

Abstract
The primary metabolic route for D-xylose, the second most abundant sugar in nature, is via the pentose phosphate pathway after a two or three step conversion to xylulose-5-phosphate. Xylulose kinase (XK; EC 2.7.1.17) phosphorylates D-xylulose, the last step in this conversion. The apo and xylulose-bound crystal structures of E. coli XK have been determined and show a dimer composed of two domains separated by an open cleft. XK dimerization was directly observed by a cryo-EM reconstruction at 36 Å resolution. Kinetic studies reveal that XK has a weak substrate-independent MgATP-hydrolyzing activity and phosphorylates several sugars and polyols with low catalytic efficiencies. Binding of pentulose and MgATP to form the reactive ternary complex is strongly synergistic. Although the steady-state kinetic mechanism of XK is formally random, a path is preferred in which D-xylulose binds before MgATP. Modeling of MgATP binding to XK and the accompanying conformational change suggests that sugar binding is accompanied by a dramatic hinge bending movement that enhances interactions with MgATP, explaining the observed synergism. A catalytic mechanism is proposed and supported by relevant site-directed mutants.
Keywords: xylulokinase, x-ray structure, mechanism, FGGY kinase, ATPase
INTRODUCTION
The ability of an organism to metabolize xylose and other pentoses is conferred by their conversion to D-xylulose-5-phosphate (X5P), a compound which is able to enter the nonoxidative branch of the pentose phosphate pathway. In eukaryotes capable of assimilating xylose, X5P is produced through the action of xylose reductase, xylitol dehydrogenase and D-xylulose kinase (XK; EC 2.7.1.17). The first two enzymes are replaced in prokaryotes with xylose isomerase which directly interconverts xylose and xylulose. There has been much interest in metabolic engineering of the two pathway variants for the past two decades because of the possibility of fermenting the vast quantities of xylose in agricultural waste products to produce ethanol 1; 2. These prospects have driven the study of the catalytic properties and substrate specificities of xylose reductase, xylitol dehydrogenase and xylose isomerase from many different sources. XK has been less well studied in general, however.
Physiological studies have shown that XK is essential for growth on xylose or xylulose and may limit the overall rate of pentose sugar utilization 3. In addition to its role in xylose metabolic assimilation, its activity with the alternative substrate 1-deoxy-D-xylulose 4 implicates it in the biosynthesis of terpenoids 5, thiamine 6 and pyridoxal 7. The enzyme has been studied from several prokaryotes and lower eukaryotes including E. coli, yeasts and fungi and XK activity has also been identified in higher eukaryotes 3; 8; 9.
Based on amino acid sequence similarity, the 53 kDa E. coli XK, encoded by the xylB gene contains an ATPase fingerprint consisting of five conserved regions found in a large group of proteins including sugar kinases, actins, and heat shock proteins 10. Structurally, superfamily members consist of two domains, domain I and domain II, which are separated by a cleft forming the active site. Generally, members bind ATP and catalyze the hydrolysis of the γ-phosphate or its transfer to a substrate such as a sugar hydroxyl group. Catalysis is preceded by a domain closure which is induced by substrate binding, as exemplified by the induced-fit mechanism of yeast hexokinase 11. Phospho group transfer is promoted by two highly conserved aspartate residues. One is located in the N-terminal region of domain I and interacts with the ATP-associated Mg2+. It is invariant across superfamily members and belongs to a signature sequence that identifies them 12. The second aspartate appears to function as a general base, activating the nucleophile for attack.
When the entire sequence is examined, XK is most similar to a family of carbohydrate kinases phosphorylating fucose, glucose, glycerol and xylulose. Of these, x-ray crystal structures of glycerol kinase (GK) from E. coli (ecGK) and Enterococcus casseliflavus have been reported and detailed relationships between structure and function have been determined for GK 13; 14. Like many other members of the family, the activity of GK is regulated by binding of small ligands (fructose-1,6-bisphosphate) as well as by interactions with other proteins (the glucose-specific phosphocarrier protein IIIGlc) 15. The enzyme from En. casseliflavus can be covalently phosphorylated at His232 resulting in a substantial activation 14.
Details surrounding XK kinetic and structural properties have often been inferred from these related enzymes. These include a substrate-induced conformational change creating a high affinity ATP binding site which has been implied but never observed in the carbohydrate kinases and which has kinetic consequences 16; 17. The oligomeric state of XK was unclear yet important since other family members such as ecGK can be regulated by effector-modulated oligomerization. The kinetic mechanism of XK has not been established and precedents within the family are mixed with some members binding substrates in an ordered manner while others are random. A comprehensive quantitative evaluation of substrate specificity for the enzyme has not been done. To shed light on these and other mechanistic issues, a combined structural and kinetic analysis of XK from E. coli is described here.
RESULTS AND DISCUSSION
Overall structure
The structure of XK in the apo form has been determined at 2.7 Å resolution using MIR phasing. The apo structure was used to phase a xylulose-bound structure at 2.1 Å. This model consists of two protein molecules (A and B) that form the asymmetric unit, each consisting of residues 1-334 and 343-484 of the 484 predicted. There was no electron density corresponding to the region from 335 to 342. There are also 310 water molecules which are observed in the asymmetric unit. The average temperature factor is 37.9 Å2 for molecule A and 44.2 Å2 for molecule B. Modeling and more detailed structural analysis therefore focused on molecule A. The r.m.s.d. between the α-carbons of each monomer is 0.95 Å with no major differences. A Ramachandran plot generated by PROCHECK indicates 90.3% of non-glycine residues in the core regions, 9.0% are in additionally allowed and 0.7% are in generously allowed areas 18. Each monomer of XK is 76 × 46 × 56 Å and is composed of two major domains (I and II) connected by a hinge segment composed of residues Ala294 to Asp299 (α-helix α11) (Fig. 1). Positive Fo-Fc density was found in the active site on both monomers which was interpreted as a xylulose molecule. Statistics associated with the final refinement are shown in Table 1.
Figure 1.
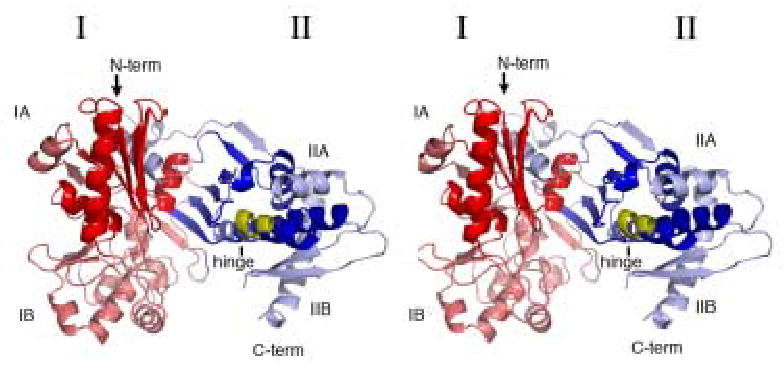
The main chain trace of XK. A) A stereo view of XK looking into the substrate binding cleft. The molecule is colored from red (domain IA), pink (domain IB) to blue (domain IIA) and light blue (domain IIB). The hinge segment is colored yellow. B) A stereo view of the side of the enzyme emphasizing the deep substrate binding groove. Figures 1 and 5 were done using PyMOL (DeLano Scientific).
Table 1.
