

Abstract
Deletions affecting the terminal end of chromosome 3p result in a characteristic set of clinical features termed 3p-syndrome. Bilateral, sensorineural hearing loss (SNHL) has been found in some but not all cases, suggesting the possibility that it is due to loss of a critical gene in band 3p25. To date, no genetic locus in this region has been shown to cause human hearing loss. However, the ATP2B2 gene is located in 3p25.3, and haploinsufficiency of the mouse homolog results in SNHL with similar severity. We compared auditory test results with fine deletion mapping in seven previously unreported 3p-syndrome patients and identified a 1.38 Mb region in 3p25.3 in which deletions were associated with moderate to severe, bilateral SNHL. This novel hearing loss locus contains 18 genes, including ATP2B2. ATP2B2 encodes the plasma membrane calcium pump PMCA2. We used immunohistochemistry in human cochlear sections to show that PMCA2 is located in the stereocilia of hair cells, suggesting its function in the auditory system is conserved between humans and mice. Although other genes in this region remain candidates, we conclude that haploinsufficiency of ATP2B2 is the most likely cause of SNHL in 3p-syndrome.
Keywords: 3p-syndrome, Hearing loss, ATP2B2, PMCA2, deafwaddler
1. Introduction
3p-syndrome is a rare disorder resulting from deletions involving the distal end of chromosome 3p. The hallmarks of the syndrome include developmental delay, growth retardation, and craniofacial manifestations (reviewed in Fernandez et al., 2004). Severe (60–75 dBnHL), bilateral sensorineural hearing loss (SNHL) has been reported in three cases (Higginbottom et al., 1982; Narahara et al., 1990; Ramer et al., 1989), but hearing loss is not a universal feature of the syndrome (Angeloni et al., 1999; Benini et al., 1999). Comparing the extent of the deletions affecting patients with SNHL and those without suggests that the loss of a critical region in band 3p25.3 may be responsible for the SNHL phenotype (Angeloni et al., 1999; Drumheller et al., 1996; Phipps et al., 1994).
To date, no genetic locus in the 3p25-pter region has been linked to dominant or recessive hearing loss in humans (Hereditary Hearing Loss Homepage). The homologous region in mice is located on chromosome 6, position 103–116 Mb (UCSC Genome Browser). Only one gene in this region has been implicated in hearing (Hereditary Hearing Impairment in Mice webpage): mutations in the Atp2b2 gene result in sensorineural deafness in several alleles of the deafwaddler mouse (Street et al., 1998). Furthermore, haploinsufficiency of the Atp2b2 gene results in bilateral SNHL of 50–60 McCullough et al., 2004). The human homolog, ATP2B2 (MIM 108733), is located in band 3p25.3; it has not been shown to cause human hearing loss, but was shown recently to have a modifier effect, increasing the severity of hearing loss caused in the first instance by mutations in other genes (Schultz et al., 2005).
ATP2B2 encodes the plasma membrane calcium ATPase, type 2 (PMCA2). In the mouse cochlea, PMCA2 is expressed in spiral ganglion neurons and hair cells of the organ of Corti (Dumont et al., 2001; Furuta et al., 1998; Wood et al., 2004). Hair cells are the receptor cells of the auditory system; mechanical deflection of their stereocilia produces the transduction current that depolarizes the cell (Holt et al., 2000). PMCA2 is localized to the stereocilia and provides the primary mechanism for extruding calcium that enters the stereocilia during transduction (Dumont et al., 2001; Lumpkin et al., 1998; Yamoah et al., 1998). Calcium in the stereocilia controls the process of adaptation; increased levels reduce the open-probability of the transduction channels (Fettiplace et al., 2006).
Here, we use deletion mapping in seven previously unreported 3p-syndrome patients to define a 1.38 Mb region of 3p25.3 that, when lost, is associated with SNHL. This region contains 18 genes, including ATP2B2. Further, we show that PMCA2 localization is conserved between humans and mice. We conclude that ATP2B2 is a strong candidate for the haploinsufficient gene responsible for hearing loss in 3p-syndrome.
2. Materials and methods
2.1. Subjects
Potential subjects were identified locally or with the help of the chromosome disorder support groups Unique and Chromosome Deletion Outreach. All subjects included in the study provided buccal swabs for DNA extraction and access to existing auditory test information. The only cytogenetic information available to us was the classification of each of the subjects as having 3p-syndrome. A family history of deafness was reported for only one subject (S4), and none of the subjects had a history suggestive of hearing loss secondary to exposure to ototoxic agents. Auditory tests were evaluated without knowledge of the deletion mapping results. Behavioral thresholds and physiological responses (i.e. auditory brainstem responses) are presented. Informed consent was obtained from all participants and/or legal guardians, and all protocols were approved by the University of Washington Institutional Review Board.
2.2. Deletion breakpoint mapping
Deletion mapping was done using quantitative real-time PCR (qPCR) as described elsewhere with minor modifications (Duno et al., 2004). Briefly, published STS markers or primer pairs designed to non-coding regions of relevant genes (see appendix) were used. Control markers from autosomal regions distant from 3p were SHGC 56873 and SHGC 24090 on the long arm of chromosomes 3 and the short arm of chromosome 4, respectively. Four replicates were used for each sample and the threshold cycle (CT) was determined for each. Bio-Rad iQ SYBR Green Supermix and iCycler were used to monitor amplicon production according to the manufacturer’s protocol. Four four-fold dilutions of control genomic DNA were used to generate a standard curve for each primer pair, which plotted CT (ordinate) by the log2(DNA dilution) (abscissa), where the greatest concentration was considered to be 1. The CT value was determined for experimental samples and converted to log2(DNA dilution) as a measure of relative genomic DNA concentration. The difference in relative DNA concentration for each 3p marker was calculated for both control markers (above). A difference of −1 (or log2[1/2]) equals a two-fold reduction in genomic DNA concentration at the 3p marker, or hemizygosity; a difference of 0 (or log21) indicates a normal, equal DNA concentration. Genomic DNA from a separate control individual was used for control data. Chromosomal band, genomic positions, and gene annotations are from the March 2006 freeze of the human genome available at the UCSC Genome Browser.
2.3. Sequencing
DNA from each subject was used as a template for PCR reactions using custom primers and Faststart Taq DNA Polymerase (Roche Applied Science, Indianapolis, IN) according to the manufacturer’s protocol. Amplicons were purified using the QuickStep 2 PCR Purification Kit (Edge BioSystems, Gaithersburg, MD). Sequencing reactions were performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) with both forward and reverse primers according to the manufacturer’s protocol. Reaction products were purified using Performa DTR Gel Filtration Cartridges (Edge BioSystems) and sequenced locally at the University of Washington Department of Biochemistry DNA Sequencing Facility. Sequences were aligned using CodonCode Aligner (CodonCode Corp., Dedham, MA). All polymorphisms were confirmed with additional PCR and sequencing.
