H37Ra (CFA, Difco Laboratories, Detroit, MI) intradermally at the base of the tail. Animals received a booster of 100 μg of CII emulsified in incomplete Freunds’ adjuvant 28 days later. Mice were monitored for the onset and progression of CIA from 3−14 weeks postimmunization. The arthritic severity of mice was evaluated as described previously with a grading system for each paw from 0−3 (Griffiths et al, 1994). The mean arthritic score was determined using arthritic animals only.
Histopathology
Mice were sacrificed after 10−12 weeks of immunization, paws decalcified and fixed. Sections were stained with H& E and examined histologically for mononuclear infiltration and bone erosion.
Anti-collagen Abs
Levels of anti-bovine and anti-mouse CII IgG were detected in sera obtained 35 days following CII immunization by ELISA (Taneja et al, 2002). Briefly, microtiter plates were coated overnight with CII (6 μg/ well in KPO4, pH 7.6) at 4°C, washed and blocked with 1% BSA in PBS/0.05% Tween 20. Sera were added in fourfold dilution (1/100−1/64000) and incubated overnight at 4°C. The plates were washed and peroxidase-conjugated goat anti-mouse IgG (Organon Teknika, West Chester, PA) was added for another overnight incubation at 4°C. After washing, O-phenylenediamine was added and the colorimetric change was measured at 410 nm. All assays were performed in duplicate and were quantified against a standard curve obtained with known positive sera, arbitrarily determined to equal 100 Ab units (U/ml).
Adoptive Transfer
B Cells from spleen of immunized DQ8 mice were isolated and washed and transferred to DQ8.umt mice immunized with CII. The study utilized 6 recipient mice receiving a total of 6−8×106 cells/mouse from donor DQ8 mice. Three mice received only sera from immunized DQ8 mice.
T cell hybridoma
Lymph node cells were harvested from peptide immunized mice. 60−100 million cells are resuspended in 10−15 mls of RPMI 1640 containing 5% horse serum supplemented with 1% anti-CD3 monoclonal antibody 500A2. After twenty-four hours of incubation the cells were harvested, washed and counted. An equal number of activated lymph node cells and BW5147.1100 cells were mixed together and pelleted. The pellet was resuspended in 1 ml of 50% PEG 1540 solution for 1 min. Five mls of medium was added to the tube and the fused cell preparation was centrifuged. The pellet was resuspended in 50 mls of culture medium. One hundred μl aliquots were added to microtiter wells (5−96 wells). Twenty-four hours later 100 μl of HAT was added. Every 2−3 days half of the medium was replaced with fresh HAT containing medium. Those wells exhibiting signs of hybridoma growth are transferred to larger wells or flasks. Antigen presentation assays were done to confirm antigen specificity.
Isolation of B cells and Dendritic cells
Spleen cells were harvested for isolation of B and dendritic cells. Dendritic and B cells were isolated by first labeling cells with micro beads coupled to CD11c and CD45R and using columns to separate the targeted cells (Miltenyi Biotech, Auburn, CA). Cells were sorted by automated cells sorter (Automacs). Cells with 99% or more purity were utilized for experiments.
Antigen presentation
Mice were immunized with 200μg of CII emulsified 1:1 with CFA (Difco). Ten days postimmunization, draining popliteal, caudal and lumbar lymph nodes were removed and prepared for in vitro culture. LNCs (1×106) were cultured in HEPES-buffered RPMI 1640 containing 5% heat inactivated horse serum and antibiotics streptomycin and penicillin in 96-well flat bottom tissue culture plates. Cells were challenged by adding 100μl of RPMI medium (negative control), Con A (20 μg/ml, positive control) and native collagen (50 μg/ml). For inhibition experiments, culture supernatant containing specific mAb (25μg/ml of antibody) GK1.5 (anti-CD4), IVD12 (anti-DQ) or Lyt2 (anti-CD8) was added to the cells challenged in vitro with CII at 50μg/ml. The cells were incubated for 48 h at 37° C. During the last 18 h the cells were pulsed with 3H-thymidine (1μCi/well). At the end of the assay, the cells were harvested using a plate harvester and incorporated radioactivity was determined using an automated counter (Micrebeta, Perkin Elmer Wallac).
Peptides derived from human type II collagen (HII) were synthesized and purified at Mayo Clinic Peptide Facility. The mice were primed with 200 μg of peptide emulsified 1:1 with CFA and challenged in vitro with 100 μg/ml of the peptide. To determine the restriction molecule for the in vitro response, blocking studies using anti HLA-DQ antibodies were performed. HII derived peptide 554−573 was used to assess the presentation of antigen by sorted B cells and dendritic cells to T cell hybridoma specific for the peptide. Also, B and DCs were incubated with peptides HII-44 and 44S for 24 hrs and then washed and cultured in the presence of hybridomas without the peptides.
