

Abstract
Prostacyclin synthase (PGIS) catalyzes an isomerization of prostaglandin H2 to prostacyclin, a potent mediator of vasodilation and anti-platelet aggregation. Here, we report the crystal structure of human PGIS at 2.15 Å resolution, which represents the first three-dimensional structure of a class III cytochrome P450. While notable sequence divergence has been recognized between PGIS and other P450s, PGIS exhibits the typical triangular prism-shaped P450 fold with only moderate structural differences. The conserved acid–alcohol pair in the I helix of P450s is replaced by residues G286 and N287 in PGIS, but the distinctive disruption of the I helix and the presence of a nearby water channel remain conserved. The side-chain of N287 appears to be positioned to facilitate the endoperoxide bond cleavage, suggesting a functional conservation of this residue in O–O bond cleavage. A combination of bent I helix and tilted B′ helix creates a channel extending from the heme distal pocket, which seemingly allows binding of various ligands; however, residue W282, placed in this channel at a distance of 8.4 Å from the iron with its indole side-chain lying parallel with the porphyrin plane, may serve as a threshold to exclude most ligands from binding. Additionally, a long “meander” region protruding from the protein surface may impede electron transfer. Although the primary sequence of the PGIS cysteine ligand loop diverges significantly from the consensus, conserved tertiary structure and hydrogen bonding pattern are observed for this region. The substrate-binding model was constructed and the structural basis for prostacyclin biosynthesis is discussed.
Keywords: prostacyclin synthase, class III cytochrome P450, hemoprotein, CYP8A1, crystal structure
Cytochromes P450 are heme-containing enzymes found in prokaryotic and eukaryotic cells. They are involved in various vital processes, including degradation of xenobiotics, biosynthesis of steroids and lipids as well as drug metabolism and carcinogenesis.1 P450s can be categorized according to their redox partners.2 Class I P450s, found in bacteria and mitochondrial membranes of eukaryotes, have two electron-transfer partners: a reductase, which is an NAD(P)H-dependent flavoprotein, and an iron-sulfur protein serving as the immediate electron donor to P450. Class II P450s, found mostly in the membranes of the endoplasmic reticulum, use only one electron transfer partner, P450 reductase. Class III P450s, such as prostacyclin synthase (PGIS), do not require any electron-transfer partner or molecular oxygen, as endoperoxides or hydroperoxides are their sole substrates. There are also a few P450s that, for example, catalyze a peroxide-dependent hydroxylation or obtain electron directly from NAD(P)H, and do not belong to the above categories but are grouped as class IV P450s. Bacterial P450s, which are mostly class I P450s, are soluble proteins and, not surprisingly, were the first few P450 crystal structures determined.3 Eukaryotic P450s, however, are mostly membrane-bound. Recently, several class II P450s were engineered to be soluble by deletion of the N-terminal transmembrane domain and an optional modification of a peripheral membrane-binding site. These endeavours circumvent the difficulty of crystallizing the membrane-bound form and their crystal structures were thus solved.4 Class IV P450s identified to date are soluble proteins and some members are structurally known.5,6 Nonetheless, there is no X-ray structure of a class III P450. Thus, the wide diversity of P450s will be helpful in the determination of tertiary structure of a class III P450 for elucidating the differences, subtle or large, in active site structure as well as providing information crucial for drug design.
PGIS catalyzes an isomerization of prostaglandin H2 (PGH2) to prostacyclin (PGI2), which is a highly potent vasodilator and inhibits platelet aggregation.7 It is expressed primarily in endothelial and smooth muscle cells, and is associated with the endoplasmic reticulum and the nuclear membrane.8,9 In tissues, the enzyme is found mainly in the aorta. PGIS cDNA was first cloned by us and Tanabe’s group from bovine aorta,10,11 and was designated as CYP8A1. The human counterpart was isolated subsequently.12
Sequence alignment of PGIS has been made with P450BM-3 by incorporating secondary structural predictions and experimental mutagenesis studies.13 Notably, the alignment identified two divergent regions in PGIS. One region is the Cys ligand loop, which consists of the consensus sequence of F(G/S)XGX(H/R)XCXG.14 In PGIS, the sequence of the Cys ligand loop is WGAGHN-HCLG. Furthermore, a highly conserved region located in the I helix with an acid–alcohol pair in the consensus sequence of (A/G)GX(E/D)(T/S) is absent from PGIS. The protein per se also exhibits unusual features. For example, PGIS has only a few known heme ligands, in contrast to many P450s, which bind various sizes and shapes of ligands. It was thus inferred that PGIS has a very limited space with a low “ceiling” at the heme distal side.15 In addition, the ferrous–CO complex of PGIS is very unstable.16 Collectively, analysis from sequence alignment and results from protein characterization suggest that PGIS is not a typical P450 with respect to the heme-binding and active site environments.
We have reported a high-yield prokaryotic system for expression of human PGIS.16 The resulting recombinant PGIS was purified to electrophoretic homogeneity with enzymatic activity higher than that of the native PGIS isolated from bovine aorta. Using this recombinant expression system, we report here the determination of the crystal structure of PGIS (Table 1) and elucidation of its exceptional features.
Table 1.