2.4. Immunohistochemistry
Human temporal bone samples were obtained from the Pathology Department, Massachusetts General Hospital (Boston, MA). Bone samples were collected at autopsy, immersed in 10% formalin, fixed for 24 hours, and decalcified in 120 mM EDTA. Samples were embedded in paraffin and sectioned at 8 μm. Immunostaining was done on thin sections using the biotin-amplified ABC method as described previously (Imamura et al., 2003). A PMCA2-specific polyclonal IgG (Upstate Biotech) was used at a dilution of 1:20,000. In this protocol, substituting normal serum for the primary antibody resulted in no signal.
3. Results
3.1. Auditory test results
Auditory test results revealed that subjects could readily be divided into affected (S1–S3) and unaffected (S4–S7) groups (Table 1). Subjects S1–S3 all exhibited moderate to severe bilateral hearing loss. The audiogram for subject S1 is given as a representative example of an affected subject (Fig. 1A; solid lines). In this example, the right (O) and left (X) ears show moderate (40–50 dBnHL) and severe (70–80 dBnHL) hearing loss, respectively. In contrast, subjects S4–S7 had hearing within normal limits on at least one occasion. Representative audiograms are presented for subjects S5 and S6 (Fig. 1A; dashed and dotted lines, respectively).
Table 1.
Summary of auditory test results for 3p-syndrome subjects
| Subject | Sex | # of Test Reports | Test Age (years) | Auditory Tests Done | Representative Thresholds (ear) | Diagnosis |
|---|---|---|---|---|---|---|
| S1 | F | 1 | 11 | ABR | 40–50 dBnHL (Right) | Mod. Bilat. SNHL |
| S2 | F | 2 | 2 | ABR | 50–70 dBnHL (Left) | Mod.-Sev. Bilat. SNHL with CHL component |
| S3 | M | 2 | 11 | SF
SD |
70–90 dBHL (Best)
60 dBHL (Best) |
Mod.-Sev. Bilat. SNHL with CHL component |
| S4 | M | 4 | 4 | SF, Tymp, DPOAE | WNL (Best) | Normal |
| S5 | M | 1 | 10 | CP, Tymp, | WNL (Both) | Normal |
| S6 | M | 4 | 3 | SF, Tymp, SD, DPOAE | WNL (Best) | Normal |
| S7 | F | 7 | 6 | SF, Tymp, ABR, DPAOE | WNL (Both) | Normal |
The number of auditory test reports available for each subject varied. The representative thresholds and hearing diagnoses were made using the best test report available for each subject (see text). The age and ear for that test are given. Abbreviations: dBnHL, dB normal hearing level; dBHL, dB hearing level (ANSI standard); WNL, Within normal limits; Mod., Moderate; Sev., Severe; SNHL, Sensorineural hearing loss; CHL, Conductive hearing loss; Bilat., Bilateral; ABR, Auditory-evoked brainstem response; SF, Sound field audiometry; CP, Conditioned play audiometry; SD, Speech detection threshold; DPOAE, Distortion product otoacoustic emissions; Tymp, Tympanogram.
Fig. 1.
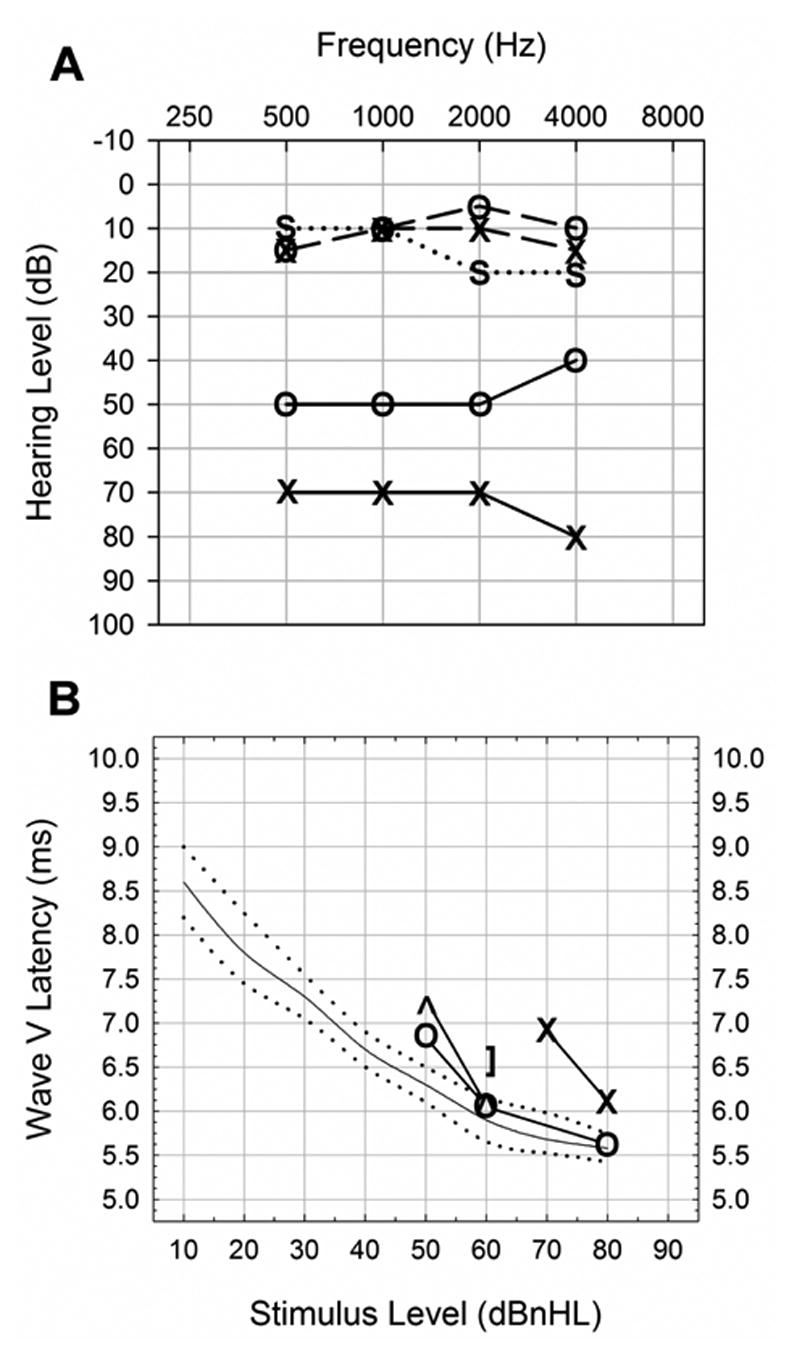
Representative auditory test results from 3p-syndrome subjects. (A) ABR audiogram from subject S1 (solid lines) showed moderate (40–50 dBnHL) and severe (70–80 dBnHL) hearing loss in the right (O) and left (X) ears, respectively. Soundfield or conditioned play audiograms from subjects S5 (dashed lines) and S6 (dotted lines) showed hearing within normal limits. Binaural sound field (S) was used for subject S6, reflecting hearing in the best ear. (B) Wave V latency plot for subject S1 showed a pattern indicative of sensorineural hearing loss, in which latencies for the right (O) and left (X) ears approached normal (solid line; dotted lines are normal range) with increasing stimulus level. No response was seen for stimulus levels below 50 dBnHL and 70 dBnHL for right and left ears, respectively. Bone conduction was also tested. Unmasked bone conduction (^) showed responses that nearly matched the right ear with air conduction (O). The right ear was then masked and bone conduction tested again to test sensorineural hearing in the left ear ( ] ). This showed a latency at 60 dBnHL that was similar to the right ear with air conduction, indicating the difference between ears was mostly due to a conductive loss.