Peptide ELISA
For B cell epitopes, sera from CII and PBS immunized mice were studied for reactivity to CII-derived peptides by ELISA. Peptides utilized were of various cyanogen bromide (CB) fragments of Human type II collagen (HII) and represented these sequences; CB6 fragment (F=44−63, G=54−73, K=94−113) CB11 fragment, (7=184−203, 17=184−303) CB8 fragment (33=444−463, 38=494−513) CB10 fragment (43=544−563, 44=554−573, 48=594−613) and CB9/7 fragment (61=724−743, 64=754−773). Briefly, Maxisorp (NUNC) plates were coated with peptides at a concentration of 10 μg/ml in 0.1M NaHCO3 buffer with a reaction volume of 100μl at 4o C overnight overnight. The non-specific sites were blocked 300ul of 3%BSA for 1hr at room temperature. Duplicate set of sera were used at a dilution of 1:100 and incubated at 4o C overnight. Plates were washed 3 times with 0.5%Tween 20 and incubated for one hr at room temperature with 100ul of anti mouse Alkaline Phophatase (Jackson Immunoresearch). Plates were washed 5 times and developed using PNPP substrate tablets (Southern Biotech). Reaction was stopped with 1M NaOH and plate read at 405nm.
Cytokines
Cytokines were measured in supernatants collected after 48 hrs from cultured splenic cells as described above. Capture ELISA was done for measuring cytokines IFNγ and IL-4 using kits (BD Pharmingen, San Diego, CA). Cells isolated from 2−3 primed mice were cultured in vitro as described above and supernatants were pooled. The experiment was repeated twice.
Statistical Analysis
Difference in incidence of arthritis between groups was analyzed using chi-square test with Yates’ correction. Antibody levels and means scores for arthritic mice were compared using two-tailed Student’s t test. Antigen presentations were analyzed using T test with unequal variance.
Results
Antibody reactivity to Collagen-derived peptides in DQ8 mice
T cell epitope mapping of overlapping HII derived peptides showed at least 16 epitopes from various CB fragments of CII, which are DQ8-restricted (Krco et al, 1999). We tested if B cell response was similar to T cell response using peptide ELISA. Known DQ8-restricted T cell epitopes were utilized. Even though B cell reactivity with the peptides was similar to T cells with most of the peptides showing reactivity to B cells, the strength of reactivity differed. Some peptides which gave a strong response with T cells had milder antibody reactivity (Fig 1, 33). There was a strong antibody response to CII in sera from immunized mice.
Figure 1.
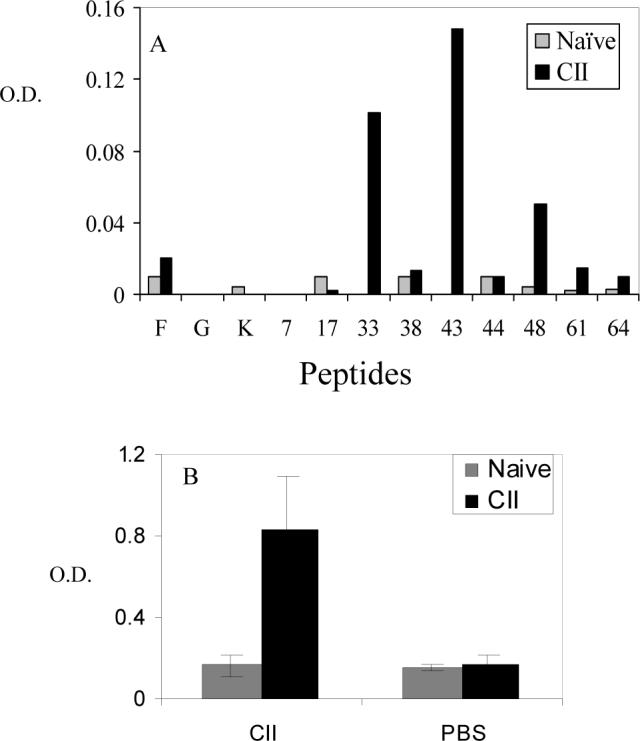
Peptide ELISA using sera from DQ8 mice showed that a differential B cell reactivity with known DQ8-restricted T cell epitopes. A) Sera from primed were tested on plates coated with known DQ8-restricted T cell epitopes of human type II collagen-derived peptides. Peptides sequences are described in methods. B) Sera from primed mice were tested for anti bovine type II antibodies.
B cells are essential for development of arthritis
DQ8, DQ8.μmt transgenic mice and negative littermate controls were immunized with BII to ascertain whether the lack of B cells would alter CIA susceptibility of DQ8 mice. As shown in Figure 2, DQ8.μmt mice were completely resistant to develop CIA (0/31), while DQ8 mice had an incidence of around 67% with severe disease. To determine if activated CII-specific cells could transfer arthritis in B cell deficient mice, splenic B cells isolated from primed DQ8 mice were transferred to DQ8.umt mice. A transient and mild arthritis (severity of 1) in 1/6 mice was observed in those receiving splenic B cells from DQ8 mice. Paws were thickened in mice when only sera were transferred. Histopathology did not show any significant inflammatory cell infiltration or pathological changes.
Figure 2.