Summary of crystallographic analysis
| Space group | P21 | ||
| Unit cell dimensions | |||
| a (Å) | 67.4 | ||
| b (Å) | 105.6 | ||
| c (Å) | 73.9 | ||
| β (deg.) | 91.9 | ||
| Data collection | Se-edge | Se-remote | Native |
| Wavelength (Å) | 0.97930 | 0.95370 | 0.980127 |
| Resolution (Å) | 30–2.48 | 30–2.48 | 30–2.15 |
| Observed reflections | 184,871 | 185,485 | 131,803 |
| Unique reflections | 35,430 | 35,364 | 51,322 |
| Completenessa (%) | 95.7 (91.1) | 96.3 (97.9) | 90.9 (93.5) |
| Multiplicity | 5.2 | 5.2 | 2.8 |
| Rsymb (%) | 9.2 (34.1) | 7.0 (27.3) | 6.3 (35.0) |
| Refinement | |||
| Resolution range (Å) | 20.15–2.15 | ||
| No. of reflection in working set (test set) | 48,702 (2601) | ||
| Rcryst a,c (%) | 19.9 (21.4) | ||
| Rfree a (%) | 26.1 (30.5) | ||
| rmsd from ideal | |||
| Bond lengths (Å) | 0.011 | ||
| Bond angles (deg.) | 1.4 | ||
Human PGIS with MAKKTSS replacing the first 18 amino acid residues and the addition of a histidine tag at the C terminus has been described.16 The recombinant PGIS was purified with modifications to the previous procedure. Briefly, bacterial expressed PGIS was harvested by sonication, solubilization with 25 mM n-octyl-β-D-glucopyranoside, and ultracentrification. It was then purified through chromatographic columns with detergent-free buffers. The successive columns are nickel nitrilotriacetate affinity column, CM-Sepharose ion-exchange column and Superdex 200 gel-filtration column. The elution buffer used for gel filtration chromatography was 20 mM Tris–HCl (pH 8.0) and 150 mM NaCl. The recombinant PGIS is a monodisperse monomer, suitable for crystallization trials. Purified PGIS has an A418/A280 ratio of 1.10 and was concentrated to 25 mg/ml for crystallization. For phase determination, selenomethionyl PGIS was prepared using E. coli strain B834. The bacteria were grown in M9 medium containing 0.25 mM L(+)-selenomethionine and 0.5 mM δ-aminolevulinic acid. Expression of PGIS was induced by 0.5 mM IPTG, and the culture was continued for ~18 h at 25 °C. Selenomethionyl PGIS was purified following the procedures used for the native recombinant PGIS, except that reducing agents were present; 4 mM β-mercaptoethanol was included in the buffers for cell disruption and nickel affinity column chromatography and 1 mM DTT for CM-Sepharose and gel-filtration column chromatography. The native recombinant PGIS (25 mg/ml in the gel-filtration buffer) was found to crystallize spontaneously into needle clusters in the Eppendorf tube when stored at 4 °C for about a week. A combination of a microseeding technique and the hanging-drop, vapor-diffusion method was then used to improve crystal quality. Specifically, seed stock was prepared by crushing these initial PGIS needle clusters with the Seed Bead Kit (Hampton Research). Following four tenfold serial dilutions of the seed stock using the gel-filtration buffer, 1 μl of diluted seeds was added into a 10 μl drop of freshly concentrated PGIS sample, and equilibrated at 4 °C against 450 μl of the gel-filtration buffer. High-quality single crystals of PGIS were most often obtained using 100-fold or 1000-fold diluted seed stock, and reached suitable size (~0.05 mm×0.05 mm×0.1 mm) within one week. Crystals of selenomethionyl PGIS were produced by the same procedure using seed stock of the native protein. Before X-ray data collection, crystals of native and selenomethionyl PGIS were transferred to the cryo-stabilization solution (the gel-filtration buffer plus additional 35% (w/v) PEG400) for approximately 10 s, and immediately flash-frozen by plunging into liquid N2. A multiwavelength anomalous diffraction (MAD) data set was collected from a single Se-labeled crystal at two different wavelengths (Se inflection and high-remote) on beamline SP12B2 (SPring8, Japan), and a data set for the native protein was collected on beamline BL13B1 (NSRRC, Taiwan). All diffraction data were processed with the HKL2000 suite of programs.35 For the MAD experiment, data from each wavelength were indexed according to the same crystal orientation matrix, but integrated and scaled independently. Initial phases were obtained at 2.48 Å using the program SOLVE.36 These preliminary phases were improved and extended to 2.15 Å by density modification with the program RESOLVE.37 The resulting experimental electron-density map was of excellent quality, which allowed RESOLVE38,39 to build 757 out of a total of 1048 residues in the asymmetric unit. The program O40 and REFMAC541 were then used for rounds of manual model rebuilding and refinement. Two PGIS molecules (designated chains A and B) are present in the asymmetric unit. No electron density is associated with residues 320–327 of chain A or residues 319–330 of chain B, and these regions were omitted from the model. The final model consists of 471 residues in chain A, 468 residues in chain B, two heme groups, one sodium ion and 457 water molecules. A total of 93.2% and 6.6% of non-glycine residues have main-chain torsion angles in the most favored and generously allowed regions of the Ramachandran plot, respectively. While the main-chain torsion angles for W39 and H130 of both chains fall in the disallowed region, they fit very well to the electron density. Superposition of the two crystallographically independent chains gives a root-mean-square difference in main-chain atoms of 0.6 Å over 467 residue pairs, indicating that their conformations are generally similar.