The hearing loss in affected subjects (S1–S3) was determined to be sensorineural in origin once confounding conductive losses due to middle ear effusions were taken into account. Middle ear effects were examined in S1 by analyzing wave V latencies (Fig. 1B). Latencies in both ears were delayed compared to the normal range (dotted lines) for a lower stimulus level, but were within the normal range at a presentation level of 60 dBnHL on the right (O), and approached the normal range at a presentation level of 80 dBnHL on the left (X). Latencies that approach normal with increasing stimulus levels indicate sensorineural hearing loss. Inter-ear differences were due to a mild conductive hearing loss in the left ear: left ear sensorineural hearing was tested using bone conduction with right ear masking and showed near-normal latencies at 60 dBnHL ( ] ). No response was obtained with left ear air conduction at this stimulus level. Auditory tests in this subject were conducted following drainage of a middle ear effusion; incomplete drainage of the middle ear may explain this residual conductive component in the left ear. Subject S2 demonstrated similar SNHL to S1, with thresholds of 50–70 dBnHL in her best ear on one occasion. On a separate testing occasion, clinically evident middle ear effusions resulted in bilateral conductive losses of similar severity to that seen in subject S1; for comparison, the representative thresholds given in Table 1 reflect the best test to most closely approximate the SNHL component. No middle ear information was available for subject S3, but the magnitude of the hearing loss (70–90 dBHL) suggests that it is at least partly sensorineural, since a maximal conductive loss is on the order of 60 dB.
Mild conductive hearing loss was also observed in subjects S4, S6, and S7 during at least one auditory test session. In each subject, the losses were accompanied by an abnormal tympanogram, suggesting a middle ear etiology. Subject S5 provided auditory test data from only one date, when hearing was within normal limits and there was no evidence of a middle ear effusion. Because the conductive losses seen in several patients were transient, representative thresholds and diagnoses (Table 1) were based on the best ear or test date to reflect most closely the sensorineural hearing component.
3.2. Deletion breakpoint mapping
We mapped the deletion breakpoints in all seven subjects using quantitative PCR to detect hemizygous markers in genomic DNA. Table 2 presents the extent of the deletions of each subject as inferred from the markers used. Six of the seven patients had terminal deletions of 3p and one (S1) retained a portion of the telomere as evidenced by two distal markers. No balanced translocations were detected.
Table 2.
Summary of deletion mapping results and corresponding hearing diagnoses in 3p- syndrome subjects
| Marker | Band | Mb | S1 | S2 | S3 | S4 | S5 | S6 | S7 |
|---|---|---|---|---|---|---|---|---|---|
| TELOMERE | |||||||||
| CHL1 | 3p26.3 | 0.21 | + | - | - | - | - | - | - |
| SHGC 84547 | 3p26.3 | 1.23 | + | - | - | - | - | - | - |
| SHGC 15249 | 3p26.1 | 7.77 | - | - | - | - | - | - | - |
| SRGAP3 (5’) | 3p25.3 | 9.27 | ND(-) | ND(-) | - | - | - | - | - |
| ARPC4 | 3p25.3 | 9.81 | ND(-) | ND(-) | - | - | - | + | + |
| IL17RE | 3p25.3 | 9.92 | ND(-) | ND(-) | - | - | ND | + | + |
| VHL | 3p25.3 | 10.16 | ND(-) | ND(-) | - | + | + | ND(+) | + |
| SHGC 19014 | 3p25.3 | 10.30 | ND(-) | ND(-) | + | ND(+) | ND(+) | ND(+) | ND(+) |
| SEC13L1 | 3p25.3 | 10.32 | ND(-) | ND(-) | + | + | ND(+) | ND(+) | + |
| WI 6061 | 3p25.3 | 10.345 | - | - | + | + | + | + | + |
| ATP2B2 (3’) | 3p25.3 | 10.347 | - | - | + | + | + | + | + |
| ATP2B2 (Middle) | 3p25.3 | 10.39 | - | - | + | ND(+) | + | ND(+) | ND(+) |
| SHGC 148056 | 3p25.3 | 11.21 | - | + | + | ND(+) | + | + | + |
| SHGC 15229 | 3p25.2 | 12.91 | + | + | ND(+) | ND(+) | ND(+) | ND(+) | ND(+) |
| WI 10567 | 3p24.2 | 26.49 | + | + | ND(+) | ND(+) | ND(+) | ND(+) | ND(+) |
| TMIE | 3p21.31 | 46.72 | + | ND(+) | ND(+) | ND(+) | ND(+) | ND(+) | ND(+) |
| CENTROMERE | |||||||||
|
| |||||||||
| SNHL present? | Yes | Yes | Yes | Normal | Normal | Normal | Normal | ||
| Hearing Level (dB) | 40–50 | 50–70 | 70–90 | WNL | WNL | WNL | WNL | ||
, Present (normal); -, Absent (hemizygous); ND, Not done. Parentheses indicate inferred status. Shaded areas illustrate deleted segments of chromosome. SNHL, Sensorineural hearing loss; WNL, Within normal limits.
Subject S1 had the most centromeric breakpoint, between markers SHGC 148056 and SHGC 15229 in 3p25.3 and 3p25.2, respectively (position 11.21–12.91 Mb). The breakpoints for all other subjects were in 3p25.3, with the most telomeric found between markers in the SRGAP3 and ARPC4 genes (position 9.27–9.81 Mb) in subjects S6 and S7.
3.3. Identification of SNHL candidate gene region
We compared the hearing phenotypes with the extent of the deletions in all subjects and found that those with SNHL (S1–S3) had deletions that extended further towards the centromere than those without SNHL (S4–S7; Table 2). Subject S4 had the largest deletion that was not associated with hearing loss. The breakpoint in S4 was between markers in the IL17RE and VHL genes (position 9.92–10.16), suggesting the loss of genetic material distal to this point is not essential for hearing function.
Subject S3 had the smallest deletion associated with SNHL in this study. To confirm and more precisely localize the breakpoint, we examined additional markers in this patient. Figure 2 shows the relative levels of genomic DNA for all markers tested in subject S3 (hollow symbols) as well as in a control subject without a 3p deletion (filled symbols). There is a two-fold relative reduction in S3 genomic DNA compared to control at markers telomeric to (above) SHGC 19014, indicating hemizygosity. These results establish the breakpoint in S3 as being between SHGC 19014 and a marker in the VHL gene (position 10.16–10.30 Mb). This represents the minimal deletion associated with SNHL in this study.
Fig. 2.