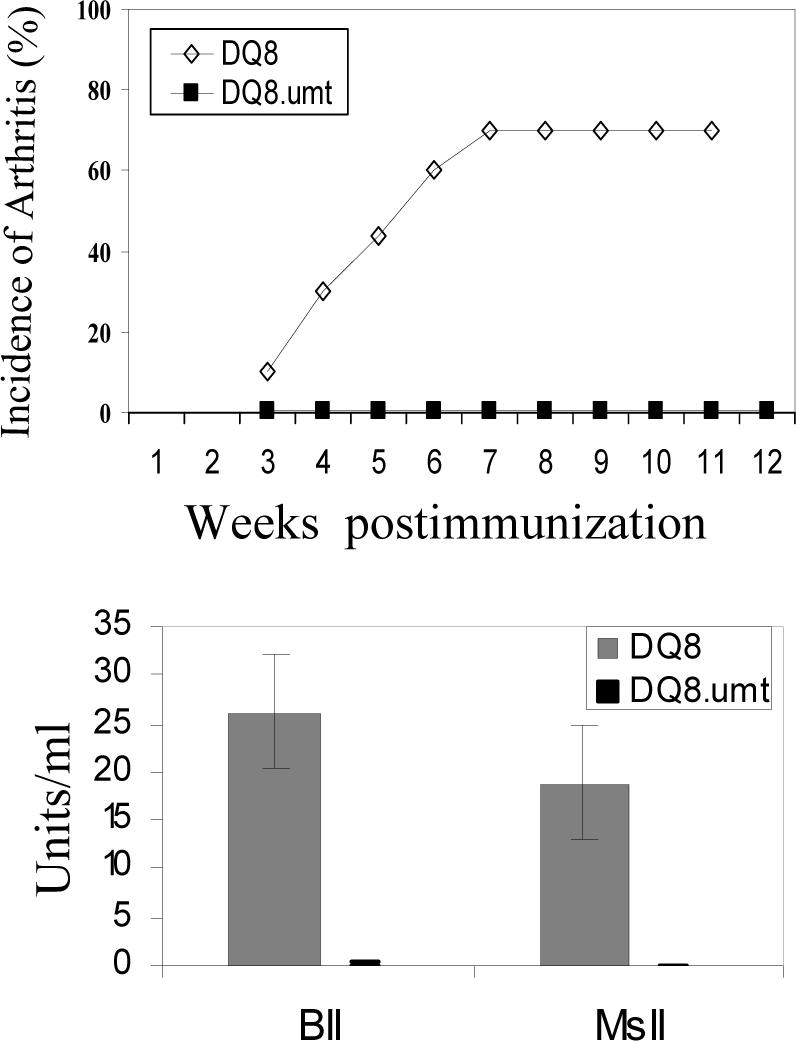
DQ8.μmt mice are resistant to develop collagen-induced arthritis. A) DQ8 and DQ8.μmt mice were immunized with CII and followed for onset of arthritis. B) Anti-CII antibodies in primed DQ8 and DQ8.umt mice, showing no detectable levels of antibodies in the latter.
B cell deficient mice did not produce anti-CII Abs to BII and mouse CII, while DQ8 mice produced high amounts of anti-BII and MsII antibodies (26.2±5.8 and 18.9±5.9 respectively).
Reduced T cell response to CII-derived peptides in μmt mice can be reconstituted by B cells from parent mice
Next we tested in vitro T cell response to CII and its derived DQ8-restricted peptide (Fig 3). LNCs isolated from mice primed with CII and known DQ8-restricted T cell peptide, 284−303, were challenged in vitro with or without the antigen. MHC-restriction study was done by addition of anti-CD4 antibody and anti-DQ antibody in in vitro culture. B cell deficient mice gave a milder in vitro response to CII and its derived peptide. Inhibition studies showed that the response in both B cell deficient and sufficient mice was DQ-restricted and mediated by CD4 cells. When anti-B cell antibody was used in vitro culture, response was reduced to 60% of the total response suggesting B cells present this peptide. DQ8.umt mice produced lower amounts of proinflammatory cytokine IFN-γ as well as IL-4 compared to DQ8 mice.
Figure 3.
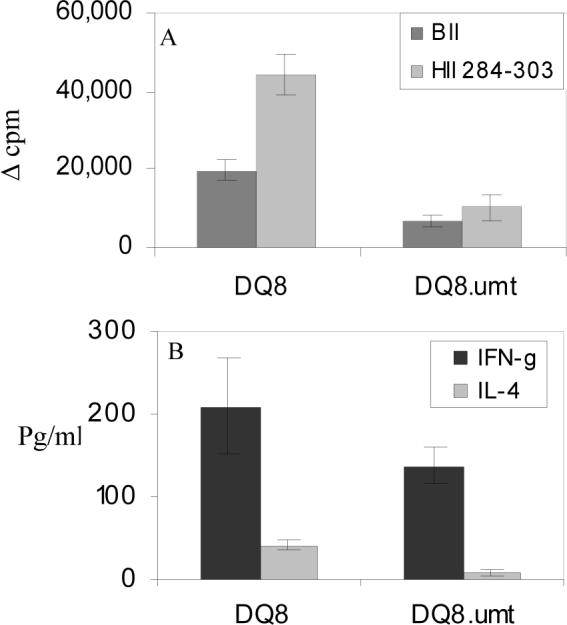
Antigen specific response in DQ8.μmt mice is lower when compared to DQ8 mice. A) In vitro T cell response to CII and CII derived peptide 284−303 in B cell sufficient and deficient DQ8 mice. DQ8.umt mice produce low amounts of cytokines B) IFNγ and IL-4.