Statistics for data from the resolution shell of 2.57–2.48 Å (Se data) and 2.21–2.15 Å (native data).
Rsym = (Σ|Ihkl−<I>|)/(ΣIhkl), where the average intensity <I> is taken overall symmetry equivalent measurements and Ihkl is the measured intensity for any given reflection.
Rcryst = (Σ||Fo|−k|Fc||)/(Σ|Fo|).
Structural characteristics of PGIS as a P450
It has been widely appreciated that, despite having low levels of sequence identity and different redox partners, class I, II and IV P450s possess the same tertiary structure and have a well-conserved heme-binding structural core. This perception is further strengthened with our finding that PGIS exhibits the typical triangular prism-shaped P450 fold with an edge length of approximately 65 Å and a thickness of approximately 40 Å. The PGIS structure contains three β-sheets and 12 α-helices for residues R23–P500 (Figure 1(a)), where the residue numbering corresponds to that of the native enzyme. A structural comparison performed by using the DALI server17 revealed that PGIS is most similar to that of CYP51 (PDB ID 1EA1),18 which catalyzes the oxidative removal of the 14α-methyl group of sterol via three successive monooxygenation reactions. The two can be aligned with a root-mean-square difference in Cα atoms of 3.4 Å over 410 structurally equivalent residue pairs.
Figure 1.
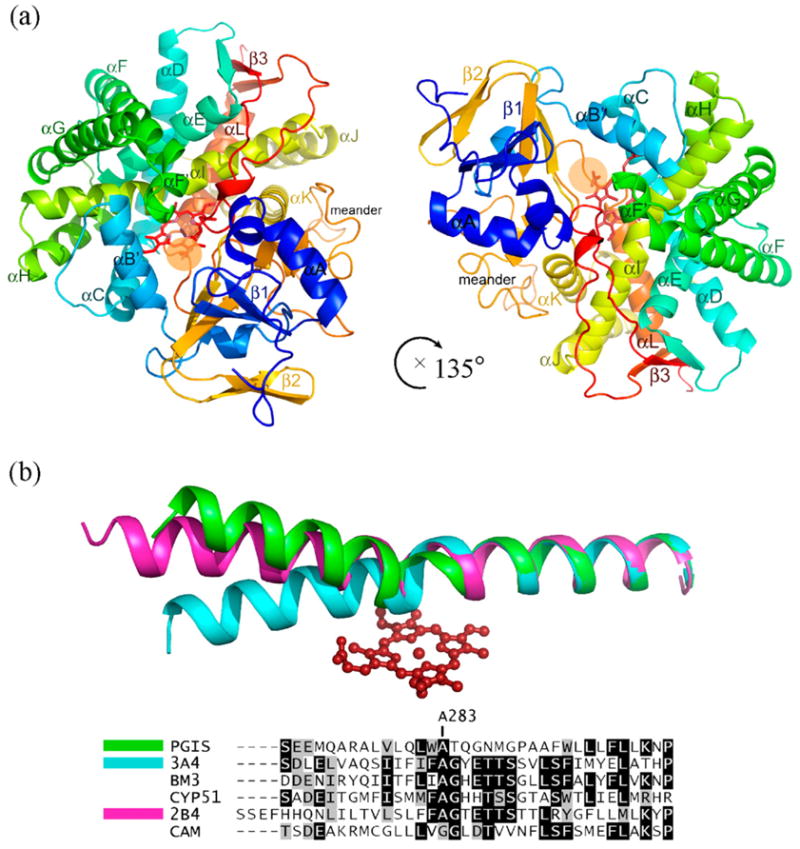
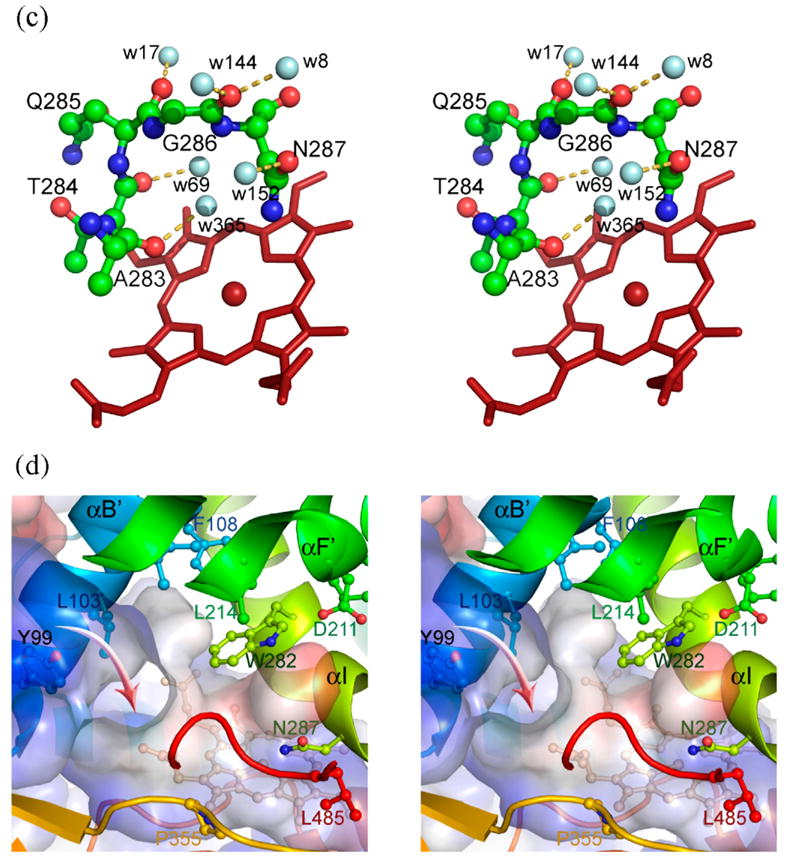
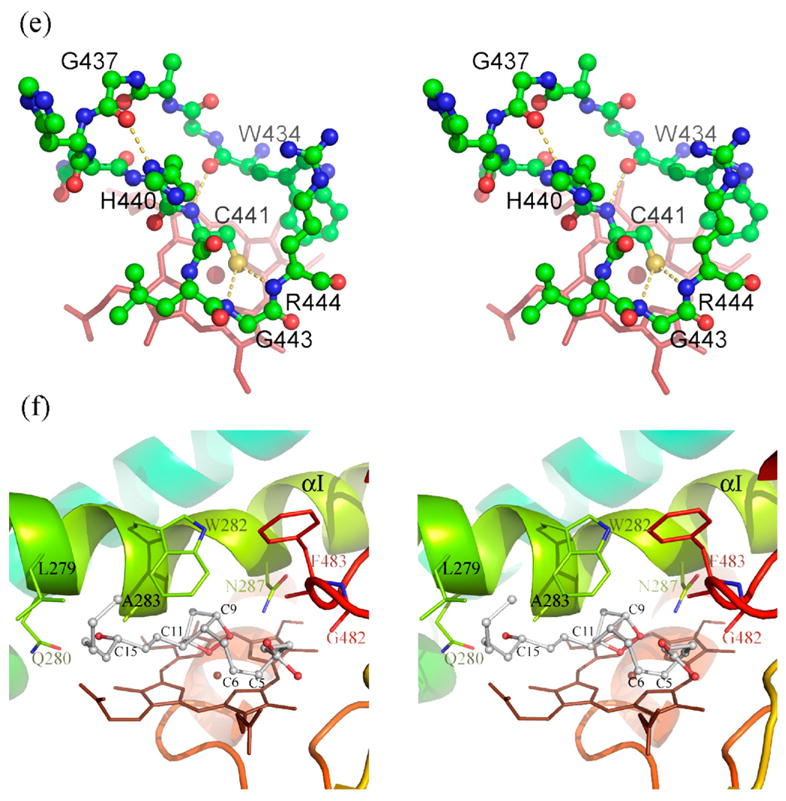