Data collection and refinement statistics. Numbers in parentheses describe values for the high resolution shell. EMTS: ethylmercurythiosalicylate
| Data Collection | Xylulose soak | apo | AgHgI3 | K2PtCl4 | EMTS(1) | EMTS(2) |
|---|---|---|---|---|---|---|
| Wavelength (Å) | 0.954 | 0.954 | 1.54 | 1.54 | 1.54 | 1.54 |
| Resolution range | 30-2.10 Å (2.18-2.10) | 25-2.7 Å (2.75-2.70) | 25-3.0 Å | 25-2.8 Å | 25-3.5 Å | 25-3.0 Å |
| Unique observations | 58,714 | 29,732 | 21,817 | 26,252 | 14,032 | 22,378 |
| Total observations | 179,132 | 294,312 | 146,366 | 140,551 | 66,934 | 112,683 |
| Completeness (%) | 99.1 (99.5) | 99.9 (100) | 99.9 (99.9) | 98.6 (98.8) | 96.6 (99.9) | 99.9(99.9) |
| Rsym | 0.081 (0.385) | 0.073 (0.350) | 0.103 (0.235) | 0.079 (0.227) | 0.132 (0.279) | 0.083 (0.154) |
| <I/σ(I)> | 10.1 (2.31) | 29.9 (5.5) | 5.9 (2.1) | 7.8 (2.5) | 5.5 (2.4) | 8.4 (3.4) |
| Refinement | Xylulose soak | Apo | ||||
|
|
||||||
| Resolution range | 30-2.1 Å | 24.88-2.7 Å | ||||
| Reflections used | 56,533 | 27,537 | ||||
| Rcryst | 0.198 | 0.205 | ||||
| Rfree | 0.259 | 0.249 | ||||
| # of protein, non- hydrogen atoms | 7,235 | 7,395 | ||||
| # of non-protein atoms | 310 | 162 | ||||
| rms bond length (Å) | 0.013 | 0.010 | ||||
| rms bond angles (o) | 1.13 | 1.745 | ||||
| Average main chain B values (Å2) | 38.1 | 33.9 | ||||
| Average side chain B values (Å2) | 47.3 | 36.6 | ||||
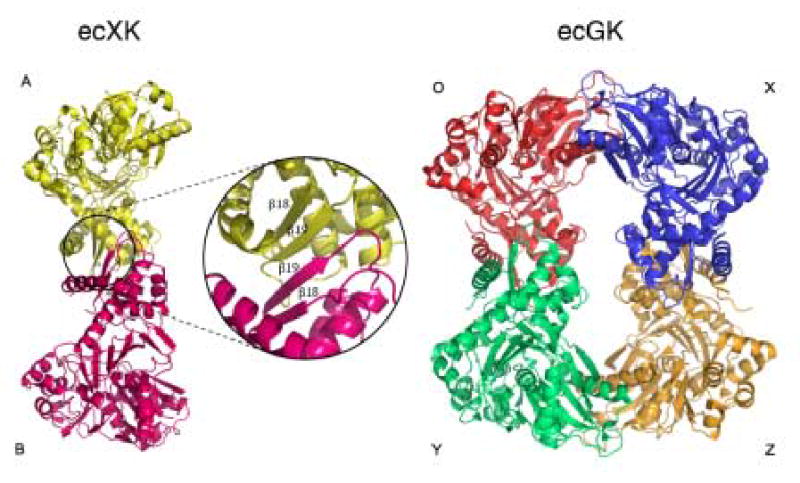
Using a VAST search to screen the Protein Data Bank (PDB), the ecGK structure was found to be the most homologous structure but also indicated XK shares structural features with the other members of the ATPase superfamily 19. The two enzymes have only 20% sequence identity, but an overlap of the domains individually indicates they are structurally homologous. An r.m.s.d. of 1.6 Å was calculated between 264 residues in domain I and 2.0 Å for 153 residues in domain II. Other structures including 3-phosphoglycerate kinase, hexokinase and NAD kinase also share the same overall ATPase scaffold with two domains linked with a hinge region 20; 21; 22. As described previously for the ecGK structure, domain I is primarily responsible for sugar substrate binding and domain II is mainly involved in ATP binding 23. Substrates bind deeply in the cleft formed between these two domains. These domains can be further divided into subdomains where IA and IIA form the ATPase core (Fig. 1A and B) of XK and subdomain IB forms part of the D-xylulose binding site. Subdomain IIB is involved in the intermolecular interface of the XK dimer, mediated by β-sheets β18 and β19 (Fig. 1A, 2A). As observed in ecGK subdomain IA contains the conserved catalytic residues and is responsible for the phospho-group transfer function.
Figure 2.
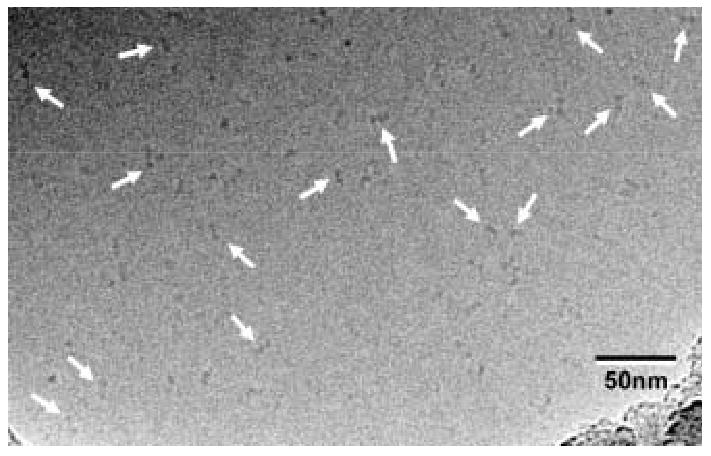
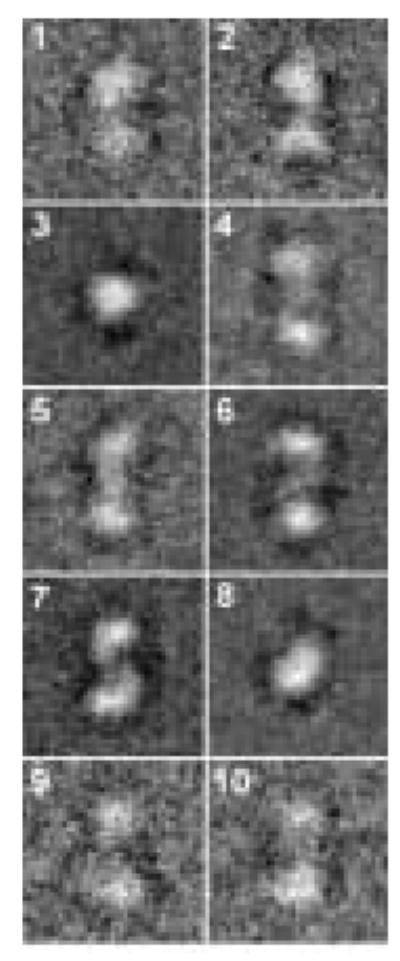
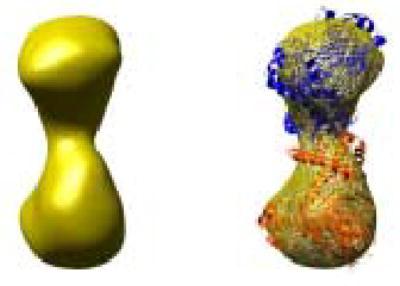
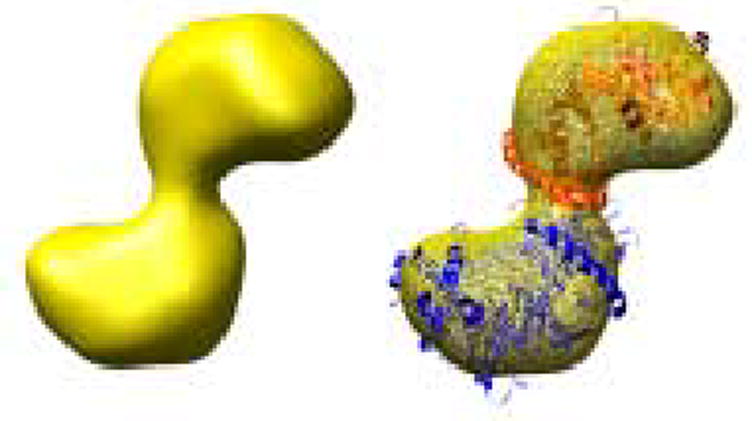
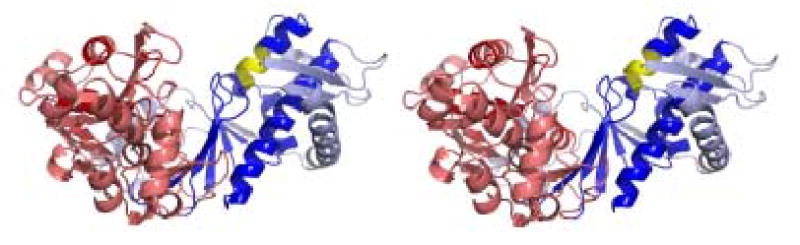
Oligomeric structure of XK. A) The dimer observed within the asymmetric unit of XK resembles the O/Y dimer found in the allosterically-inhibited ecGK tetramer. A zoomed view of XK dimeric interface is shown and illustrates the interactions between β-interactions responsible for mediating the interface. B) A cryo-EM image of xylulose kinase particles in vitrified buffer. Arrows indicate dimeric XK particles. C) Some class averages. Number of particles in these classes with the panel size of 19.2 x 19.2 nm2: panel 1: 11; panel 2: 10; panel 3: 31; panel 4: 20; panel 5: 13; panel 6: 33; panel 7: 34; panel 8: 36; panel 9: 6; panel 10: 9. D) and E) Two views of the surface-rendered density map from cryo-EM limited to 36 Å resolution with and without the dimeric crystal structure superimposed. The surface threshold for the display of the 3D density map were chosen so that the included volume corresponds to 105.2 kDa, assuming a protein density of 0.82 Da/Å3 (1.35g/ml) and displayed using UCSF Chimera 55.
Regulation of carbohydrate kinase members
In ecGK, fructose 1,6-bisphosphate (FBP) inhibits the enzyme by creating an inactive tetramer from two active dimers by mediating interactions between an extended loop from each domain I across the O/X and Y/Z interfaces (Fig. 2A) 15. The binding site is primarily formed by Gly233, Gly234 and Arg236 located on each side. XK has no known allosteric regulators and a comparison shows that the O/X or Y/Z interfaces are different between ecGK and a hypothetical XK tetramer 14. In XK, the corresponding region is structurally different from GK with a short α-helix substituted for the loop (Figs. 2A and 6). Also none of the residues involved in the tetramer formation in GK are found in XK and especially the Arg236 (Thr224 in XK) described to be important for the tetramerization 15 (Fig. 6).