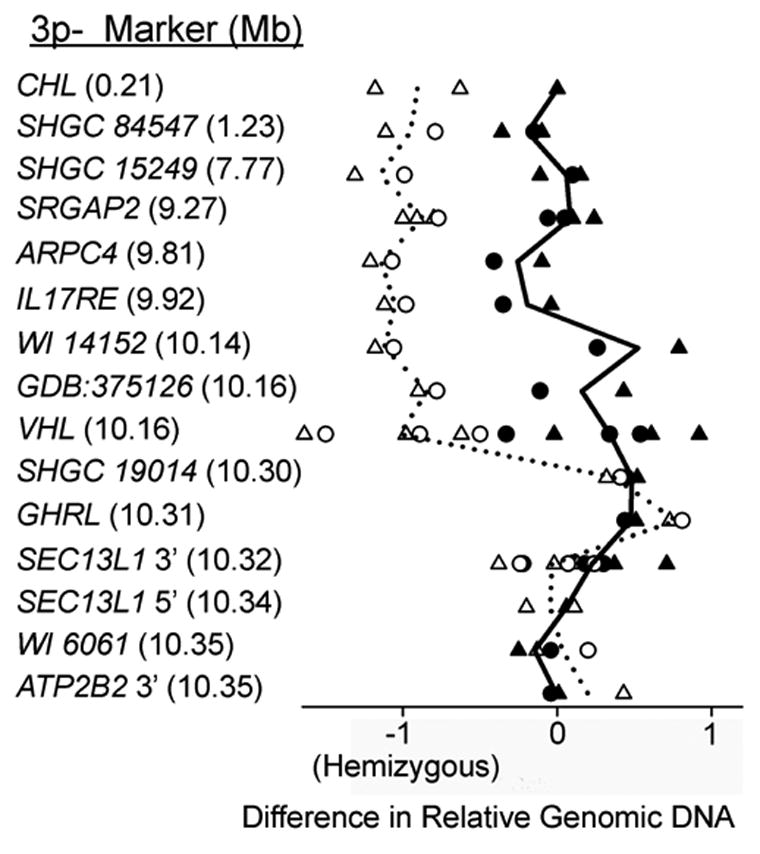
qPCR deletion mapping in subject S3. The difference in relative genomic DNA from unlinked (Chrs. 3q and 4p) control marker SHGC 56873 (triangles) or SHGC 24090 (circles) at each 3p marker tested in S3 (left column) is plotted. Data from subject S3 (open symbols) are compared with a normal hearing control without a deletion (filled-in symbols). Lines are drawn to illustrate the mean value for all data points for each marker in each subject. Values on the abscissa are log2(x), where x is the relative DNA calculated from a standard curve. A difference of −1 in relative genomic DNA indicates a two-fold reduction or hemizygosity (see methods). The decrease in CT at markers telomeric (above) to SHGC 19014 in S3 indicates the deletion breakpoint is between a marker in the VHL gene (position 10.16 Mb) and SHGC 19014 (position 10.30 Mb).
Based on our deletion mapping results, the loss of genetic material centromeric to the IL17RE gene in 3p25.3 corresponded with the presence of SNHL. While it is possible that the smallest 3p deletion associated with SNHL may point to the gene or genes responsible, it is also possible that the regulation of nearby genes is affected. Chromosomal aberrations, such as deletions or translocations, have demonstrated the importance of long-range, cis-acting regulatory elements on gene expression; reductions in gene transcription and haploinsufficiency phenotypes have been shown to be caused by loss or translocation of genetic material distant to the gene of interest (reviewed in Kleinjan et al., 2005; Merla et al., 2006). Therefore, we have defined a region for the candidate gene or genes responsible for SNHL in 3p-subjects that includes the smallest interval associated with SNHL (IL17RE to SHGC 19014), as well as an additional 1 Mb in the direction of the centromere (position 9.92–11.30 Mb). There are 18 genes located in the region we have defined (Table 3).
Table 3.
Sensorineural hearing loss candidate genes
| Location (Mb) | Gene | Function |
|---|---|---|
| 9.93–9.95 | IL17RC | Cytokine receptor |
| 9.95–9.96 | CRELD1 | Cell adhesion |
| 9.96–9.97 | PRRT3 | Unknown |
| 9.98–10.00 | TMEM111 | Unknown |
| 10.00–10.02 | AK092352 | Unknown |
| 10.03–10.04 | CIDEC | Unknown |
| 10.04–10.12 | FANCD2 | Tumor suppressor |
| 10.10–10.12 | LOC115795 | Unknown |
| 10.13–10.14 | C3orf10 | Unknown |
| 10.16–10.17 | VHL | Tumor suppressor |
| 10.18–10.26 | IRAK2 | Cytokine receptor kinase |
| 10.27–10.30 | TATDN2* | DNase (Predicted) |
| 10.30–10.31 | GHRL* | Growth hormone secretagogue |
| 10.32–10.34 | SEC13L1* | Unknown |
| 10.34–10.47 | ATP2B2* | Plasma membrane Ca2+ pump |
| 10.83–10.96 | SLC6A11* | GABA transporter |
| 11.01–11.06 | SLC6A1* | GABA transporter |
| 11.27–11.28 | HRH1* | Histamine receptor |
Denotes genes within candidate gene region but not lost in all patients with SNHL (see text)
The ATP2B2 gene is included on this list of candidate genes. The gene itself was partially or completely removed in subjects S1 and S2, both of which had SNHL. Subject S3, however, demonstrated SNHL but did not lose the ATP2B2 transcription unit: the breakpoint was 45–185 kb distal.
3.4. Genomic sequencing for confounding mutations
To consider the possibility that subject S3 had a separate point mutation in ATP2B2 that accounted for his SNHL, we sequenced the entire ATP2B2 open reading frame in him and in a subject with normal hearing, S7. We identified one single nucleotide polymorphism (SNP) in subject S3 and three in subject S7, all of which were heterozygous, silent mutations previously identified and listed online in dbSNP (Table 4A and NCBI-dbSNP). Since the SNP found in S3 (3357G>A) was also found in S7, we conclude that it is not responsible for the SNHL found in S3.
Table 4.
Polymorphisms identified in subjects by genomic sequencing of selected genes.
| A. ATP2B2* | |||
|
| |||
| Subject | Nucleotide | Amino Acid | Genotype |
|
| |||
| S3 | 3357G>A | Silent | +/ATP2B23357G>A |
| S7 | 603C>G | Silent | +/ATP2B2603C>G |
| 1437C>T | Silent | +/ATP2B21437C>T | |
| 3357G>A | Silent | +/ATP2B23357G>A | |
|
B. GJB2
| |||
| Subject | Nucleotide | Amino Acid | Genotype |
|
| |||
| S1 | None | +/+ | |
| S2 | None | +/+ | |
| S3 | 79G>A | V27I | +/GJB2V27I |
| 341A>G | E114G | +/GJB2E114G | |
| S4 | 35delG | Frame-shift, Premature stop | +/GJB235delG |
| S5 | None | +/+ | |
| S6 | None | +/+ | |
| S7 | None | +/+ | |
denotes “wild-type.”
ATP2B2 was sequenced only in subjects S3 and S7.