Structure of human prostacyclin synthase. (a) Ribbon representation of PGIS viewed from substrate entrance channel (beige circle). The structure is rainbow-colored with the N terminus in blue and the C terminus in red. The heme is shown as a stick model in the center of the molecule. Major helices and β sheets are labeled. (b) Structural overlay (top panel) and sequence alignment (bottom panel) of the I helix in known P450 structures. Each structure was superimposed pairwise with PGIS at the C-terminal half. The highly conserved alanine residue is indicated in accordance with PGIS sequence. Black-shaded or gray-shaded residues in the bottom panel correspond to those that share more than 50% of identity or similarity, respectively. (c) Stereoview of the central I helix region of PGIS. The color coding is: oxygen, red; nitrogen, blue; carbon, green. Water molecules are depicted as cyan spheres. H-bonds are shown as broken lines. (d) Stereoview of the substrate-binding channel and active-site chamber of PGIS. The coloring scheme is the same as that in (a). Structural elements and a part of residues constituting the active site are indicated. The solvent-accessible molecular surface is shown as a semitransparent envelope in cyan. The curved arrow indicates the entrance of the substrate-binding channel. (e) Stereoview of the PGIS cysteine ligand loop. Residues are shown. The color coding is the same as that in (c) and broken lines indicate H-bonds. (f) Stereoview of the substrate-binding model. PGH2 is depicted in ball and stick; the carbon atoms are shown in gray and oxygen atoms are in red. The numberings of key carbon atoms in PGH2 are also indicated. The Figure was generated using PyMol [http://www.pymol.org].