To determine whether the reduced T cell response to antigens in B cell deficient mice was due to lack of antigen presenting function of B cell in this model, we tested the ability of B cells from DQ8 mice to induce antigen-specific proliferative responses in vitro. T cells in various concentrations from primed DQ8.umt mice were co cultured with B cells isolated from DQ8 primed mice (Figure 4). Reconstitution of B cell compartment from DQ8 mice restored the response of T cells from DQ8.umt mice to the peptide HII-284−303 similar to DQ8 B cell sufficient mice as shown in Figure 3. In vitro cytokine analysis of the supernatant showed restoration of IFN-γ and IL-4 production that was similar to DQ8 mice (Figure 3). These findings suggest that B cells not only produce autoantibodies but can also present antigens in vivo.
Figure 4.
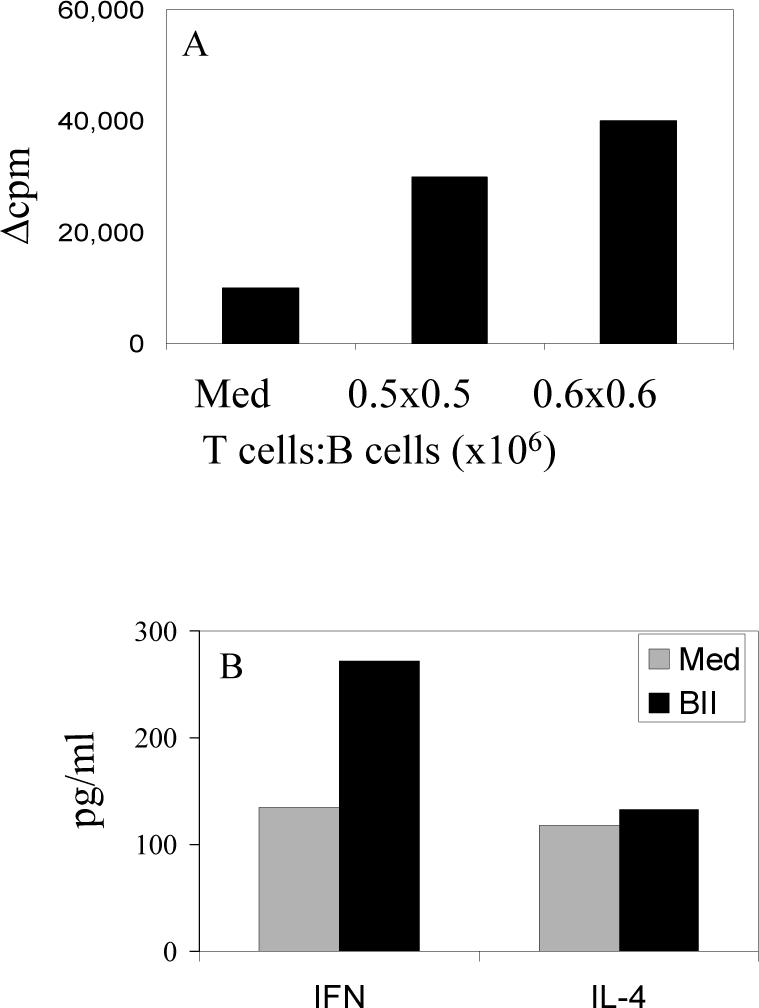
B cells from parent strain can restore in vitro antigen specific response. A) T cells from CII-derived peptide 284−303 primed DQ8.μmt mice co-cultured in vitro with B cells isolated from primed DQ8 mice in 2 different concentrations. The response was restored to the level of B cell sufficient mice as shown in Figure 3. B) Cytokine, IFN-γ and IL-4, levels were also restored to that of DQ8 mice as shown in Figure 3. Cytokines were measured by ELISA using supernatants from cultures in 4A.
B cells can present antigen to antigen specific hybridomas.
T cell epitope, HII derived peptide 554−573 (HII-44), is known to be presented by DQ8 in transgenic mice (Krco et al, 1999). As shown in Figure 1, B cells showed strong reactivity to HII 544−563 (HII-43) with milder response to HII-44 (554−573). To test the efficiency of B cells to present in antigen-specific manner we compared it with dendritic cells in presenting antigen to specific hybridomas. First we used truncations of the HII-44 peptide to determine the minimal T cell epitope presented by DQ8 using transgenic mice (Figure 5A). HII 556−564 (44S) was the minimal epitope that could be presented by LNCs of DQ8 mice. Sorted B and dendritic cells from primed mice were compared for their ability to present minimal epitope and HII-44 (554−573) to specific T cell hybridomas (Figure 5B). B cells from HII-44 primed mice were able to present both HII-44 and the minimal epitope, 44S, to the T cell hybridoma specific for HII-44 as suggested by IL-2 production after 24 hrs in vitro cultures. Interestingly a higher production of IL-2 was observed when B and DCs were isolated from female mice in both experiments (data not shown). However, dendritic cells produced higher amounts of IL-2 compared to B cells suggesting a much more efficient presentation of the antigen by dendritic cells. Next we compared presentation of already loaded antigen by DCs and B cells. Both B and dendritic cells were first incubated with the peptides and washed and then cultured with hybridoma in the absence of peptide. Both DCs and B cells presented less efficiently than in the presence of the peptide. Cells sorted from naïve mice did not produce much IL-2.