Mutations in the GJB2 gene (MIM 121011), which encodes the connexin 26 (Cx26) protein, are known to be the most common cause of hereditary hearing loss (reviewed in Kenneson et al., 2002). To minimize the potential for confounding, hearing loss-causing mutations from this locus, we sequenced the entire open reading frame of GJB2 in all seven subjects. We identified three sequence variants in two subjects, S3 and S4 (Table 4B, see also the Connexin-deafness homepage). Subject S3 was heterozygous for a conservative V27I amino acid change and a non-conservative E114G change. Both of these changes have been reported previously and are not believed to cause hearing loss in the heterozygous state (Choung et al., 2002; Kudo et al., 2000; Santos et al., 2005); however, congenital profound hearing loss was reported in two members of a Korean family that were homozygous for the E114G change, and variable hearing loss was seen in related carriers, suggesting a modifier may be involved (Park et al., 2000). We therefore conclude that changes seen in GJB2 in S3 are unlikely to be the primary cause of the SNHL observed in this subject although a cumulative or modifier effect may contribute to hearing loss in this subject. Subject S4 was found to be heterozygous for a 35delG mutation, which results in a frame shift and premature stop codon in Cx26. This single nucleotide deletion (also known as 30delG) is one of the most common causes of GJB2-associated, recessive deafness (Denoyelle et al., 1997). As S4 had hearing within normal limits, we conclude that this individual is an unaffected heterozygous carrier of the mutation. Of note, this subject did report a family history of hearing loss.
We also screened all subjects for the previously identified V586M mutation in ATP2B2 (Schultz et al., 2005). None of the subjects in this study contained this mutation.
3.5. PMCA2 staining in human cochlear sections
If ATP2B2 is involved in SNHL in humans—as Atp2b2 is known to be in mice—we would predict that PMCA2 might be similarly distributed in the two species. In mice, PMCA2 has been shown to be localized to neurons and to hair cell stereocilia where it is the primary calcium transporter (Dumont et al., 2001; Lumpkin et al., 1998; Yamoah et al., 1998). To examine the localization of PMCA2 in the human auditory system, we used a PMCA2-specific antibody to stain human cochlear sections. Fig. 3A shows PMCA2 staining in the stereocilia and cuticular plates of outer hair cells (OHC; arrows). PMCA2 was not detected in other cells of the organ of Corti (OC). Isolated dark spots were present in the stria vascularis (SV), but this appeared to be due to pigment in melanocytes under higher magnification (not shown). Fig. 3B shows a higher magnification view of the organ of Corti at a more apical location in the cochlea. In this section, the stereocilia and cuticular plates of OHC (arrows) and inner hair cells (IHC; arrowhead) were labeled. IHC staining was less robust and only apparent in some sections. This expression pattern is similar to that seen in mice (Dumont et al., 2001; Wood et al., 2004).
Fig. 3.
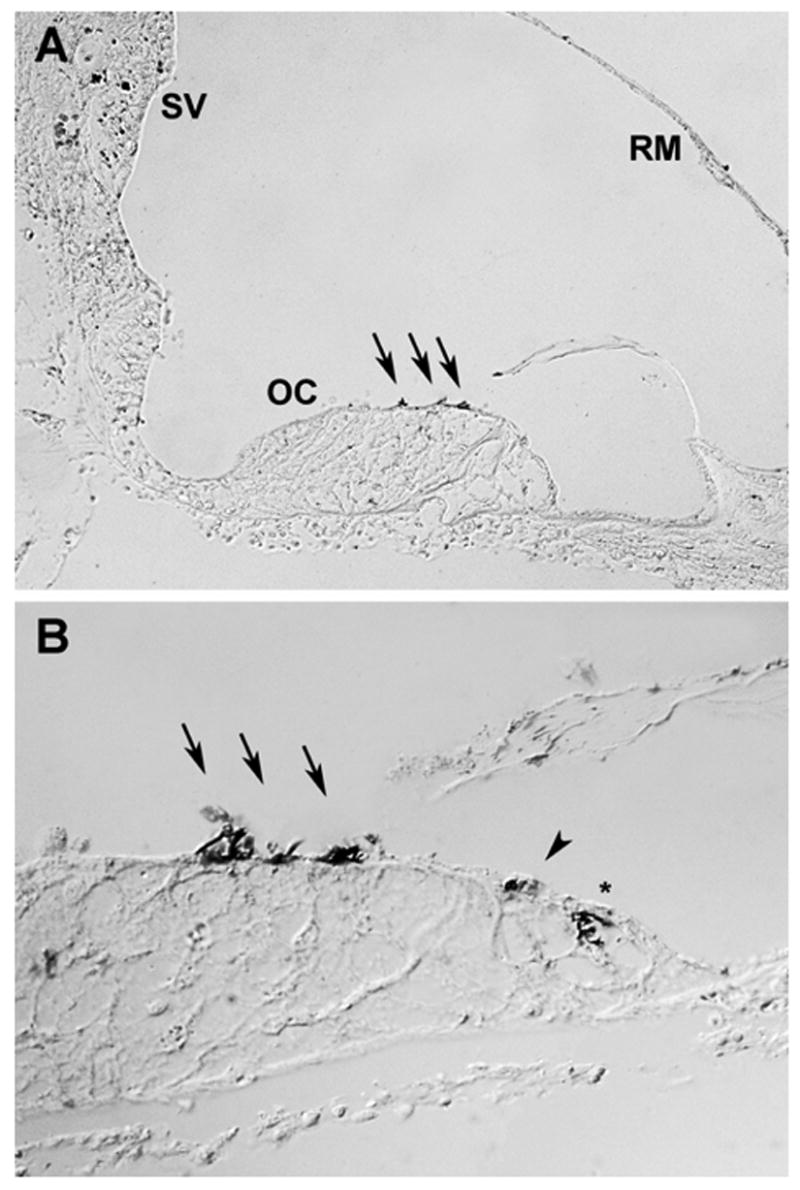
PMCA2 immuno-labeling in human cochlear sections. (A) A cross-section of the scala media from a basilar portion of the cochlea showed PMCA2 labeling in the stereocilia and cuticular plates of the outer hair cells (arrows). PMCA2 staining was not evident in other cells of the organ of Corti (OC), stria vascularis (SV), or Reissner’s membrane (RM). (B) A higher magnification view of the organ of Corti (OC) from a more apical portion of the same cochlea showed PMCA2 labeling in the stereocilia and cuticular plates of both inner (arrowhead) and outer hair cells (arrows). The asterisk indicates an artifact that was not observed in any other section of this cochlea or others. Both sections shown were from the same donor, a 55 year old male for whom no auditory history was available. Similar staining was observed in a second set of sections from a 49 year old male donor.
4. DISCUSSION
In this study, we investigated the genetic basis for the sensorineural hearing loss (SNHL) found in some patients with 3p-syndrome. By comparing auditory test results with deletion mapping in seven previously unreported subjects, we identified a 1.38 Mb region of 3p25.3 that, when lost, was associated with the presence of moderate to severe, bilateral SNHL.