The I helix and its functional aspects
Although the overall fold is similar to other P450s, PGIS differs in the location of several helices, heme environment and the meander region. The significant differences include the B′, F, G and I helices, which are important for substrate binding. The I helix (residues E270–L300) in PGIS is disrupted in the middle such that the N-terminal portion bends upward from the porphyrin plane (Figure 1(b)). The bent helix pushes up the F/G loop, while the B′ helix leans toward the I-helix, thus creating a channel that connects the heme distal pocket to the protein surface (Figure 1(a)). Mutagenesis studies have suggested that I67, V76, P355, E360, and L384 in PGIS likely line the substrate-binding pocket.13 As residues P355, E360, and L384 are located in this channel with their side-chains forming part of the channel lining, it is straightforward to speculate that this would be the substrate-binding channel for PGIS. The I helix plays a critical role in P450 catalysis because it shapes the distal pocket, and a conserved acid–alcohol pair is involved in the hydroxylation reaction.19 A structural feature in the region of the acid–alcohol pair is a disruption of the helix. Together with several neighboring water molecules, the residues in the acid–alcohol region are involved in protonation of the ferric peroxo intermediate in P450 catalysis.20,21 PGIS has no acid–alcohol pair in the helix. Instead, G286–N287 are present in the corresponding positions (Figure 1(c)). The carbonyl oxygen atom of G286 forms hydrogen bonds to two water molecules (Wat144 and Wat8, Figure 1(c)), which in turn form H-bonds to D211 located at the F helix and L485 at the β3-2 strand. This bridge structure is very similar to that in P450terp, in which the carboxylate group of the acidic residue (i.e. D270) forms an H-bond to Q185 (at the F helix) and a salt-bridge with K419 (at the β3-2 strand).22 In addition, an H-bond between Wat17 and Q285 of PGIS has its structural counterpart in P450terp. Two PGIS water molecules, Wat365 and Wat69, form H-bonds to the carbonyl oxygen atom of A283 and the side-chain OH of T284, respectively. Residue A283, whose carbonyl oxygen atom is only 4 Å from the iron, is homologous to A264 of P450BM3 and A267 of P450terp. In both P450s, the carbonyl oxygen atom of the alanine forms an H-bond to the distal water ligand. The Wat69-mediated disruption of PGIS I helix resembles the case in P450cam in which the carbonyl oxygen atom of G249, the second residue preceding the acid–alcohol pair of D251–T252, is H-bonded to a water molecule bound at the “back” of the helix.23 Collectively, positions of several well-ordered water molecules in the vicinity of the I helix are maintained in PGIS, suggesting that the enzyme is positioned for protonation of the ferric peroxo intermediate, an earlier step of hydroxylation. It is worth noting that thromboxane synthase, a class III P450, was found to catalyze hydroxylation of prostaglandin H2 analogs in the presence of iodosylbenzene.24 For residue N287, the side-chain carbonyl oxygen atom forms H-bond to Wat152, and the side-chain amide group is positioned 4.7 Å from the iron. Interestingly, P450cin, which catalyzes the mono-oxygenation of 1,8-cineole, also contains asparagine at this position with its side-chain amide group H-bonding to the ether oxygen atom of the substrate.25 However, the structure of ordered water molecules is absent and I helix kink is not obvious, suggesting that P450cin adopts different structural elements for mono-oxygenation. Instead, the role of the asparagine residue in PGIS appears to be homologous to N137 of coral allene oxide synthase,26 a catalase-type protein that converts the fatty acid hydroperoxide to an epoxide. In coral allene oxide synthase, this side-chain amide group binds the distal oxygen atom of the hydroperoxide, causing homolysis of the peroxide bond that subsequently produces an alkoxyl radical. Previous studies have shown that PGIS catalyzes homolysis of the peroxide bond of a hydroperoxide, accompanied by the formation of an alkoxyl radical and a heme intermediate with a compound II-like optical spectrum.16 Therefore, the functional role of the threonine/serine residue in P450 mono-oxygenases, to be involved in O–O bond cleavage, is likely conserved as N287 in PGIS.
Substrate-binding region
The B′ helix (residues H97–F108) is long and oriented almost perpendicular to the heme, as opposed to a short B′ helix that mostly runs parallel or downward in other P450s. Like microsomal P450s, which usually have longer F and G helices than prokaryotic P450s,27 PGIS also has long F and G helices. The F helix in PGIS is interrupted by one turn of a 310 helix and is therefore divided into an F helix (residues H190–D211) and an F′ helix (residues L214–R219). The F′ helix lies above and is almost parallel with the porphyrin plane, making it a good candidate for interacting with substrate. Supporting this notion are NMR studies that showed the residues L214 and P215 in a short peptide probe contacted a PGH2 analog.28 Following the F′ helix, the direction of the main chain changes abruptly, causing the F/G loop (residues G220–S223) to lie almost perpendicular to the porphyrin plane. The G helix is disrupted by a coil near the C terminus as well, and is divided into a G helix (residues V224–L237) and a G′ helix (residues P243–L246).
The PGIS structure reveals a well-defined distal heme pocket (Figure 1(d)), which is surrounded by residues from the regions commonly used by other P450s for substrate binding: the I helix (Q280, L281, W282, A283, T284, Q285 and N287), the loop between the K helix and β1-4 strand (A353, A354, P355 and F356), and the loop between two β3 strands at the C terminus (G482, F483, G484 and L485). Given that the size of PGIS substrate is large, two additional regions (the B′ and F′ helices) that are located at the substrate entrance channel but slightly farther from the heme may also contribute to substrate binding. Residues whose side-chains facing the channel in the B′ helix are Y99, L103, I107 and F108, whereas those in the F′ helix are D211, L214 and P215. The residues from these five substrate-binding sites constitute a highly hydrophobic cavity with only two residues near the heme being hydrophilic; Q280 and N287. Notably, residue W282 sits 8.4 Å from the iron with its aromatic side-chain nearly parallel with the porphyrin plane. As a result of this low threshold, the active site volume of PGIS, determined by the LIGSITE algorithm,29 is only about 355 Å3, as compared to CYP3A430 of 520 Å3 and CYP2C931 of 670 Å3. The low active site volume may explain why PGIS has a limited number of ligands. Furthermore, the position of W282 also supports the “low ceiling” hypothesis proposed by Ullrich,15 and may also explain the instability of ferrous–CO complex found in PGIS,16 as mutation at this residue greatly stabilizes the ferrous–CO complex (H.-C Y. and L.-H W., unpublished results).