Figure 5.

Presentation of a CII-derived peptide, HII-44 (554−573), and its minimal epitope by B and dendritic cells to antigen-specific and DQ8-restricted T cell hybridoma shows B cells can present the peptide. A) T cell proliferation in vitro with peptide 554−573 and its minimal epitope presented by APCs. B) B cells and dendritic cells isolated from naïve mice and mice primed with HII-44 and minimal peptide 44S were cultured in vitro with T cells hybridoma specific for HII-44 in the presence or absence of the peptides. IL-2 levels were measured by ELISA from the in vitro culture supernatants after 24 hrs. Also, B and dendritic cells were pre-incubated with the peptides HII-44 and HII-44S for 24 hrs, washed and then used as antigen presenting cells to specific hybridoma without the presence of peptide. *p<0.05.
DQ8.μmt mice have higher number of CD3+ cells
To understand if there was an immunological defect in DQ8.μmt mice, we analyzed various cells by FACS in the naïve and CII primed mice. There was an increase in CD3+ cells in B cell deficient mice compared to DQ8 mice (P=0.03), although CD4 and CD8 cell number were not significantly different suggesting an increase in CD3+DN cells (Figure 6). Also, an increase in Mac1+ cells was also observed in DQ8.μmt mice compared with DQ8 mice, p=0.01. Similar to naïve mice, primed DQ8.umt mice also showed an increased number of CD3 and Mac1+ cells when compared to DQ8 mice. In primed DQ8 mice, CD8+CD3+ cells proliferated more than in DQ8.umt mice (p=0.002). Costimulatory molecule CD40+ cells were significantly decreased with a concomitant increase in CD28+ cells (P=.001 and p=.06 respectively). However, when CD4+CD28+ cells were analyzed, DQ8.umt mice had much lower frequency than DQ8 mice did, p=.02. Transgenic mice with B cell deficiency had higher number of cells expressing CD95 than those sufficient in B cells.
Figure 6.
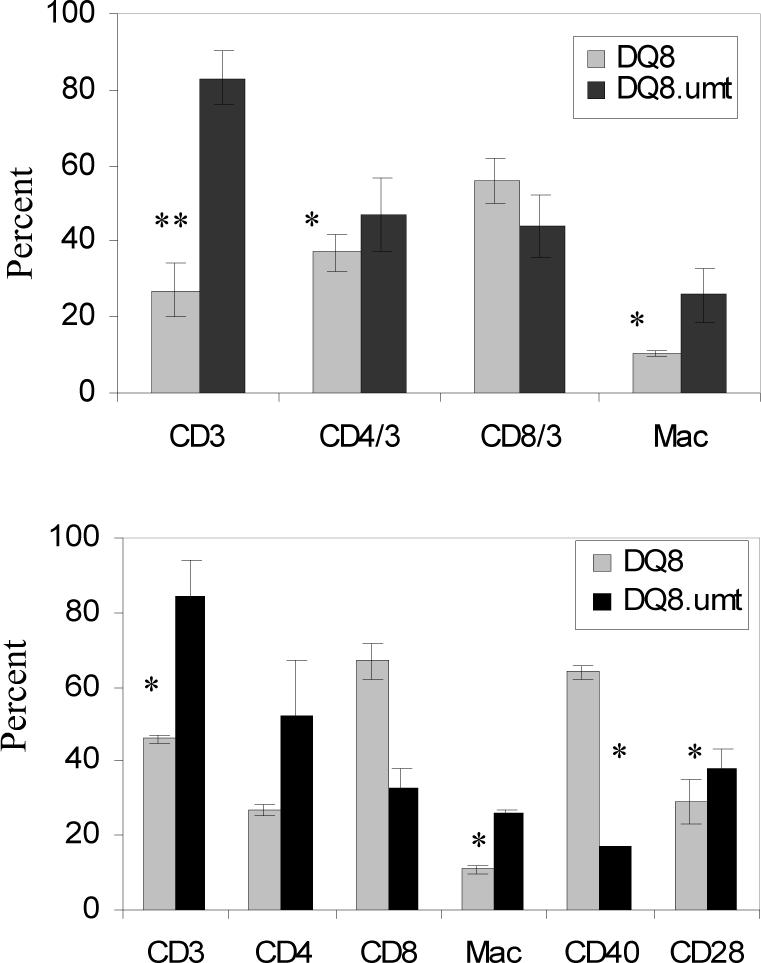
Cell profile of A) naïve and B) immunized mice show that DQ8.μmt mice have higher frequency of CD3+ and Mac1+ cells. After priming, a significant difference was observed in frequency of cells with costimulatory molecules. *p<0.05, ** p<0.01.