Figure 6.
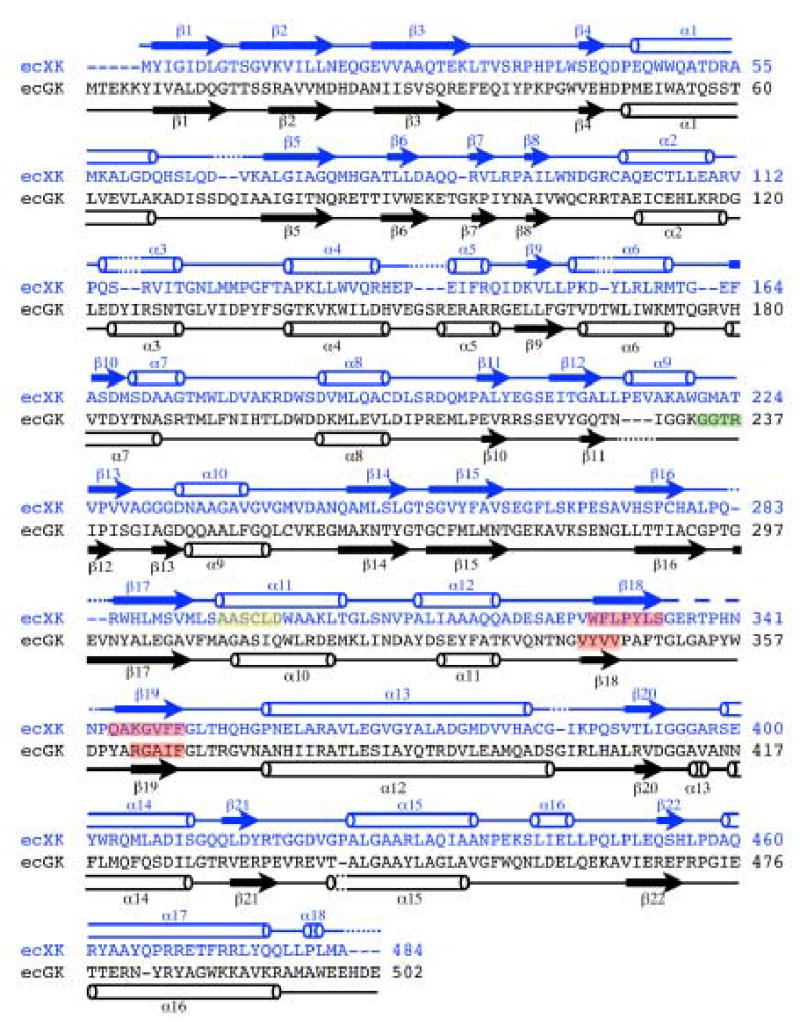
Pairwise sequence alignment of XK and ecGK. Green designates residues involved in tetramer formation in ecGK; yellow, the XK hinge segment; red, the dimeric interface of both XK end ecGK. Residues composing each XK domain and subdomain are as follows: domain I, 1-293; subdomain IA, (1-76, 151-161, 224-242); subdomain IB (77-105, 162-223, 243-245); domain II, 246-484; subdomain IIA (246-264, 282-307, 358-418); subdomain IIB (265-281, 308-357, 419-484).
The two molecules in the crystallographic asymmetric unit do clearly maintain the dimeric contact found between the O/Y and X/Z ecGK dimers which are present along the vertical axes in Fig. 2A 15. The XK dimeric interface is primarily composed of an antiparallel β-sheet contact composed of the segment from 345 to 359 (Figs. 2A and 6). This is structurally homologous with what is found in ecGK subdomain IIB. Residues composing the β-sheet and mediating the interface are 80% identical between ecGK and XK. This is also a well-conserved region across the kinase family implying other members are dimeric as well.
There are alternate modes of regulation found in other sugar kinase members, particularly GK. Phosphorylation of His232 in En. casseliflavus GK, which is present in a loop mediating the O/X interface (Fig. 2A), has been found to activate the enzyme. The phosphotransferase system is known to allosterically regulate ecGK via interactions with the phosphocarrier protein IIAGlc. Xylose and xylulose are not recognized by this system and the IIAGlc binding site on the carboxyl terminus of GK is not conserved in sequence or structure in XK.
Oligomeric structure
Since XK appeared to be a tenuously associated dimer in the crystal structure, cryo electron microscopy with 3D map reconstruction were used to confirm this arrangement in solution. Figure 2B shows projection structures of XK in different orientations. The low molecular weight of protein made particle selection difficult, but the class averages in Fig. 2C clearly document the dimeric nature of XK. In order to increase the signal to noise ratio, class averages were calculated and used for the 3D reconstruction, (Fig. 2B–E). The final reconstruction was resolution limited to 36 Å resolution (Fig. 2D and 2E). The docking of the XK dimer (Fig. 2D, E) into the 3D density map strongly supports the dimeric arrangement observed in the crystal structure. A real space correlation coefficient of 85.5% was calculated by overlapping 36 Å maps and indicates a high level of agreement. The XK dimeric state was further confirmed by dynamic light scattering experiment which shows a molecular weight of 110 kDa (data not shown).
Conservation of the XK ATPase Core Structure
Subdomains IA and IIA form the ATPase core and they each contain a five stranded β-sheet flanked by three α-helices (Fig. 1). This architecture has previously been observed in other superfamily members such as ecGK, yeast hexokinase, actin and the DNAse domains of hsc70 and DnaK 12; 13; 24; 25. Catalytic residues are also well conserved among the superfamily including two conserved aspartate, glutamate, or glutamine residues at the active site which have key roles in the catalytic mechanism (Table 2). The first of these functions as a general base, assisting the removal of a proton from the attacking hydroxyl group (Asp233 in XK). The second is an aspartate residue in domain I which interacts with the Mg2+ ion complexed with the nucleotide (Asp6 in XK). The positions of the aspartates are very similar when XK is overlayed with structures of the rabbit actin (PDB accession number 1QZ5), the yeast apo-hexokinase PII (1IG8), the E. coli DnaK (1DKG) or ecGK (1GLF). Residues expected to be responsible for specificity are spatially divergent and none of the residues involved in the xylulose binding in XK are found in the human hexokinase type 1 complexed with D-glucose (1HKC).
Table 2.
Multiple ATPase sequence alignment with catalytic residues shown in red.

|
ecXK, Escherichia coli xylulose kinase; ecGK, Escherichia coli glycerol kinase; ecDNAK, Escherichia coli prokaryotic heat shock protein; hACTS, Human Alpha-actin-1; scHXKB, Saccharomyces cerevisiae Hexokinase-B; ecFUCK Escherichia coli fucokinase
Substrate specificity
Apparent kinetic parameters of XK for the phosphorylation of D-xylulose, D-ribulose, D-arabitol and xylitol are summarized in Table 3. In the absence of substrate, XK has a weak but significant ATPase activity for which kinetic parameters are also shown. This ATPase activity, recorded at a fixed MgATP concentration of 5 mM, was strongly (13-fold) inhibited in the presence of the non-phosphorolyzable inhibitor 5-F-xylulose (0.18 mM), suggesting that hydrolysis of ATP in the absence of substrate is an inherent property of XK and does not result because of contamination of the enzyme preparation with another ATPase. The results in Table 3 reveal a relatively relaxed substrate specificity of the enzyme. However, in kcat/Km terms D-xylulose is either preferred or strongly preferred over the other substrates tested. Up to 1300-fold changes in catalytic efficiency are observed in response to alterations of substrate structure (Fig. 3) and reflect mainly increases in the apparent Michaelis constant. The value of kcat, in contrast, was almost constant across the substrate series, suggesting a common rate-determining step in the reaction of XK with the different compounds. The marked 50-fold decrease in kcat/Km caused by inversion of C3 chirality in D-xylulose to D-ribulose shows the high stereochemical selectivity of XK (Table 3). Replacement of the C2 carbonyl group of D-xylulose by an L-configured hydroxyl group in xylitol brought about an even larger 460-fold decrease in catalytic efficiency. In addition, it caused a complete loss of synergism in the apparent binding of substrate and MgATP. Using the apparent Michaelis constant for the ATPase reaction of XK as reference, MgATP was 41-fold more tightly bound when D-xylulose was present at a saturating concentration. The model of the XK ternary complex provides explanations for these kinetically determined effects, showing a sugar-dependent domain closure.
Table 3.