Previous studies of 3p-syndrome patients have reported auditory test results and deletion breakpoint mapping similar to our own. Five published case reports provided or alluded to hearing measurements: three patients had severe (60–75 dBnHL), bilateral SNHL, whereas the other two did not show any SNHL (Angeloni et al., 1999; Benini et al., 1999; Higginbottom et al., 1982; Narahara et al., 1990; Ramer et al., 1989). The deletion breakpoints were mapped in two of these patients, one with SNHL and one without. The breakpoint in the patient with SNHL (subject LD) was located between markers D3S1317 and D3S601 (position 10.21–10.43 Mb) while that of the unaffected patient (subject A) was located further towards the telomere, between marker D3S18 and the VHL gene (position 8.77–10.16 Mb) (Angeloni et al., 1999; Drumheller et al., 1996; Phipps et al., 1994; Ramer et al., 1989). Thus, the patient with SNHL had a measurably larger deletion whose extent is in line with the candidate region for SNHL we defined in our study (position 9.92–11.30 Mb).
These data support the hypothesis that haploinsufficiency of one or more genes in 3p25.3 is responsible for the SNHL seen in 3p-patients. To date, no gene in this region of the genome has been linked to dominant or recessive hearing loss in humans (Hereditary Hearing Loss Homepage). Thus, this represents the identification of a novel hearing loss locus in human.
The region we have defined in 3p25.3 includes 18 candidate genes (Table 3). Of these, the function is known or predicted for eleven, and auditory data from human or mouse exist for only two, ATP2B2 and VHL (discussed below). Considering these limitations, we propose that ATP2B2 is a strong candidate for the responsible gene.
Functional genomic analysis of the mouse homolog, Atp2b2, has demonstrated a hearing phenotype similar to that seen in 3p-patients. Mice heterozygous for a functional null Atp2b2 allele show moderate, bilateral SNHL (McCullough et al., 2004). The protein product of this gene, PMCA2, is essential for proper hearing because it is the primary calcium transporter in the stereocilia of hair cells in the cochlea (Dumont et al., 2001; Lumpkin et al., 1998; Yamoah et al., 1998).
To consider the role of PMCA2 in human hearing, we used an antibody against PMCA2 in human temporal bone sections to compare expression with mice. Results demonstrated that PMCA2 is localized similarly to the stereocilia of hair cells in the human cochlea, suggesting its function in the auditory system is conserved between humans and mice. Expression in the spiral ganglion neurons in human cochlea could not be evaluated because of the somewhat necrotic state of both specimens examined.
Recent evidence also indicates ATP2B2 is involved in human hearing. A partial loss-of-function allele (ATP2B2V586M) was shown to act as a dominant modifier of hearing loss in patients with a deafness-causing recessive mutation in the CDH23 gene or a dominant mutation in the MYO6 gene (MIM 605516 and 600970, respectively). The ATP2B2V586M allele was insufficient to cause hearing loss in carriers without the accompanying mutations in CDH23 or MYO6, and no patients were identified that were homozygous for ATP2B2V586M (Schultz et al., 2005). Of note, no subjects in our study carried the ATP2B2V586M allele. In mice, a similar modifier effect has been reported between Atp2b2 and Cdh23, the mouse homolog of CDH23: hearing loss was increased in mice heterozygous for the functional null Atp2b2dfw2J allele when they were also homozygous for a hypomorphic allele of Cdh23 (ahl1) (Noben-Trauth et al., 2003; Noben-Trauth et al., 1997). These data suggest ATP2B2 and CDH23 act genetically as reciprocal modifiers. Additional parallels, including similarities in PMCA2 localization and functional data showing SNHL in heterozygous null Atp2b2dfw2J when in a good hearing background (McCullough et al., 2004), further support the hypothesis that haploinsufficiency of ATP2B2 is causing the SNHL in 3p-patients.
Of the other ten candidate genes with known or predicted function, hearing loss has only been associated with mutations in VHL (MIM 608537) (Hinney et al., 2002; Manski et al., 1997; Megerian et al., 2002). Hearing loss in these patients, however, is secondary to the development of endolymphatic sac tumors as part of von Hippel Lindau disease (VHL; MIM 193300), not as a primary genetic disease. These tumors arise in the temporal bone surrounding the cochlea, are locally invasive, and are amenable to surgical resection to preserve hearing (Lonser et al., 2004; Manski et al., 1997; Megerian et al., 2002). VHL disease has not been found in 3p-patients (Drumheller et al., 1996), suggesting VHL is not the cause of the hearing loss in 3p-patients.
Mutations have been reported in CRELD1 and GHRL, but hearing ability was not reported. Missense mutations in CRELD1 were associated with atrioventricular septal defects in three patients, one of which was said to have no extracardiac abnormalities (Robinson et al., 2003). Mutations in GHRL were described in two patients and were not associated with an observable phenotype (Hinney et al., 2002). Loss of function or targeted deletions of the mouse homologs of HRH1, GHRL, FANCD2, and VHL have been characterized, but no auditory data were reported for these mice (Houghtaling et al., 2003; Kleymenova et al., 2004; Ma et al., 2002; Wortley et al., 2004). The remaining five genes with described functions include two genes involved in cytokine signaling, two GABA transporters, and a DNase (UCSC Genome Browser).
We cannot rule out the possibility that one of the 17 genes aside from ATP2B2 is at least partly responsible for the SNHL in 3p-patients. In particular, the deletion in one subject with affected hearing in our study (S3) left intact the ATP2B2 transcription unit, the sequence of which showed only one benign SNP (Table 4A). It is possible that expression of ATP2B2 in this subject could be affected by loss of cis-acting regulatory elements or by cis-acting suppression of expression by the novel telomeric chromatin structure. Long-range regulatory elements have been shown to be essential for proper expression of numerous genes, particularly those with restricted spatial or temporal expression patterns (reviewed in Kleinjan et al., 2005; Merla et al., 2006). Alternatively, the two functional SNPs in GJB2 may have contributed to the SNHL in S3; although both SNPs identified have been reported to be benign, one (E114G) has been associated with SNHL in a Korean family (Park et al., 2000). Furthermore, the contribution of conductive hearing loss in this subject could not be assessed, so SNHL could be a minor component of the total hearing loss. If, based on these ambiguities in the etiology of the hearing loss in S3, we exclude S3 from the analysis, the critical region for SNHL in our 3p-syndrome subject pool would include the same 18 genes considered above, but both of the patients demonstrating significant SNHL (S1 and S2; Table 2) would have been unambiguously hemizygous for ATP2B2 function.
In conclusion, we have used deletion mapping in 3p-syndrome patients to identify a novel human hearing loss locus in 3p25.3. The region we have defined contains 18 candidate genes. Considering the mouse and human literature, we suggest that haploinsufficiency of ATP2B2 is the most likely cause of the observed phenotype. Further, we predict that loss-of-function mutations specific to ATP2B2 would cause bilateral SNHL.