Heme environment and its functional aspects
PGIS in solution has a six-coordinated iron with thiolate and water as the axial ligands in the substrate-free form.16 Surprisingly, the PGIS crystal structure with C441 as the axial ligand is five-coordinated, lacking a bound water ligand at the sixth position. The six- to five-coordination conversion was also reported in the crystal structures of CYP2C931 and CYP3A430 and is probably due to the process of crystallization or X-ray induced reduction. The heme is embedded between the I and L helices but the interactions of the propionate groups with PGIS apoprotein are distinct from other P450s. Most P450s have either the two propionate groups pointing toward the proximal side or one to the proximal and the other to the distal side. Moreover, the two propionate groups each form two to three H-bonds with the protein directly or through water molecules. While the heme iron and the four pyrrole rings have unambiguous electron density, the positions of propionate groups in the PGIS crystal are ill-defined and show conformational heterogeneity in both monomers. A-ring propionate forms an H-bond bridge with the protein backbone through a water molecule and points toward the distal side, whereas D-ring propionate is less defined. Given that it would clash with a lysine residue located at the proximal side, the D-ring propionate most likely points toward the distal pocket. Thus, the direct interactions between propionate groups and the protein are largely absent from PGIS, indicating a somewhat greater plasticity of the heme lodging in PGIS. Weaker interactions between propionate groups and the PGIS apoprotein may mean that the enzyme is deficient in the mono-oxygenation reaction, as computational analyses have shown that these interactions are crucial for abstraction of a hydrogen atom from the substrate by the oxyferryl intermediate, the final step of hydroxylation.32
Despite structural differences between P450s in functionally important regions, the most conserved element of P450 structure is near the heme-binding pocket. Although the primary sequence of Cys ligand loop in PGIS is divergent, the tertiary structure of the Cys ligand loop is conserved (Figure 1(e)). The carbonyl oxygen atom of W434 is H-bonded to the backbone NH of C441 to enclose the loop. G437 serves two roles; allowing a formation of β-hairpin turn and forming an H-bond to the Nδ of H440. Moreover, the sulfur atom of C441 is 3.4 Å from the backbone NH groups of G443 and R444. The thiolate group is thus stabilized by the partial positive charges on the backbone, an arrangement similar to that found in other P450s. The PGIS structure also reveals an unusual meander region (residues R393–P433) located on the proximal side of the heme. This coil region is markedly longer (by more than ten residues) compared to other P450s, and the coil structure protrudes to the protein surface. It is thus plausible that the meander region in PGIS interferes the electron transfer via a steric effect, as it has long been thought that the P450 redox partner delivers its electron from the proximal side.33 Unfortunately, knowing the PGIS structure at present gives no clear information as to why PGIS accepts no electrons from the reductase. Analysis of surface potentials of PGIS reveals no noticeable difference from other P450s in charged amino acid residues on the proximal side; nor the dipole moment along the molecule axis, which exerts a long-range electrostatic attraction with the redox partner (data not shown).33 As the P450 electron transfer system is complicated, the enigma of PGIS in electron transfer remains unsolved.
Substrate-binding model and mechanistic implications
To gain further insights into how PGH2 is converted into PGI2, we investigated the potential enzyme–substrate interactions by manually docking a PGH2 molecule into the active site of PGIS with the C11 oxygen atom coordinating to the iron (Figure 1(f)), ready for the subsequent homolytic scission of the endoperoxide bond.34 While the active-site access channel is quite large, it is evident that the channel shrinks considerably toward the sixth coordination position of the iron in the substrate-free PGIS. As the result, steric clashes are observed between the bicyclic ring and residues from the I-helix when the PGH2 molecule is present in the active site, implying that conformational changes around this region are needed for PGH2 to bind productively. It should be noted that even more steric clashes are observed when the C9 oxygen atom was modeled to coordinate to the iron, indicating that PGIS preferentially selects the C11 oxygen atom as its distal ligand. The carboxylate group and the alkyl side-chains of PGH2, nonetheless, can be fitted in the wider part of the channel. The current PGH2-binding model, as shown in Figure 1(f), is the one with the largest number of favorable interactions for the PGH2 side-chains (the 15-OH of PGH2 H-bonds to L279 and Q280, and the carboxylate group of PGH2 H-bonds to G482) while minimizing the repulsive interactions of the bicyclic ring (C9 O against N287, C11 O against the iron, and C10, C11, and C13 against A283). The model places C9 O in close proximity to the side-chain amide nitrogen atom of N287 (~3 Å), suggesting a potential involvement of N287 in the endoperoxide bond cleavage. Notably, the carboxylate side-chain is molded to a spiral in which C9 is only 3.4 Å from C6, and C5 is located right above the edge of porphyrin A-ring. This conformation fits very well to the proposed reaction mechanism,34 in which PGH2 binding to the heme iron via the C11 oxygen atom is the initial step. Subsequently, homolysis of endoperoxide takes place, giving rise to C11-O-Fe (IV) and C9 alkoxy radical. The alkoxyl radical then undergoes cyclization with the C5–C6 double bond to form the five-member ring. Our model suggests that the resultant C9 alkoxy radical would be adjacent to C6, making the cyclization the only available option for this labile radical intermediate. The cyclization yields a C5 radical, which is then oxidized by Fe(IV) to form the carbocation. Our model again shows that C5 can be very close to porphyrin (~3 Å), making the radical donate an electron readily to the Fe(IV)-porphyrin. Finally, the enzyme restores its resting state and the C11 oxygen atom abstracts a proton from the carbocation to produce PGI2. The biosynthesis of PGI2 is thus controlled by the structural constraint of PGH2 within a hydrophobic environment that favors the radical reactions, in contrast to the more hydrophilic active sites in the peroxidases.