Apparent kinetic parameters for several XK substrates. Constants were calculated by fit of equation 1 to initial rate data.
| Substrate | KmSubstrate (mM) | KmMgATP(mM) | kcat(s−1) | kcat/KmSubstrate (s−1mM−1) | kcat/KmMgATP (s−1mM−1) |
|---|---|---|---|---|---|
| D-xylulose | 0.29 ± 0.02 | 0.15 ± 0.01 | 255 ± 5 a | 880 ± 70 | 1700 ± 120 |
| D-ribulose | 14 ± 1 | 0.19 ± 0.01 | 235 ± 6a | 17 ± 1 | 1240 ± 70 |
| xylitol | 127 ± 7 | 3.0 ± 0.4 | 237 ± 14 b | 1.9 ± 0.2 | 79 ± 12 |
| D-arabitol | 141 ± 8 | 8.5 ± 1.6 | 105 ± 13c | 0.7 ± 0.1 | 12 ± 3 |
| ATPase | - | 6.1 ± 0.5 | 0.07 ± 0.01 | - | 0.011 ± 0.002 |
measured at 5 mM ATP, varied sugar concentrations;
measured at 1 M xylitol, varied ATP concentrations;
measured at 0.6 M arabitol, varied ATP concentrations;
Figure 3.
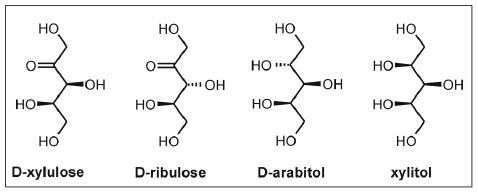
Sugar substrates used in substrate specificity studies
A significant level of synergism was also observed in the binding of the dead-end inhibitor 5-F-xylulose and MgATP. The 5-F-xylulose is a linear competitive inhibitor with respect to D-xylulose. The value of Kic for the inhibitor decreased ≈ 6-fold in response to a 30-fold increase in the fixed concentration of MgATP from the KmATP level to a saturating level (Table 4). Interestingly, binding of D-xylulose and AMPPNP, a linear competitive inhibitor with respect to MgATP, was not synergistic (Table 4), suggesting that the dead-end inhibitor differs from MgATP in properties essential for the ligand-induced crosstalk of the XK domains. The substrate selectivities and the resulting synergistic binding of ATP are explained by results of modeling experiments in which the ternary complex was modeled with both ATP and xylulose using the structure of the ecGK ternary complex as the basis (described below).
Table 4.
Inhibition patterns and constants from inhibition studies with dead-end inhibitors.
| Inhibitor | Varied substrate | Fixed substrate | Ki (mM) | Eq. | Inhibition pattern |
|---|---|---|---|---|---|
| 5-F-xylulose | xylulose | ATP
0.167 mM |
0.15 ± 0.02 | 3 | competitive |
| 5-F-xylulose | xylulose | ATP
5 mM |
0.026 ± 0.002 | 3 | competitive |
| 5-F-xylulose | ATP | xylulose
0.28 mM |
- | substrate inhibition by ATP | |
| AMPPNP | ATP | xylulose
0.28 mM |
1.1 ± 0.1 | 3 | competitive |
| AMPPNP | ATP | xylulose
4.25 mM |
0.71 ± 0.10 | 3 | competitive |
| AMPPNP | xylulose | ATP
0.167 mM |
1.9 ± 0.09 | 4 | uncompetitive |
Kinetic Mechanism of XK from Initial Rate and Dead End Inhibition Patterns
The steady-state kinetic mechanism of XK was examined by determining initial rate patterns. A double reciprocal plot of initial rates recorded at varied concentrations of D-xylulose at several constant concentrations of MgATP shows an intersecting pattern (Figure 4A) implying that D-xylulose and MgATP must bind to the enzyme before the first product is released. Dead-end inhibitor binding studies were used to determine the order of substrate addition in the sequential kinetic mechanism of XK (Table 4). As described above, 5-F-xylulose and AMPPNP are competitive inhibitors with respect to D-xylulose and ATP respectively. AMPPNP is a linear uncompetitive inhibitor with respect to D-xylulose concentration, measured at Km levels of ATP (Figure 4B). The presence of 5-F-xylulose induced substrate inhibition by MgATP under conditions in which initial rates were measured at a varied concentration of MgATP and a constant Km concentration of D-xylulose, as shown in Figure 4C. These results suggest a kinetic mechanism of XK. The presence of ATPase activity in the absence of substrate indicates that the mechanism is probably formally random but a path in which substrate binds before MgATP is strongly preferred. In a predominantly ordered mechanism, binding of a dead-end inhibitor (I = AMPPNP) that is competitive with respect to the second substrate (B = MgATP) is expected to give uncompetitive inhibition with respect to the first substrate (A = D-xylulose). The proposed mechanism explains substrate inhibition by MgATP induced by 5-F-xylulose due to formation of an abortive E-I-B (I= 5-F-xylulose) complex that prevents release of the inhibitor, as shown in Figure 4D. In a fully random mechanism, substrate inhibition by B is not observed under the conditions in Figure 4C because the inhibitor readily dissociates from the E-I-B complex, resulting in E-B, which is free to combine with A to yield products. The results reveal that XK shares kinetic properties with E. coli ribulokinase 26 and yeast hexokinase 27. ecGK was reported to have a random kinetic mechanism 28 and ribulokinase from Klebsiella aerogenes appears to use a steady-state random mechanism 29.
Figure 4.
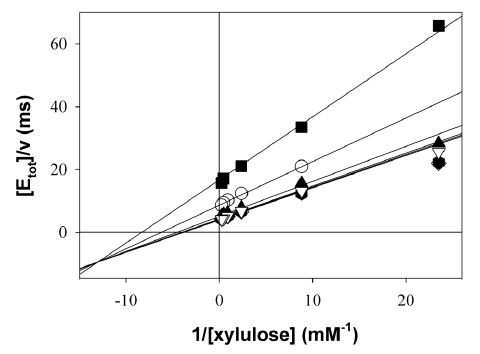
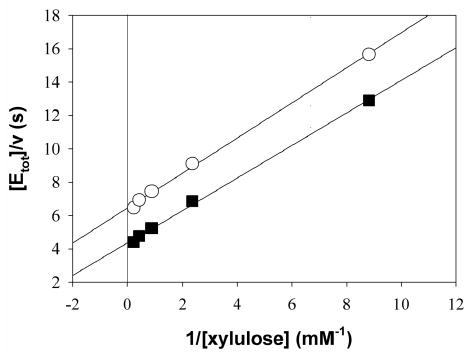
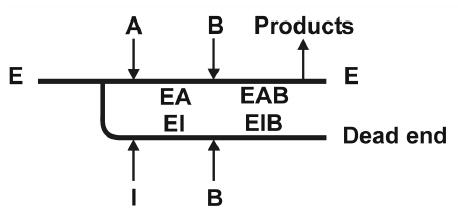
Kinetic results and proposed kinetic mechanism. A) Double-reciprocal plot of initial rate data for XK with D-xylulose varied at MgATP concentrations of 0.05 mM, squares; 0.13 mM, circles; 0.49 mM, triangles up; 1.46 mM, diamonds; 2.44 mM, hexagons; 4.88 mM, triangles down. Lines are calculated from a non linear fit of the data to eq. 2 B) Uncompetitive inhibition of 0.99 mM AMPPNP (circles) versus D -xylulose at 0.17 mM ATP. C) Substrate inhibition by ATP at 0.28 mM xylulose; squares uninhibited, circles 0.184 mM 5-F-xylulose. D) The kinetic mechanism for XK appears to involve ordered substrate binding with the sugar (A) binding first and MgATP (B) second. The formation of a dead-end EIB complex results in substrate inhibition by B.
Initial rates shown in Figure 4A were fitted to eq. 2 and kinetic parameters are summarized in Table 5. The apparent dissociation constants for the binary complexes of XK with D-xylulose (KiA) and 5-F-xylulose (Kic determined at a non-saturating MgATP level) are similar, indicating that the fluorine can substitute well for the reactive hydroxy group in the initial substrate binding recognition. This is an important result in view of the proposed mechanistic scenario for XK and related members of this family of carbohydrate kinases (see below for details) probably involves activation of the 5-OH through proton abstraction by a catalytic base. The fluorine obviously could not replace the original hydroxyl group if binary complex interactions involved a hydrogen bond from the 5-OH to the base. The findings suggest that formation of the ternary complex is required to correctly align the catalytic groups on the enzyme and the reactive parts of the substrates.
Table 5.
Kinetic constants for E. coli xylulokinase at pH 7.4 from fit of equation 2 to initial rate data.
| kcat | 263 ± 3 s−1 | |
| KiA | 0.074 ± 0.031 mM | A : xylulose |
| KmA | 0.27 ± 0.01 mM | B: MgATP |
| KmB | 0.17 ± 0.01 mM |
The predominantly ordered mechanism of XK implies that the value of kcat/Km essentially reflects the bimolecular rate constant of substrate binding (kon) to the free enzyme. kon therefore governs the sugar substrate selectivity (Table 3), probably through a conformational mechanism. The relative constancy of kcat under conditions when kon changes more than 5000-fold suggests that ternary complex interconversion is presumably the rate-determining step of the reaction (Table 3 and Fig. 4).