Electronic Resources
Hereditary Hearing Loss Homepage: http://webhost.ua.ac.be/hhh/
Connexin-deafness Homepage: http://davinci.crg.es/deafness/
Hereditary Hearing Impairment in Mice webpage: http://www.jax.org/hmr/map.html
UCSC Genome Browser: http://genome.ucsc.edu/
NIH-NCBI dbSNP database: http://www.ncbi.nih.gov/SNP
Unique: http://www.rarechromo.org/
Chromosome Deletion Outreach: http://www.chromodisorder.org/
Acknowledgments
We wish to thank all patients and families for their participation in this study. We thank Beverly Searle, Ph.D. of Unique and Linda Sorg of Chromosome Deletion Outreach for their assistance in finding participants. We also thank Valerie Street for reviewing the manuscript and help with IRB issues; Glen MacDonald for help with images; and Anda Cornea for help in the preparation of this manuscript. This work was supported by a dual-degree NRSA fellowship F30-DC005861 to BJM, R01-DC003929 to JCA, and P30-DC004661 and R01-DC002739 to BLT.
Abbreviations
- ABR
auditory-evoked brainstem response
- bp
base pair
- CT
threshold cycle
- dBHL
dB hearing level (ANSI standard)
- dBnHL
dB for normal hearing level (ABR)
- IHC
inner hair cell
- kb
kilobase (1,000 base pairs)
- Mb
megabase (1 million base pairs)
- MIM
Mendelian inheritance in man
- OC
organ of Corti
- OHC
outer hair cell
- PMCA2
plasma membrane calcium ATPase type 2
- qPCR
Quantitative (real-time) polymerase chain reaction
- RM
Reissner’s membrane
- SNHL
sensorineural hearing loss
- SNP
single nucleotide polymorphism
- SPL
sound pressure level
- STS
sequence tagged site
- SV
stria vascularis
- VHL
von Hippel Lindau Disease
Appendix
Primer sequences for deletion mapping markers.
| Marker | Forward Primer | Reverse Primer |
|---|---|---|
| CHL* | GCGGATGGTCGTAGCATATT | GTCGCCAATCTGCTTTTCTC |
| SHGC 84547 | GGTGTGAGGACGACATTAGGAAA | TCTCAGCTGTTAAAGGAGGATGC |
| SHGC 15249 | TTATGTTCTGTCCACACATTTGC | TTGAACTCATATATCATTGGCCC |
| SRGAP2 (5’)* | TGCTTGCAGTGAGTTCTGCT | GGTGAACACAAGGCTGGACT |
| ARPC4* | CTGAAGCTGGGCTGTACCAT | CACCTAGGGCTGTCTCGGTA |
| IL17RE* | GCACTCCAGCCTGGGTAATA | ATGGAGCATTTGAGCAGCTT |
| WI 14152 | TTTATTAGGGAAATATGAGAGGCA | TGTGGCTGCCAGAGAATGTT |
| GDB:375126 | ACAGTAACGAGTTGGCCTAG | CTGCGTGCGCGCTCCCGAGT |
| VHL* | CAACTACTCGGGAGGCTGAG | TTTTCGCCCCTCTAAGGTTT |
| SHGC 19014 | CACTTTATTTTCAGAAAAAGATGGC | AGTGAAACCTCACCTGTGTGG |
| GHRL* | CACCTGGCAGAAATGACTGA | GAGCCAGGCTGGTGATTCTA |
| SEC13L1* (3’) | CCACTTTGGCTCCATGTCTT | GCAATGGGGAAGGGAGTTAT |
| SEC13L1* (5’) | AGCTGCAGTGGGGTAGAAGA | AGCTGGAGATGGTCACTGCT |
| WI 6061 | GACACCGACCTGGAAGAAGA | AGCTAAAGCGACGTCTCCAG |
| ATP2B2 (3’)* | CTTCAGCGGAGGAAACTGAG | AAAACGAGGCTGGAATCAGA |
| ATP2B2 (Middle)* | GTCCATCAGCTCCCAGATGT | CTCCTACAAACCCAGCCTGA |
| SHGC 148056 | AGAAAGGCTAGGCTTCTCCTGAT | TGGTCACTAAAAGAGCGAGTGTG |
| SHGC 15229 | AACGCACTCCTTTCCTCTCA | TCTTTTGCCTCCGAGTTGTT |
| WI 10567 | AGCGATGAAATTTATATGTTATGCC | AGGGTTAAATTCTTGCCGCT |
| TMIE* | CAGGGTCCTGGCTTAGACTG | AAGAAGCCAGCATCTTCAGC |
| SHGC 56873 | CCATGTTCTGCTATGGTGCC | TGCCAAGCTATGCACTCTCTG |
| SHGC 24090 | CTTGTAGGCTTCCCACATTAGG | TGCATTTCCCTCAGTATCAGG |
Primers were designed to non-coding regions of the indicated genes using Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Brendan J. McCullough, Email: brenmcc@u.washington.edu.
Joe C. Adams, Email: Joe_Adams@meei.harvard.edu.
Dustin J. Shilling, Email: dustinj@u.washington.edu.
M. Patrick Feeney, Email: pfeeney@u.washington.edu.
Kathleen C. Y. Sie, Email: Kathleen.Sie@seattlechildrens.org.
Bruce L Tempel, Email: bltempel@u.washington.edu.
References
- Angeloni D, Lindor NM, Pack S, Latif F, Wei MH, Lerman MI. CALL gene is haploinsufficient in a 3p-syndrome patient. Am J Med Genet. 1999;86:482–5. doi: 10.1002/(sici)1096-8628(19991029)86:5<482::aid-ajmg15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Benini D, Vino L, Vecchini S, Fanos V. 46, XY, del (3) (pter → p25) syndrome: further delineation of the clinical phenotype. Eur J Pediatr. 1999;158:955–7. doi: 10.1007/s004310051256. [DOI] [PubMed] [Google Scholar]
- Choung YH, Moon SK, Park HJ. Functional study of GJB2 in hereditary hearing loss. Laryngoscope. 2002;112:1667–71. doi: 10.1097/00005537-200209000-00026. [DOI] [PubMed] [Google Scholar]
- Denoyelle F, Weil D, Maw MA, Wilcox SA, Lench NJ, Allen-Powell DR, Osborn AH, Dahl HH, Middleton A, Houseman MJ, Dode C, Marlin S, Boulila-ElGaied A, Grati M, Ayadi H, BenArab S, Bitoun P, Lina-Granade G, Godet J, Mustapha M, Loiselet J, El-Zir E, Aubois A, Joannard A, Petit C, et al. Prelingual deafness: high prevalence of a 30delG mutation in the connexin 26 gene. Hum Mol Genet. 1997;6:2173–7. doi: 10.1093/hmg/6.12.2173. [DOI] [PubMed] [Google Scholar]
- Drumheller T, McGillivray BC, Behrner D, MacLeod P, McFadden DE, Roberson J, Venditti C, Chorney K, Chorney M, Smith DI. Precise localisation of 3p25 breakpoints in four patients with the 3p-syndrome. J Med Genet. 1996;33:842–7. doi: 10.1136/jmg.33.10.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont RA, Lins U, Filoteo AG, Penniston JT, Kachar B, Gillespie PG. Plasma membrane Ca2+-ATPase isoform 2a is the PMCA of hair bundles. J Neurosci. 2001;21:5066–78. doi: 10.1523/JNEUROSCI.21-14-05066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duno M, Hove H, Kirchhoff M, Devriendt K, Schwartz M. Mapping genomic deletions down to the base: a quantitative copy number scanning approach used to characterise and clone the breakpoints of a recurrent 7p14.2p15.3 deletion. Hum Genet. 2004;115:459–67. doi: 10.1007/s00439-004-1174-y. [DOI] [PubMed] [Google Scholar]
- Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, Lifton RP, State MW. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2004;74:1286–93. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fettiplace R, Hackney CM. The sensory and motor roles of auditory hair cells. Nat Rev Neurosci. 2006;7:19–29. doi: 10.1038/nrn1828. [DOI] [PubMed] [Google Scholar]