Discussion
A critical role of B cells has been demonstrated in rheumatoid arthritis by using depletion antibodies for B cells. In CIA model, a role of B cells has been documented by generating disease using serum transfer containing autoantibodies. However our understanding of how B cells contribute to disease susceptibility besides producing antibodies is incomplete. The present work provides evidence that B cells are critical for the development of arthritis and contribute in antigen presentation and autoantibody production. Our study showing restoration of T cell response of μmt mice by adding B cells confirms that one of the major functions of B cells in CIA is antigen presentation to autoreactive T cells. DQ8.μmt mice have increased number of CD3+ cells and slightly higher number of macrophages. This could simply be compensation due to the lack of B cells. However presentation by professional APCs in these mice is not sufficient to lead to the development of arthritis. Lack of autoantibodies due to absence of B cells can not be ruled out as one strong reason for no disease. A recent study showed that in the absence of CD19 and CD21, mice can produce antibody responses to type II collagen but are impaired in the ability to activate T cells (Del Nagro et al, 2005). Since antibodies alone can induce mild and transient arthritis, the present data along with available literature suggests that B cells can contribute towards pathogenesis by both antigen presentation and antibody production.
Our result showing low in vitro response in DQ8.μmt mice are in confirmation with the previous reports that activated and memory T cells are dependent on B cells (Constant et al, 1995, Linton et al, 2000). T cell response to CII and its derived DQ8-restricted peptide was restored when B cells were substituted in vitro from wild type mice further substantiating that B cells can be effective antigen presenting cells. Clonal expansion of OVA specific T cells in vivo and initiating antigen-presenting cells in lymph nodes has been demonstrated to involve direct presentation of antigen by B cells (Ron and Sprent, 1987, Kurt-Jones et al, 1988, Janeway et al, 1987). Using proteoglycan induced arthritis model, it was shown that antigen-specific B cells could restore autoreactive T cell activation (O’Neil et al, 2005). In the present data, arthritis could not be transferred efficiently in DQ8.μmt mice when B cells isolated from spleen of primed DQ8 mice were transferred. One reason could be that autoreactive T cells in DQ8.μmt were not primed efficiently and B cells that were transferred may not have enough antigen-specific B cells that could lead to expansion of autoreactive T cells resulting in pathogenesis. Using murine model of lupus, an antibody independent role of B cells was shown (Kim et al, 1999). In contrast to in vivo data, our in vitro data suggested that B cells can efficiently present antigen to T cells since primed T cells could produce cytokines in response to the antigen. DQ8 mice produced IFN-γ and IL-4 when challenged in vitro with HII-284−303 while DQ8.μmt mice had a lower in vitro response with low levels of cytokines. When B cells were substituted in vivo, mice did not develop arthritis though production of IFN-γ and IL-4 was restored suggesting that memory CD4 priming was restored. There is data to suggest that only IL-4 is secreted when B cells present antigen to naïve T cells while antigen-specific B cells have been shown to produce IL-4 which can result in antibody responses (Stockinger et al, 1996, Macaulay et al, 1997). Our results of increased IFN-γ and IL-4 in mice reconstituted with B cells suggest that B cells are presenting antigen to autoreactive T cells.
B cells have been shown to efficiently present antigens to T cells due to the antigen-specific receptors that allow concentrating antigen (Baird and Parker, 1996). To test the efficiency of B cells in presenting antigen, we compared antigen presentation by B cells to the other relevant APCs like DCs. B cells were able to present antigen to T cell hybridomas even though DCs were more efficient, further confirming that B cells can contribute towards pathogenesis by presenting a pathogenic antigen. Using a spontaneous model of arthritis it was shown that B cells are less potent APCs to naïve T cells in comparison to DCs, however activated B cells induced T cell response similar to that of DCs (Shih et al, 2006). In our model, mice were immunized with the antigen thus B cells were activated. Our data is in line with another study demonstrating that B cell blasts stimulate CD4+ T cells fourfold less effectively than dendritic cells (Cassell and Schwartz, 1994). Reconstitution of DQ8.umt mice with activated B cells from primed mice restored T cell response and cytokine production but did not lead to development of disease in recipients raising a possibility that B cells can present only some of the DQ8-restricted peptides with greater efficiency. Antibody response to DQ8 restricted peptides confirms that B cell epitopes vary in strength from T cell response suggesting that B cells can present some peptides better than other peptides. Our data is in confirmation with another study showing antigen-specific B cells can present antigen to an autoreactive T cell hybridoma in murine model of proteoglycan induced arthritis (Brennan et al, 1995) and further provides evidence that B cells are important as antigen presenting cells in arthritis development in this model.
To understand if the resistance to develop CIA in B cell deficient mice was due to abnormality in cell constitution, CD3+ cells and its subsets and mac-1+ cells were compared in naïve mice. DQ8.umt mice had higher frequency of CD3+, CD4+ and Mac-1+ cells suggesting it was not due to a defect in cell numbers. For T cells to get activated, costimulatory molecules are required as a second signal in addition to peptide binding. We studied costimulatory molecules in primed mice and found no difference in the frequency of CD28+ cells in both strains. However CD40+ cells were significantly decreased in DQ8.μmt mice compared to DQ8 mice. Mac-1 + positive cells are in higher frequency but that does not compensate for the lack of B cells that would have expressed CD40. A lower frequency of CD40+ cells could be another reason for milder in vitro response to CII and its peptide in DQ8.μmt mice.