Conformational Changes and Substrate Binding
The subject of substrate-induced conformational changes in sugar kinases has been implied by the observed cooperative binding of substrates in many of them and the observation that there is restricted solvent accessibility in structures of bound substrates. Domain motions on the order of only 7º have been observed when comparing wild-type and mutant GK14 which are different from the 12º movement observed for the more distantly related hexokinase 30. The apo and xylulose-bound structures coupled with modelling indicate that the domain closure in XK is dramatically greater than movements seen in closely related carbohydrate kinases before. Overlaying the N-terminal domains of the unbound and xylulose-bound XK shows that xylulose binding induces a 12.2º closure which increases when ATP binds in modelling experiments (see below). The second molecule in the asymmetric unit of the apoenzyme structure is actually closed 1.0º relative to the xylulose-bound form indicating that there is conformational flexibility. Similar flexibility is also found in the case of human glucokinase which exists in three different forms: super-open, open and closed 31. The enzyme undergoes a conformational change from the super-open to open-form when glucose binds and then to closed-form in the presence of ATP. Assuming that the mechanism of synergistic binding described for human glucokinase can be applied to XK, the apo form may represent the super-open form and the xylulose-soaked structure corresponds to the open form.
A comparison of the apo and xylulose-bound forms of XK shows that the helical segment 294-299 (in α11) in domain IIA (Fig.1A, 1B, 5E and 6) acts as a hinge. Although there are no significant structural effects of hinge movement on domain I (Cα r.m.s. deviation between apo and xylulose bound structures is only 0.75Å), effects are seen on domain II (rmsd of 3.7 Å). The movements in domain II involve a rigid-body motion toward the domain I along with a slight 5º yaw across an axis between the N-term to C-term of the structure (Fig. 5A). The domain motion also induces a closure at the bottom of the cleft by bending the four parallel β-strands of domain IIA which are part of the interface between domain IA and IIA (Fig1A and B, Fig 5A).
Figure 5.

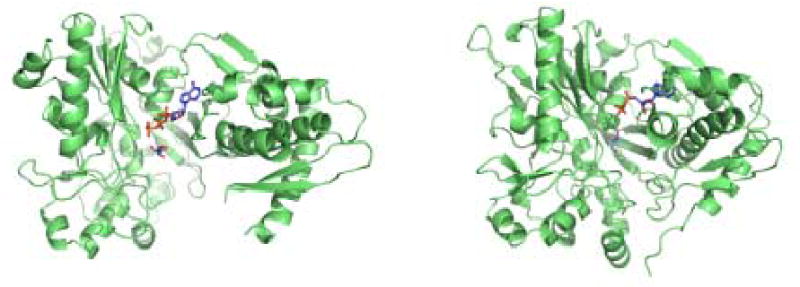
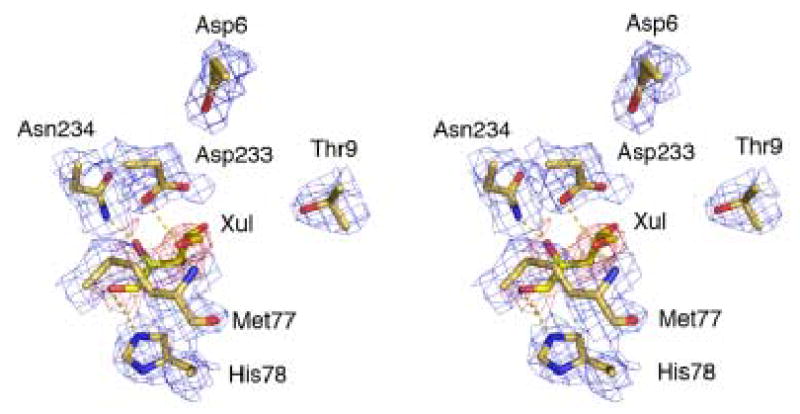
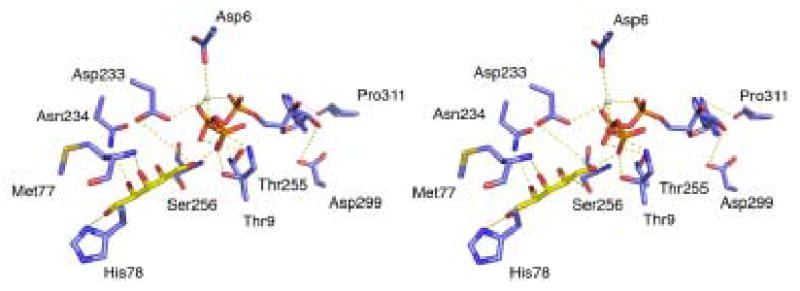
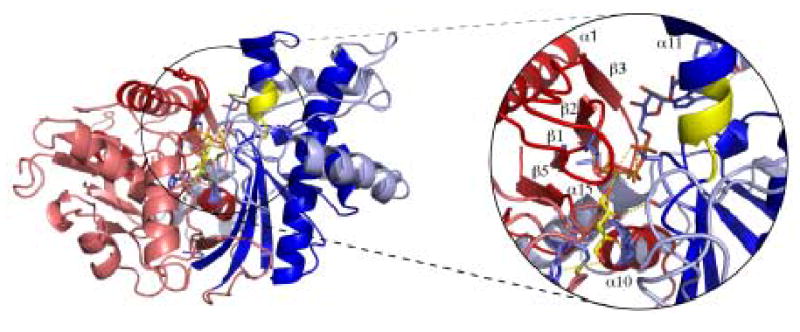
Substrate-induced conformational changes in XK and modeling of the ternary structure using ecGK. A) A stereo view of domains I overlaps from apo XK (blue), xylulose bound XK (green) and the ecGK ternary complex (red) shows a ~37° opening of the former relative to the latter. The domain motion is indicated. B) Model of the XK ternary complex in open conformation (left) and closed ternary structure of ecGK (right) both in the same relative orientation. C) 2Fo-Fc electron density maps of the xylulose binding site contoured at 1.2σ (blue) and Fo-Fc electron density at 2σ for xylulose (red). Hydrogen bonds are shown with a dashed yellow line. D) Interactions details of both xylulose and MgATP in the XK active site model in closed-conformation. Hydrogen bonds are shown with a dashed yellow line. Xylulose carbons are shown in yellow. E) XK ternary complex model in closed conformation. A zoomed view of the active site is shown to illustrate interactions from various secondary structural elements.
To determine conformational changes associated with ternary complex formation, XK crystals were soaked with xylulose and ATP-γS but rapidly dissolved, probably reflecting a change disrupting the lattice. Modeling was done to close xylulose-bound XK using the closed, ternary complex of ecGK as a template. This yielded a model in which the domains are now 37º closed relative to the apo form. Hydrogen bonding interactions mediating xylulose binding come from the sidechains of His78, Asp233, Asn234 and the Met77 backbone nitrogen, all belonging to the domain IA. Xylulose bridges the cleft by interacting with a mobile Ser256 which resides on a loop via a hydrogen bond from domain IIA. Non-polar interactions involving the side chain of Trp96 also contribute to xylulose binding in the active site. Compared to xylulose, xylitol is a much poorer substrate and is not synergistic with ATP binding (Table 3). Modeling indicates it is only stabilized by 2 hydrogen bonds (Thr9 and backbone nitrogen from Met77) and polar interactions with Trp96 and Asp233. It does not seem to bridge the cleft by interacting with Ser256 or any residues on the flexible loop from domain IIA as observed for the xylulose binding
The catalytic residues (Asp6, Thr9 and Asp233) are conserved across the ATPase superfamily including ecGK (Table 2, Fig. 5C) but the residues in the triphosphate binding site are more variable and none of the ATP binding residues are conserved in XK 23. Modeling ATP binding is therefore more difficult but plausible hydrogen bonds Asp6, Thr9 and Asp233 in domain IA and Thr255, Asp299 and Pro311 in domain IIA may bridge the cleft (Fig. 5D). In the model of the closed ternary complex, 4% of the 284 Å2 surface of the sugar is solvent accessible and this increases to 14% when the ATP molecule is removed, consistent with the kinetically ordered mechanism.
Catalytic Mechanism of XK
Structural overlays indicate Asp6 and Asp233 of XK are homologous in their positions to Asp10 and Asp245 of ecGK 23. A crucial role of Asp6 and Asp233 in the XK catalytic mechanism can be inferred from biochemical studies of relevant mutants (D10N, D245N) of ecGK as well as from the conservation pattern of the two residues in members of the carbohydrate kinase family 32. Analysis of kinetic consequences in D6A and D233A variants confirmed that both Asp residues are essential for XK activity. Enzymatic rates of phosphorylation of D-xylulose were at the limit of detection, ~4.5 orders of magnitude below the level of wild-type activity. The substrate-independent ATPase activity seen in the wild-type (Table 3), however, was much less affected than the phospho-group transfer activity in D233A mutant (kcat = 0.13 ± 0.01 s−1; KmATP = 8.2 ± 1.2 mM) which displays a ~2-fold increase in the original value of kcat for this activity. The ATPase activity of the D6A mutant (kcat ≈ 0.0038 s−1), by contrast, was significantly (19-fold) decreased, compared with the wild-type activity.