In summary, we describe the first crystal structure of a class III P450. The PGIS structure closely resembles other P450s, although some notable differences are apparent. While the F/G loop, B′, F and G helices in PGIS appear to be responsible for binding different ligands, a wider substrate entrance opening is created, thanks to the bent I helix and tilted B′ helix. These structural features, which should have favored binding of PGIS with ligands of various sizes and shapes, were restricted by the presence of the residue W282 which likely serves as a ceiling to exclude most ligands. PGIS also reveals an unusual meander structure protruding from the protein surface, which may impede electron transfer. Another important feature of the PGIS structure is the plasticity of the heme propionate groups that may have an important implication in understanding the enzymatic catalysis. The presence of N287 in the active site and its potential role in peroxide cleavage warrants further experimental investigation. The PGIS structure thus should provide an avenue to better understand the structure/function relationship of the atypical P450.
Protein Data Bank accession code
The atomic coordinates and structure factors of PGIS have been deposited in the RCSB Protein Data Bank with ID code 2IAG.
Acknowledgments
We thank Dr H.-Y. Chen for providing chemicals, Y.-C. Li, P.-C. Ho, H.-T. Chang, L.-Y. Lin, W.-L. Li, and the PX teams at the National Synchrotron Radiation Research Center (NSRRC BL13B1, Taiwan; SPring8 BL12B2, Japan) for assistance during data collection. This work is supported by National Science Council grant NSC95-2113-M-005-003 (to N.-L. C.) and the National Institutes of Health grant HL60625 (to L.-H. W.).
Abbreviations used
- PGIS
prostacyclin synthase
- PGH2
prostaglandin H2
- PGI2
prostacyclin
- MAD
multiwavelength anomalous diffraction
References
- 1.Coon MJ. Cytochrome P450: nature’s most versatile biological catalyst. Annu Rev Pharmacol Toxicol. 2005;45:1–25. doi: 10.1146/annurev.pharmtox.45.120403.100030. [DOI] [PubMed] [Google Scholar]
- 2.Mestres J. Structure conservation in cytochromes P450. Proteins: Struct Funct Genet. 2005;58:596–609. doi: 10.1002/prot.20354. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Poulos TL. Crystallization of cytochromes P450 and substrate-enzyme interactions. Curr Top Med Chem. 2004;4:1789–1802. doi: 10.2174/1568026043387205. [DOI] [PubMed] [Google Scholar]
- 4.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Microsomal cytochrome P450 2C5: comparison to microbial P450s and unique features. J Inorg Biochem. 2000;81:183–190. doi: 10.1016/s0162-0134(00)00102-1. [DOI] [PubMed] [Google Scholar]
- 5.Lee DS, Yamada A, Sugimoto H, Matsunaga I, Ogura H, Ichihara K, et al. Substrate recognition and molecular mechanism of fatty acid hydroxylation by cytochrome P450 from Bacillus subtilis. Crystallographic, spectroscopic, and mutational studies. J Biol Chem. 2003;278:9761–9767. doi: 10.1074/jbc.M211575200. [DOI] [PubMed] [Google Scholar]
- 6.Park SY, Shimizu H, Adachi S, Nakagawa A, Tanaka I, Nakahara K, et al. Crystal structure of nitric oxide reductase from denitrifying fungus Fusarium oxysporum. Nature Struct Biol. 1997;4:827–832. doi: 10.1038/nsb1097-827. [DOI] [PubMed] [Google Scholar]
- 7.Vane JR, Botting RM. Pharmacodynamic profile of prostacyclin. Am J Cardiol. 1995;75:3A–10A. doi: 10.1016/s0002-9149(99)80377-4. [DOI] [PubMed] [Google Scholar]
- 8.Smith WL, DeWitt DL, Allen ML. Bimodal distribution of the prostaglandin I2 synthase antigen in smooth muscle cells. J Biol Chem. 1983;258:5922–5926. [PubMed] [Google Scholar]
- 9.Ullrich V, Castle L, Weber P. Spectral evidence for the cytochrome P450 nature of prostacyclin synthetase. Biochem Pharmacol. 1981;30:2033–2036. doi: 10.1016/0006-2952(81)90218-5. [DOI] [PubMed] [Google Scholar]
- 10.Hara S, Miyata A, Yokoyama C, Inoue H, Brugger R, Lottspeich F, et al. Isolation and molecular cloning of prostacyclin synthase from bovine endothelial cells. J Biol Chem. 1994;269:19897–198903. [PubMed] [Google Scholar]
- 11.Pereira B, Wu KK, Wang LH. Molecular cloning and characterization of bovine prostacyclin synthase. Biochem Biophys Res Commun. 1994;203:59–66. doi: 10.1006/bbrc.1994.2148. [DOI] [PubMed] [Google Scholar]
- 12.Miyata A, Hara S, Yokoyama C, Inoue H, Ullrich V, Tanabe T. Molecular cloning and expression of human prostacyclin synthase. Biochem Biophys Res Commun. 1994;200:1728–1734. doi: 10.1006/bbrc.1994.1652. [DOI] [PubMed] [Google Scholar]
- 13.Shyue SK, Ruan KH, Wang LH, Wu KK. Prostacyclin synthase active sites. Identification by molecular modeling-guided site-directed mutagenesis. J Biol Chem. 1997;272:3657–3662. doi: 10.1074/jbc.272.6.3657. [DOI] [PubMed] [Google Scholar]