Conclusion
In conclusion the data presented here shows an important role of B cells in antigen presentation along with antibody production in collagen induced arthritis model and thereby suggest a similar role in human RA.
Acknowledgement
Authors acknowledge Julie Hanson and Tad Trejo for maintaining transgenic mice and Michele Smart for screening the transgenic mice.
Funding source: The study was supported by grants from National Arthritis Foundation Investigator Award to Veena Taneja and NIH AR 30752. The HLA Class II transgenic mice were produced with the support from NIH grant AI 14764
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992;358:337–41. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 2.Amigorena S, Lankar D, Briken V, Gapin L, Viguier M, Bonnerot C. Type II and III receptors for immunoglobulin G (IgG) control the presentation of different T cell epitopes from single IgG-complexed antigens. J. Exp. Med. 1998;187:505–15. doi: 10.1084/jem.187.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird AM, Parker DC. Analysis of low zone tolerance induction in normal and B-cell deficient mice. J Immunol. 1996;157:1833–39. [PubMed] [Google Scholar]
- 4.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 5.Brennan FR, Mikecz K, Buzas EI, Ragasa D, Cs-Szabo G, Negroiu G, Glant TT. Antigen-specific B cells present cartilage proteoglycan (aggrecan) to an autoreactive T cell hybridoma derived from a mouse with proteoglycan-induced arthritis. Clin Exp Immunol. 1995;101:414–21. doi: 10.1111/j.1365-2249.1995.tb03128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol. Rev. 1999;169:107–21. doi: 10.1111/j.1600-065x.1999.tb01310.x. [DOI] [PubMed] [Google Scholar]
- 7.Cassell DJ, Schwartz RH. A quantitative analysis of antigen-presenting cell function: activated B cells stimulate naïve CD4 T cells but are inferior to dendritic cells in providing costimulation. J Exp Med. 1994;180:1829–1840. doi: 10.1084/jem.180.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Constant S, Schweitzer N, West J, Ranney P, Bottomly K. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 1995;15:3734–41. [PubMed] [Google Scholar]
- 9.Del Nagro CJ, Kolla RV, Rickert RC. A critical role for complement C3d and the B cell coreceptor (CD19/CD21) complex in the initiation of inflammatory arthritis. J. Immunol. 2005;175:5379–89. doi: 10.4049/jimmunol.175.8.5379. [DOI] [PubMed] [Google Scholar]
- 10.Falcone M, Lee J, Patstone G, Yeung B, Sarvetnick N. B lymphocytes are crucial antigen-presenting cells in the pathogenic autoimmune response to GAD65 antigen in nonobese diabetic mice. J. Immunol. 1998;161:1163–8. [PubMed] [Google Scholar]
- 11.Goldstein R, Arnett FC. The genetics of rheumatic disease in man. Rheum Dis Clin N Am. 1987;13:587–510. [PubMed] [Google Scholar]
- 12.Gonzalez-Quintial R, Baccala R, Pope RM, Theofilopoulos AN. Identification of clonally expanded T cells in rheumatoid arthritis using a sequence enrichment nuclease assay. J Clin Invest. 1996;97:1335–43. doi: 10.1172/JCI118550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Griffiths MM, Eichwald EJ, Martin JH, Smith CB, DeWitt DW. Imunogenetic control of experimental type II collagen induced arthritis. Arthritis Rheum. 1981;24:781–89. doi: 10.1002/art.1780240605. 1981. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths MM, Nabozny GH, Hanson J, Harper JS, McCall S, Moder KG, Cannon GW, Luthra HS, David CS. Collagen induced arthritis and TCRs in SWR and B10.Q mice expressing EaK transgene. J Immunol. 1994;153:2758–68. [PubMed] [Google Scholar]
- 15.Hamano Y, Arase H, Saisho H, Saito T. Immune complex and Fc receptor-mediated augmentation of antigen presentation for in vivo Th cell responses. J. Immunol. 2000;164:6113–19. doi: 10.4049/jimmunol.164.12.6113. [DOI] [PubMed] [Google Scholar]
- 16.Janeway CA, Jr, Ron J, Katz ME. The B cell is the initiating antigen-presenting cell in peripheral lymph nodes. J Immunol. 1987;138:1051–5. [PubMed] [Google Scholar]
- 17.Kim HJ, Krenn V, Steinhausen G, Berek C. Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J. Immunol. 1999;162:3053–62. [PubMed] [Google Scholar]
- 18.Krco C, Watanabe S, Harders J, Griffiths MM, Luthra HS, David CS. Identification of T cell determinants on human type II collagen recognized by HLA-DQ8 and HLA-DQ6 transgenic mice. J Immunol. 1999;163:1661–5. [PubMed] [Google Scholar]
- 19.Kurt-Jones EA, Liano D, Hay Glass KA, Benacerraf B, Sy MS, Abbas AK. The role of antigen-presenting B cells in T cell priming in vivo. Studies of B cell-deficient mice. J. Immunol. 1988;140:3773–78. [PubMed] [Google Scholar]