Results of structural analysis, modeling and site-directed mutagenesis suggest a mechanism for XK which is consistent with mechanisms for other family members such as ecGK but also reveals new details (Fig. 7). A conserved role of the side chain of Asp6 in coordinating and positioning the MgATP for catalysis, is supported for XK. An additional function served by Asp6 (in concert with Mg2+ ligated by it) might be the stabilization of the ADP leaving group during phospho-group transfer to sugar or water.
Figure 7.
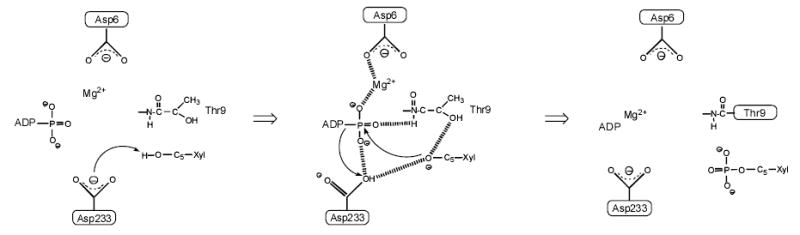
The proposed catalytic mechanism for XK.
The role of Asp233 is to deprotonate the D-xylulose hydroxyl at the 5 position, activating it for nucleophilic attack on the phosphate. The main chain NH group from Thr9 could stabilize the transition state and the associated negative charge development on O5 through a hydrogen bond (Fig. 7). The relative timing of bond forming and bond cleaving steps in the phospho-group transfer catalyzed by kinases and hydrolases as well as the question of whether the transition state has a rather dissociative or associative character have attracted much attention among enzymologists and is still under debate. The extent to which base catalysis contributes to stabilization of the transition state depends on the nature of bonding in it. Bond cleavage in the leaving group at little or no bond formation in the incoming nucleophile dominates in the dissociative transition state which is characterized by a decrease in the combined bond order to the incoming and departing groups. In that extreme case, base catalysis to the removal of a proton would provide no significant advantage. The 104.5-fold loss of activity in D233A and the charge-stabilizing hydrogen bond from the backbone amide of Thr9 would suggest a significant nucleophilic participation of the 5-OH in the transition state. The replacement of the putative catalytic base in related kinases showed a range of effects, but typically ≥ 102.7-fold losses of activity irrespective of the character of the residue introduced by the mutation. Although it is possible that Asp233 and its homologues in other kinases are involved in the correct positioning of the reactive hydroxyl group for nucleophilic attack, the magnitude of the functional disruption caused in the mutants suggests direct participation in catalysis, as a base, to be the major role of them.
MATERIALS AND METHODS
Materials
D-xylulose (≥ 95% pure) was produced by microbial oxidation of D-arabitol 33. 5-F-D-xylulose was synthesized by a reported protocol 34 and kindly provided by Prof. Arnold Stütz from the Institute of Organic Chemistry, Graz University of Technology. All other reagents were from Sigma or Merck.
Cloning, Protein Expression and Purification
The XK gene (xylB) was PCR-amplified from E. coli genomic DNA using the primers 5′-GCTAGTCCATATGTATATCGGGATAGATCTT-3′ and 5′-ACTGCCCGGGCGCCATTAATGGCAGAAGTTG-5′ (NdeI and SmaI sites are underlined). The resulting fragment was inserted into the plasmid pTYB2 (New England Biolabs) to yield the final bacterial expression vector which was sequenced to ensure no mutations were present. Recombinant XK was produced in fusion with plasmid-derived intein and chitin binding domains in the E. coli expression strain BL21*. Wild-type protein was expressed in LB with 100 μg/ml ampicillin and induced with 500 μM IPTG for 12 h at 15 °C. Cells were harvested 12 hours after induction, lysed using a microfluidizer and the lysate was clarified by centrifugation. The fusion protein were passed over a chitin column and washed with at least 20 column volumes of Buffer A (20 mM Tris, pH 8.0, 500 mM NaCl, 0.1 mM EDTA) supplemented with 0.1% Triton X-100 followed by 20 column volumes of Buffer A alone. Protein was cleaved from the column by incubating for 12 h in Buffer A with 50 mM 2-mercaptoethanol. XK was eluted in Buffer A, dialyzed into 20 mM Tris, pH 8.0, and further purified by anion exchange chromatography on a quaternized polyethyleneimine HQ column using a BioCad Sprint system. A 0 - 3 M NaCl gradient was applied and XK eluted at a salt concentration of 0.45 M. The purified protein was then exchanged into 20 mM Hepes, pH 7.4, with 20 mM NaCl and concentrated to 2 mg/ml (for kinetic studies) or 26 mg/ml (for crystallization). Preparations of wild-type and mutant enzymes migrated in denaturating SDS-PAGE as single protein bands to identical positions that were fully consistent with the expected molecular mass of 53 kDa for the full-length XK protomer (data not shown).
Calibrated gel filtration experiments were carried out on a BioCad Sprint chromatography system using a Superdex 200 HR 10/30 column. The column was equilibrated with PBS buffer containing 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.3 and loaded with purified XK at 0.3 mg/ml. Elution was carried out at a flow rate of 0.5 ml/min.
Site-directed Mutagenesis
The D6A and D233A mutations were created with a two-stage PCR protocol using the expression vector as the template 35. In the first step, two separate primer extension reactions were performed using the forward oligonucleotide primers: 5′-ATGTATATCGGGATAGCGCTTGGCACCTCG-3′ (for D6A) and 5′-GCAGGCGGTGGCGCGAATGCAGCTGGTGCAGTTGG-3′ (for D233A) and their complementary sequences respectively (mismatched base pairs are underlined). Each 50 μL reaction mixture contained 30 ng of template DNA, 15 pmol of each primer, 240 μM each dNTP and 3 U of Pfu DNA polymerase (Promega) in reaction buffer. Four cycles of separate amplification (95°C for 50 s, 60°C for 50 s and 68°C for 10 minutes) were followed by 18 identical cycles of amplification of the combined reaction mixtures. The final extension phase was 7 min at 68°C. After digestion of the template DNA with DpnI, the amplified plasmid vectors (10 μL) were used to transform E. coli TOP10 cells. Plasmid DNA from positive clones was entirely sequenced. Production and purification of XK mutants was performed as described above for the wild-type.
Crystallization and Data Collection
Purified XK was crystallized at 25° C by the hanging drop vapor diffusion method. Drops containing 1 μl of 26 mg/ml protein solution were mixed with 1 μl of the precipitant solution (1.5 M ammonium sulfate, 50 mM sodium citrate, pH 6, 1% w/v t-butanol) and suspended over a 1 ml reservoir containing the precipitant solution. Crystals used in data collection were transferred to a cryoprotectant solution consisting of 75% v/v precipitant solution, 25% v/v ethylene glycol and flash-cooled in a stream of liquid nitrogen at 110 K. XK crystals belonged to the space group P212121 with unit cell dimensions of a = 61.5 Å, b = 112.3 Å, and c = 142.4 Å. Similar conditions were used for the xylulose-bound form with the exception that 0.5 M xylulose was added to the harvest buffer. Native data for both enzyme forms was collected at beamline 9-1 at the Stanford Synchrotron Radiation Laboratory (SSRL) at 110 K using an ADSC CCD detector (Table 1).
Phasing, Structure Determination and Refinement
Attempts at molecular replacement using various unmodified and modified GK models as a search object were not successful for phasing the apo structure so de novo phasing was pursued. Derivative data were collected on a Rigaku R-AXIS IV. All of the data were reduced using the programs DENZO and SCALEPACK and non-negative intensities were used for phasing and refinement 36. Heavy atom refinement, density modification and phasing calculations were performed using the CCP4 package 37. Figures of merit before and after solvent flattening were 0.55 and 0.66 respectively at 3.0 Å.
Before refinement, 4% of the reflections were flagged for the calculation of Rfree. Water molecules were automatically picked in REFMAC5 and manually checked. Several rounds of crystallographic refinement and manual fitting and refitting using the programs CNS, REFMAC5, O and COOT produced the final model 38; 39; 40 with an Rcryst of 0.197 and Rfree of 0.259. The statistics associated with the final round of refinement are summarized in Table 1.
Dynamic light scattering
Dynamic light scattering experiments were performed on a Protein Solutions DynaPro 99 molecular sizing instrument. The protein concentration was approximately 1 mg/mL in 10 mM Hepes, pH 7.4.