- 14.Graham-Lorence S, Peterson JA. P450s: structural similarities and functional differences. FASEB J. 1996;10:206–214. doi: 10.1096/fasebj.10.2.8641554. [DOI] [PubMed] [Google Scholar]
- 15.Ullrich V, Brugger R. Prostacyclin and thromboxane synthase: new aspects of hemethiolate catalysis. Angew Chem Int Ed Engl. 1994;33:1911–1919. [Google Scholar]
- 16.Yeh HC, Hsu PY, Wang JS, Tsai AL, Wang LH. Characterization of heme environment and mechanism of peroxide bond cleavage in human prostacyclin synthase. Biochim Biophys Acta. 2005;1738:121–132. doi: 10.1016/j.bbalip.2005.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 18.Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14alpha-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci USA. 2001;98:3068–3073. doi: 10.1073/pnas.061562898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasemann CA, Kurumbail RG, Boddupalli SS, Peterson JA, Deisenhofer J. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3:41–62. doi: 10.1016/s0969-2126(01)00134-4. [DOI] [PubMed] [Google Scholar]
- 20.Poulos TL. Structural biology of heme monooxygenases. Biochem Biophys Res Commun. 2005;338:337–345. doi: 10.1016/j.bbrc.2005.07.204. [DOI] [PubMed] [Google Scholar]
- 21.Sligar SG, Makris TM, Denisov IG. Thirty years of microbial P450 monooxygenase research: peroxo-heme intermediates–the central bus station in heme oxygenase catalysis. Biochem Biophys Res Commun. 2005;338:346–354. doi: 10.1016/j.bbrc.2005.08.094. [DOI] [PubMed] [Google Scholar]
- 22.Hasemann CA, Ravichandran KG, Peterson JA, Deisenhofer J. Crystal structure and refinement of cytochrome P450terp at 2.3 Å resolution. J Mol Biol. 1994;236:1169–1185. doi: 10.1016/0022-2836(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 23.Poulos TL, Finzel BC, Howard AJ. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- 24.Hecker M, Baader WJ, Weber P, Ullrich V. Thromboxane synthase catalyses hydroxylations of prostaglandin H2 analogs in the presence of iodosylbenzene. Eur J Biochem. 1987;169:563–569. doi: 10.1111/j.1432-1033.1987.tb13646.x. [DOI] [PubMed] [Google Scholar]
- 25.Meharenna YT, Li H, Hawkes DB, Pearson AG, De Voss J, Poulos TL. Crystal structure of P450cin in a complex with its substrate, 1,8-cineole, a close structural homologue to D-camphor, the substrate for P450cam. Biochemistry. 2004;43:9487–9494. doi: 10.1021/bi049293p. [DOI] [PubMed] [Google Scholar]
- 26.Oldham ML, Brash AR, Newcomer ME. The structure of coral allene oxide synthase reveals a catalase adapted for metabolism of a fatty acid hydroperoxide. Proc Natl Acad Sci USA. 2005;102:297–302. doi: 10.1073/pnas.0406352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulos TL, Johnson EF. Structures of cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. Plenum; New York: 2005. pp. 87–114. [Google Scholar]
- 28.Deng H, Wu J, So SP, Ruan KH. Identification of the residues in the helix F/G loop important to catalytic function of membrane-bound prostacyclin synthase. Biochemistry. 2003;42:5609–5617. doi: 10.1021/bi026749z. [DOI] [PubMed] [Google Scholar]
- 29.Hendlich M, Rippmann F, Barnickel G. LIGSITE: automatic and efficient detection of potential small molecule-binding sites in proteins. J Mol Graph Model. 1997;15:359–363. doi: 10.1016/s1093-3263(98)00002-3. [DOI] [PubMed] [Google Scholar]
- 30.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, et al. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 31.Williams PA, Cosme J, Ward A, Angove HC, Matak Vinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 32.Guallar V, Baik MH, Lippard SJ, Friesner RA. Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proc Natl Acad Sci USA. 2003;100:6998–7002. doi: 10.1073/pnas.0732000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sevrioukova IF, Li H, Zhang H, Peterson JA, Poulos TL. Structure of a cytochrome P450-redox partner electron-transfer complex. Proc Natl Acad Sci USA. 1999;96:1863–1868. doi: 10.1073/pnas.96.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecker M, Ullrich V. On the mechanism of prostacyclin and thromboxane A2 biosynthesis. J Biol Chem. 1989;264:141–150. [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallog sect D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallog sect D. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Terwilliger TC. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallog sect D. 2003;59:38–44. doi: 10.1107/S0907444902018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terwilliger TC. Automated side-chain model building and sequence assignment by template matching. Acta Crystallog sect D. 2003;59:45–49. doi: 10.1107/S0907444902018048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallog sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallog sect D. 1994:764–767. [Google Scholar]