- 20.Lanzavecchia A. Antigen uptake and accumulation in antigen-specific B cells. Immunol. Rev. 1987;99:39–51. doi: 10.1111/j.1600-065x.1987.tb01171.x. [DOI] [PubMed] [Google Scholar]
- 21.Linton P, Harbertson J, Bradley LM. A critical role for B cells in the development of memory CD4 cells. J. Immunol. 2000;165:5558–65. doi: 10.4049/jimmunol.165.10.5558. [DOI] [PubMed] [Google Scholar]
- 22.Macaulay AE, De Kruyff RH, Goodnow CC, Umetsu DT. Antigen-specific B cells preferentially induce CD4+ T cells to produce IL-4. J Immunol. 1997;158:4171–9. [PubMed] [Google Scholar]
- 23.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat. Immunol. 2002;3:360–65. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 24.Nepom GT. Determinants of genetic susceptibility in HLA-associated autoimmune disease. Clin.Immunol Immunopathol. 1989;53(2Pt2):S53–S62. doi: 10.1016/0090-1229(89)90070-6. [DOI] [PubMed] [Google Scholar]
- 25.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkamans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–6. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 26.O’Neil SK, Shlomchik MJ, Glant TT, Cao Y, Doodes PD, Finnegan A. Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe rehumatoid arthritis. J Immuno. 2005;174:3781–88. doi: 10.4049/jimmunol.174.6.3781. [DOI] [PubMed] [Google Scholar]
- 27.Panayi GS. B cells: a fundamental role in the pathogenesis of rheumatoid arthritis? Rheumatology Suppl. 2005;2:ii3–ii7. doi: 10.1093/rheumatology/keh616. [DOI] [PubMed] [Google Scholar]
- 28.Ron Y, Sprent J. T cell priming in vivo: a major role for B cells in presenting antigen to T cells in lymph nodes. J. Immunol. 1987;138:2848–56. [PubMed] [Google Scholar]
- 29.Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Immunol. 1998;161:3912–18. [PubMed] [Google Scholar]
- 30.Shih FF, Racz J, Allen PM. Differential MHC class II presentation of a pathogenic autoantigen during health and disease. J Immunol. 2006;176:3438–3448. doi: 10.4049/jimmunol.176.6.3438. [DOI] [PubMed] [Google Scholar]
- 31.Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D. The role of B cells in lpr/lpr-induced autoimmunity. J. Exp. Med. 1994;180:1295–306. doi: 10.1084/jem.180.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther. 2003;5(suppl4):S1–S6. doi: 10.1186/ar1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stockinger B, Zal T, Zal A, Gray D. B cells solicit their own help from T cells. J Exp Med. 1996;183:891–89. doi: 10.1084/jem.183.3.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strober S, Holoshitz J. Mechanisms of immune injury in rheumatoid arthritis: evidence for the involvement of T cells and heat-shock proteins. Immunol Rev. 1990;118:233–55. doi: 10.1111/j.1600-065x.1990.tb00818.x. [DOI] [PubMed] [Google Scholar]
- 35.Stuart JM, Dixon FJ. Serum transfer of collagen-induced arthritis in mice. J Exp Med. 1983;158:378–92. doi: 10.1084/jem.158.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svensson L, Jirholt J, Holmdahl R, Jansson L. B cell-deficient mice do not develop type II collagen-induced arthritis (CIA) Clin. Exp. Immunol. 1998;11:521–26. doi: 10.1046/j.1365-2249.1998.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taneja V, Taneja N, Paisansinsup T, Behrens M, Griffiths M, Luthra HS, David CS. CD4 and CD8 T cells in susceptibility protection to collagen-induced arthritis in HLA-DQ8-transgenic mice: implications for rheumatoid arthritis. J Immunol. 2002;168:5867–75. doi: 10.4049/jimmunol.168.11.5867. [DOI] [PubMed] [Google Scholar]
- 38.Trentham DE, Townes AS, Kang AH. Autoimmunity to type II collagen: an experimental model of arthritis. J Exp Med. 1977;146:857–868. doi: 10.1084/jem.146.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Boxel JA, Paget SA. Predominantly T-cell infiltrate in rheumatoid synovial membranes. N Engl J Med. 1975;293:517–20. doi: 10.1056/NEJM197509112931101. [DOI] [PubMed] [Google Scholar]
- 40.Vidard L, Kovacsovics-Bankowski M, Kraeft SK, Chen LB, Benacerraf B, Rock KL. Analysis of MHC class II presentation of particulate antigens of B lymphocytes. J. Immunol. 1996;156:2809–18. [PubMed] [Google Scholar]
- 41.Wooley PH, Luthra HS, Singh SK, Huse AR, Stuart JM, David CS. Passive transfer of arthritis to mice by injection of human anti-type II collagen antibody. Mayo Clin Proc. 1984;59:737–43. doi: 10.1016/s0025-6196(12)65583-9. [DOI] [PubMed] [Google Scholar]
- 42.Wooley PH, Luthra HS, Stuart JM, David CS. Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J Exp Med. 1981;154:688–700. doi: 10.1084/jem.154.3.688. [DOI] [PMC free article] [PubMed] [Google Scholar]