Kinetic Assays
All initial rate measurements were performed with a Beckman Coulter DU 800 UV/Vis spectrophotometer equipped with a temperature-controlled cell holder. XK activity was determined with a continuous assay (at 25 ± 1°C) in which the XK-catalyzed production of ADP was coupled to the oxidation of NADH via reactions catalyzed by pyruvate kinase (PK) and lactate dehydrogenase (LDH). The depletion of NADH was monitored, typically for 10 min, as the decrease in absorbance at 340 nm (εNADH = 6220 M−1cm−1). Unless stated otherwise, reactions were performed in 50 mM Hepes buffer, pH 7.4, containing 10 mM MgCl2, 50 mM KCl, pH 7.4, 0.3 mM NADH, 1 mM phosphoenolpyruvate, 3 U/ml PK and 5.4 U/ml LDH. BSA (1 mg/ml) was added as an essential stabilizer of XK concentrations of the monomer were adjusted to 50 – 260 nM to give rates in the range 0.005 and 0.1 ΔA340/min. The standard assay contained 5 mM ATP and 4.3 mM D-xylulose. For the determination of kinetic parameters, substrate concentrations were varied as indicated in the results section. The ATP-hydrolyzing activity of XK was measured using ATP, at 10 mM or in the indicated varied concentrations, as the sole substrate. Reactions were always started by adding 10 μL of XK solution to 490 μL of preincubated substrate solution and blank rates were not significant under all conditions used. Dead-end inhibition studies used the above described assay and recorded initial rates in the presence of the reversibly binding inactive substrate analogues 5-F-xylulose orβ,γ-imidoadenosine 5′-triphosphate (AMPPNP).
Data fitting was performed using unweighted non-linear least-squares regression with the Sigmaplot program version 7. Apparent kinetic parameters were obtained using fits to eq. 1 of initial rates recorded under conditions in which one substrate concentration was varied and the other substrate concentration was constant and saturating. Eq. 2 describes an intersecting initial rate pattern for an ordered bi bi reaction. In eqs. 1 and 2, v is the initial rate, kcat is the catalytic center activity, [E] is the XK concentration; [A] and [B] are substrate concentrations; KA is an apparent Michaelis constant; KiA is the apparent dissociation constant for the binary complex of E and A; KmA and KmB are the Michaelis constants for A and B respectively.
| (eq. 1) |
| (eq. 2) |
Competitive and uncompetitive dead-end inhibition patterns were fitted to eqs. 3 and 4 respectively where [I] is the inhibitor concentration and Kic and Kiuc are the corresponding inhibition constants.
| (eq. 3) |
| (eq. 4) |
Molecular Modeling and Protein Domain Motion Analysis
The XK holo model with both ATP and xylulose was generated based on the E. coli glycerol kinase (ecGK) crystal structure in the closed conformation, bound with adenosine-5′-(β,γ -difluoromethylene) triphosphate, glycerol and Mg2+ (PDB: 1BWF). COOT was first used to overlay domain I of the xylulose-bound XK in the open conformation and holo ecGK structure in the closed conformation 39; 41; 42. This domain contains the ecGK catalytic residues Asp10, Thr13, Arg17 and Asp245 which were aligned with the analogous residues in XK: Asp6, Thr9, Lys13 and Asp233 15; 23; 43; 44; 45. ATP was manually positioned using COOT in the holo XK model based on this structural overlay. The xylulose was held in place based on the refined XK xylulose-bound open conformation structure. To generate a model of the open ternary complex, three rounds of both Cartesian restrained dynamics at 200 K and restrained energy minimization cycles using CNS were performed until ΔE < 0.05 kJ mol−1 Å−138.
A model of the XK ternary complex in closed conformation was created using SwissPDB-viewer with ecGK (PDB: 1BWF) as template 45. A pair-wise alignment of both XK and ecGK sequences with ClustalW 1.83 was used as a guide to superimpose the XK open structure onto the ecGK closed structure 19. Two rounds of energy minimization using CNS were performed to relieve possible steric clashes and overlaps. The xylulose position was derived from the refined XK open conformation structure. The same procedure as described above was applied to generate models of both opened and closed XK-ATP-xylitol complex.
Dyndom software from the CCP4 package was used to determine domains, hinge axes and hinge bending residues in apo XK apo by comparing with the crystal structure of ecGK in closed conformation (PDB: 1BWF) 46. The Hingefind algorithm was used with VMD 1.8.4 (MacOS X) to estimate the rotation angle of domain II toward domain I of XK 47; 48. Since the Hingefind algorithm requires two structures with the same number of atoms, only XK open structure and XK closed (model) was used. This gave a good estimate of the possible domain motion closure assuming XK and ecGK close in a similar manner. Accessible surface and volume calculations were done using both CNS v1.1 and areaimol (CCP4 package ) with a solvent probe radius of 1.4 Å 38.
Electron microscopy and image processing
3.5 μl of 0.5 mg/ml XK was applied to a holey carbon film coated electron microscopy grid (Quantifoil Micro Tools GmbH). The grid was blotted and quick-frozen by plunging into liquid ethane. The vitrified sample was mounted in a Gatan cryo sample holder and transferred to the microscope, a JEOL JEM 2100F operated at 200 keV 49. Images were recorded at a nominal magnification of 50,000× under minimum dose procedures on a Tietz 4096 × 4096 pixel CCD camera. A total of 487 particles were selected from 26 micrographs using the EMAN Boxer program 50. All the subsequent image processing was performed with the SPIDER program 51. The first 3D model was generated from two side views and one top view which came from reference-free alignment and classification 52 and then was blurred, thresholded and projected into a set of angle directions to produce references. The particles were then iteratively re-aligned onto the 50 class averages with a multi-reference alignment procedure, class-internally reference-free aligned and averaged. Class averages from the 39 classes that included more than three particles were used for the following 3D reconstruction, applying a two-fold symmetry to the reconstructed volume. In the first 5 iterations of 3D reconstruction, the model was masked by a sphere with the radius of 8 nm and 12~18 class average images were excluded due to their lower correlation coefficients to the initial model. In the next 30 iterations, a lower correlation coefficient threshold was applied so that only two class average images were excluded in the final 14 iterations, due to their lowest correlation coefficients to the model. The 3D model converged to a stable final model. The resolution was determined by Fourier shell correlation 53; 54 between two reconstructions from two half sets of class averages, employing a 0.5 criterion. The x-ray structure was not used during the entire electron microscopy image processing, except to determine the handedness of the final reconstruction.
Acknowledgments
Initial crystallization of XK by Youzhong Wan is gratefully acknowledged. This work was supported by the National Institutes of Health (GM66135 to D.K.W.), the Austrian Science Funds (FWF projects 15208 and 18275 to B.N.) and the Keck Foundation.
ABBREVIATIONS
- AMPPNP
5′-adenylyl-β, γ-imidodiphosphate
- XK
E. coli xylulose kinase
- ecGK
E. coli glycerol kinase
- NAD+
NADH: nicotinamide adenine dinucleotide (oxidized, reduced)
- NADP+
NADPH: nicotinamide adenine dinucleotide (oxidized,reduced)
- rmsd
root mean square deviation
- X5P
xylulose-5-phosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hahn-Hagerdal B, Wahlbom CF, Gardonyi M, van Zyl WH, Cordero Otero RR, Jonsson LJ. Metabolic engineering of Saccharomyces cerevisiae for xylose utilization. Adv Biochem Eng Biotechnol. 2001;73:53–84. doi: 10.1007/3-540-45300-8_4. [DOI] [PubMed] [Google Scholar]
- 2.Jeffries TW, Jin YS. Metabolic engineering for improved fermentation of pentoses by yeasts. Appl Microbiol Biotechnol. 2004;63:495–509. doi: 10.1007/s00253-003-1450-0. [DOI] [PubMed] [Google Scholar]
- 3.Lawlis VB, Dennis MS, Chen EY, Smith DH, Henner DJ. Cloning and sequencing of the xylose isomerase and xylulose kinase genes of Escherichia coli. Appl Environ Microbiol. 1984;47:15–21. doi: 10.1128/aem.47.1.15-21.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wungsintaweekul J, Herz S, Hecht S, Eisenreich W, Feicht R, Rohdich F, Bacher A, Zenk M. Phosphorylation of 1-deoxy-D-xylulose by D-xylulokinase of Escherichia coli. Eur J Biochem. 2001;268:310–316. doi: 10.1046/j.1432-1033.2001.01875.x. [DOI] [PubMed] [Google Scholar]
- 5.Eisenreich W, Schwarz M, Cartayrade A, Arigoni D, Zenk MAB. The deoxyxylulose phosphate pathway of terpenoid biosynthesis in plants and microorganisms. Chem Biol. 1998;5:R221–R233. doi: 10.1016/s1074-5521(98)90002-3. [DOI] [PubMed] [Google Scholar]
- 6.David S, Estramareix B, Fischer J-C, Therisod M. 1-deoxy-D-threo-2-pentulose: The precursor of the five-carbon chain of the thiazole of thiamine. J Am Chem Soc. 1981;103:7341–7342. [Google Scholar]
- 7.Hill RE, Sayer BG, Spenser JD. Biosynthesis of vitamin B6: incorporation of D-1-deoxyxylulose. J Am Chem Soc. 1989;111:1916–1917. [Google Scholar]
- 8.Rodriguez-Pena JM, Cid VJ, Arroyo J, Nombela C. The YGR194c (XKS1) gene encodes the xylulokinase from the budding yeast Saccharomyces cerevisiae. FEMS Microbiol Lett. 1998;162:155–160. doi: 10.1111/j.1574-6968.1998.tb12993.x. [DOI] [PubMed] [Google Scholar]
- 9.Jin YS, Jones S, Shi NQ, Jeffries TW. Molecular cloning of XYL3 (D-xylulokinase) from Pichia stipitis and characterization of its physiological function. Appl Environ Microbiol. 2002;68:1232–9. doi: 10.1128/AEM.68.3.1232-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc Natl Acad Sci USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett WS, Jr, Steitz TA. Glucose-induced conformational change in yeast hexokinase. Proc Natl Acad Sci U S A. 1978;75:4848–52. doi: 10.1073/pnas.75.10.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flaherty KM, McKay DB, Kabsch W, Holmes KC. Similarity of the three-dimensional structures of actin and the ATPase fragment of a 70-kDa heat shock cognate protein. Proc Natl Acad Sci U S A. 1991;88:5041–5. doi: 10.1073/pnas.88.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hurley JH, Faber HR, Worthylake D, Meadow ND, Roseman S, Pettigrew DW, Remington SJ. Structure of the regulatory complex of Escherichia coli IIIGlc with glycerol kinase. Science. 1993;259:673–7. [PubMed] [Google Scholar]
- 14.Yeh JI, Charrier V, Paulo J, Hou L, Darbon E, Claiborne A, Hol WG, Deutscher J. Structures of enterococcal glycerol kinase in the absence and presence of glycerol: correlation of conformation to substrate binding and a mechanism of activation by phosphorylation. Biochemistry. 2004;43:362–73. doi: 10.1021/bi034258o. [DOI] [PubMed] [Google Scholar]
- 15.Ormo M, Bystrom CE, Remington SJ. Crystal structure of a complex of Escherichia coli glycerol kinase and an allosteric effector fructose 1,6-bisphosphate. Biochemistry. 1998;37:16565–72. doi: 10.1021/bi981616s. [DOI] [PubMed] [Google Scholar]
- 16.Flachner B, Varga A, Szabo J, Barna L, Hajdu I, Gyimesi G, Zavodszky P, Vas M. Substrate-Assisted Movement of the Catalytic Lys 215 during Domain Closure: Site-Directed Mutagenesis Studies of Human 3-Phosphoglycerate Kinase. Biochemistry. 2005;44:16853–65. doi: 10.1021/bi051726g. [DOI] [PubMed] [Google Scholar]
- 17.Zecchinon L, Oriol A, Netzel U, Svennberg J, Gerardin-Otthiers N, Feller G. Stability domains, substrate-induced conformational changes, and hinge-bending motions in a psychrophilic phosphoglycerate kinase. A microcalorimetric study. J Biol Chem. 2005;280:41307–14. doi: 10.1074/jbc.M506464200. [DOI] [PubMed] [Google Scholar]
- 18.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 19.Panchenko AR, Bryant SH. A comparison of position-specific score matrices based on sequence and structure alignments. Protein Sci. 2002;11:361–70. doi: 10.1110/ps.19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernstein BE, Michels PA, Hol WG. Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature. 1997;385:275–8. doi: 10.1038/385275a0. [DOI] [PubMed] [Google Scholar]
- 21.Kuser PR, Krauchenco S, Antunes OA, Polikarpov I. The high resolution crystal structure of yeast hexokinase PII with the correct primary sequence provides new insights into its mechanism of action. J Biol Chem. 2000;275:20814–21. doi: 10.1074/jbc.M910412199. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Lou Y, Yokota H, Adams PD, Kim R, Kim SH. Crystal structures of an NAD kinase from Archaeoglobus fulgidus in complex with ATP, NAD, or NADP. J Mol Biol. 2005;354:289–303. doi: 10.1016/j.jmb.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 23.Feese MD, Faber HR, Bystrom CE, Pettigrew DW, Remington SJ. Glycerol kinase from Escherichia coli and an Ala65-->Thr mutant: the crystal structures reveal conformational changes with implications for allosteric regulation. Structure. 1998;6:1407–18. doi: 10.1016/s0969-2126(98)00140-3. [DOI] [PubMed] [Google Scholar]
- 24.Zhu X, Zhao X, Burkholder WF, Gragerov A, Ogata CM, Gottesman ME, Hendrickson WA. Structural analysis of substrate binding by the molecular chaperone DnaK. Science. 1996;272:1606–14. doi: 10.1126/science.272.5268.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson RC, Turbedsky K, Kaiser DA, Marchand JB, Higgs HN, Choe S, Pollard TD. Crystal structure of Arp2/3 complex. Science. 2001;294:1679–84. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 26.Lee LV, Gerratana B, Cleland WW. Substrate specificity and kinetic mechanism of Escherichia coli ribulokinase. Arch Biochem Biophys. 2001;396:219–24. doi: 10.1006/abbi.2001.2613. [DOI] [PubMed] [Google Scholar]
- 27.Yang VW, Jeffries TW. Regulation of phosphotransferases in glucose- and xylose-fermenting yeasts. Appl Biochem Biotechnol. 1997;63–65:97–108. doi: 10.1007/BF02920416. [DOI] [PubMed] [Google Scholar]
- 28.Pettigrew DW, Yu GJ, Liu Y. Nucleotide regulation of Escherichia coli glycerol kinase: initial-velocity and substrate binding studies. Biochemistry. 1990;29:8620–7. doi: 10.1021/bi00489a018. [DOI] [PubMed] [Google Scholar]
- 29.Neuberger MS, Hartley BS, Walker JE. Purification and properties of D-ribulokinase and D-xylulokinase from Klebsiella aerogenes. Biochem J. 1981;193:513–24. doi: 10.1042/bj1930513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett WS, Jr, Steitz TA. Structure of a complex between yeast hexokinase A and glucose. II. Detailed comparisons of conformation and active site configuration with the native hexokinase B monomer and dimer. J Mol Biol. 1980;140:211–230. doi: 10.1016/0022-2836(80)90103-5. [DOI] [PubMed] [Google Scholar]
- 31.Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure. 2004;12:429–38. doi: 10.1016/j.str.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Pettigrew DW, Smith GB, Thomas KP, Dodds DC. Conserved active site aspartates and domain-domain interactions in regulatory properties of the sugar kinase superfamily. Arch Biochem Biophys. 1998;349:236–45. doi: 10.1006/abbi.1997.0444. [DOI] [PubMed] [Google Scholar]
- 33.Mayer G, Kulbe KD, Nidetzky B. Utilization of xylitol dehydrogenase in a combined microbial/enzymatic process for production of xylitol from D-glucose. Appl Biochem Biotechnol. 2002;98–100:577–589. doi: 10.1385/abab:98-100:1-9:577. [DOI] [PubMed] [Google Scholar]
- 34.Hadwiger P, Mayr P, Nidetzky B, Stütz AE, Tauss A. Synthesis of 5,6-di-modified open-chain D-fructose derivatives and their properties as substrates of bacterial polyol dehydrogenase. Tetrahedron: Asymmetry. 2002;11:607–620. [Google Scholar]
- 35.Wang W, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange Site-Directed Mutagenesis. Biotechniques. 1999;26:680–2. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-Ray Diffraction Data Collected in Oscillation Model. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 38.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 40.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 41.Bystrom CE, Pettigrew DW, Branchaud BP, O’Brien P, Remington SJ. Crystal structures of Escherichia coli glycerol kinase variant S58-->W in complex with nonhydrolyzable ATP analogues reveal a putative active conformation of the enzyme as a result of domain motion. Biochemistry. 1999;38:3508–18. doi: 10.1021/bi982460z. [DOI] [PubMed] [Google Scholar]
- 42.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 43.Mao C, Ozer Z, Zhou M, Uckun FM. X-Ray structure of glycerol kinase complexed with an ATP analog implies a novel mechanism for the ATP-dependent glycerol phosphorylation by glycerol kinase. Biochem Biophys Res Commun. 1999;259:640–4. doi: 10.1006/bbrc.1999.0816. [DOI] [PubMed] [Google Scholar]
- 44.Guex N, Diemand A, Peitsch MC. Protein modelling for all. Trends Biochem Sci. 1999;24:364–7. doi: 10.1016/s0968-0004(99)01427-9. [DOI] [PubMed] [Google Scholar]
- 45.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–23. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 46.Hayward S, Berendsen HJ. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998;30:144–54. [PubMed] [Google Scholar]
- 47.Wriggers W, Schulten K. Protein domain movements: detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins. 1997;29:1–14. [PubMed] [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 49.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimans. Quart Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 50.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 51.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–9. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 52.Frank J. Three-dimensional electron microscopy of macromolecular assemblies. Ch.4. Academic Press; San Diego, CA: 1996. [Google Scholar]
- 53.van Heel MJ. Similarity measures between images. Ultramicroscopy. 1987;21 [Google Scholar]
- 54.Unser M, Trus BL, Steven AC. A new resolution criterion based on spectral signal-to-noise ratios. Ultramicroscopy. 1987;23:39–52. doi: 10.1016/0304-3991(87)90225-7. [DOI] [PubMed] [Google Scholar]
- 55.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]