- Furuta H, Lin L, Hepler K, Ryan AF. Evidence for differential regulation of calcium by outer versus inner hair cells: plasma membrane Ca-ATPase gene expression. Hear Res. 1998;123:10–26. doi: 10.1016/s0378-5955(98)00091-4. [DOI] [PubMed] [Google Scholar]
- Higginbottom MC, Mascarello JT, Hassin H, McCord WK. A second patient with partial deletion of the short arm of chromosome 3: karyotype 46,XY,del(3)(p25) J Med Genet. 1982;19:71–3. doi: 10.1136/jmg.19.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinney A, Hoch A, Geller F, Schafer H, Siegfried W, Goldschmidt H, Remschmidt H, Hebebrand J. Ghrelin gene: identification of missense variants and a frameshift mutation in extremely obese children and adolescents and healthy normal weight students. J Clin Endocrinol Metab. 2002;87:2716. doi: 10.1210/jcem.87.6.8672. [DOI] [PubMed] [Google Scholar]
- Holt JR, Corey DP. Two mechanisms for transducer adaptation in vertebrate hair cells. Proc Natl Acad Sci U S A. 2000;97:11730–5. doi: 10.1073/pnas.97.22.11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, Grompe M. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–35. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura S, Adams JC. Changes in cytochemistry of sensory and nonsensory cells in gentamicin-treated cochleas. J Assoc Res Otolaryngol. 2003;4:196–218. doi: 10.1007/s10162-002-2037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: a HuGE review. Genet Med. 2002;4:258–74. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleymenova E, Everitt JI, Pluta L, Portis M, Gnarra JR, Walker CL. Susceptibility to vascular neoplasms but no increased susceptibility to renal carcinogenesis in Vhl knockout mice. Carcinogenesis. 2004;25:309–15. doi: 10.1093/carcin/bgh017. [DOI] [PubMed] [Google Scholar]
- Kudo T, Ikeda K, Kure S, Matsubara Y, Oshima T, Watanabe K, Kawase T, Narisawa K, Takasaka T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population. Am J Med Genet. 2000;90:141–5. doi: 10.1002/(sici)1096-8628(20000117)90:2<141::aid-ajmg10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Kim HJ, Butman JA, Vortmeyer AO, Choo DI, Oldfield EH. Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med. 2004;350:2481–6. doi: 10.1056/NEJMoa040666. [DOI] [PubMed] [Google Scholar]
- Lumpkin EA, Hudspeth AJ. Regulation of free Ca2+ concentration in hair-cell stereocilia. J Neurosci. 1998;18:6300–18. doi: 10.1523/JNEUROSCI.18-16-06300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma RZ, Gao J, Meeker ND, Fillmore PD, Tung KS, Watanabe T, Zachary JF, Offner H, Blankenhorn EP, Teuscher C. Identification of Bphs, an autoimmune disease locus, as histamine receptor H1. Science. 2002;297:620–3. doi: 10.1126/science.1072810. [DOI] [PubMed] [Google Scholar]
- Manski TJ, Heffner DK, Glenn GM, Patronas NJ, Pikus AT, Katz D, Lebovics R, Sledjeski K, Choyke PL, Zbar B, Linehan WM, Oldfield EH. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease JAMA. 1997;277:1461–6. doi: 10.1001/jama.277.18.1461. [DOI] [PubMed] [Google Scholar]
- McCullough BJ, Tempel BL. Haplo-insufficiency revealed in deafwaddler mice when tested for hearing loss and ataxia. Hear Res. 2004;195:90–102. doi: 10.1016/j.heares.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Megerian CA, Haynes DS, Poe DS, Choo DI, Keriakas TJ, Glasscock ME., 3rd Hearing preservation surgery for small endolymphatic sac tumors in patients with von Hippel-Lindau syndrome. Otol Neurotol. 2002;23:378–87. doi: 10.1097/00129492-200205000-00026. [DOI] [PubMed] [Google Scholar]
- Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet. 2006;79:332–41. doi: 10.1086/506371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahara K, Kikkawa K, Murakami M, Hiramoto K, Namba H, Tsuji K, Yokoyama Y, Kimoto H. Loss of the 3p25.3 band is critical in the manifestation of del(3p) syndrome: karyotype-phenotype correlation in cases with deficiency of the distal portion of the short arm of chromosome 3. Am J Med Genet. 1990;35:269–73. doi: 10.1002/ajmg.1320350225. [DOI] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35:21–3. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Zheng QY, Johnson KR, Nishina PM. mdfw: a deafness susceptibility locus that interacts with deaf waddler (dfw) Genomics. 1997;44:266–72. doi: 10.1006/geno.1997.4869. [DOI] [PubMed] [Google Scholar]
- Park HJ, Hahn SH, Chun YM, Park K, Kim HN. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535–8. doi: 10.1097/00005537-200009000-00023. [DOI] [PubMed] [Google Scholar]
- Phipps ME, Latif F, Prowse A, Payne SJ, Dietz-Band J, Leversha M, Affara NA, Moore AT, Tolmie J, Schinzel A, et al. Molecular genetic analysis of the 3p-syndrome. Hum Mol Genet. 1994;3:903–8. doi: 10.1093/hmg/3.6.903. [DOI] [PubMed] [Google Scholar]
- Ramer JC, Ladda RL, Frankel C. Two infants with del(3)(p25pter) and a review of previously reported cases. Am J Med Genet. 1989;33:108–12. doi: 10.1002/ajmg.1320330115. [DOI] [PubMed] [Google Scholar]
- Robinson SW, Morris CD, Goldmuntz E, Reller MD, Jones MA, Steiner RD, Maslen CL. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am J Hum Genet. 2003;72:1047–52. doi: 10.1086/374319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RL, Wajid M, Pham TL, Hussan J, Ali G, Ahmad W, Leal SM. Low prevalence of Connexin 26 (GJB2) variants in Pakistani families with autosomal recessive non-syndromic hearing impairment. Clin Genet. 2005;67:61–8. doi: 10.1111/j.1399-0004.2005.00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JM, Yang Y, Caride AJ, Filoteo AG, Penheiter AR, Lagziel A, Morell RJ, Mohiddin SA, Fananapazir L, Madeo AC, Penniston JT, Griffith AJ. Modification of human hearing loss by plasma-membrane calcium pump PMCA2. N Engl J Med. 2005;352:1557–64. doi: 10.1056/NEJMoa043899. [DOI] [PubMed] [Google Scholar]
- Street VA, McKee-Johnson JW, Fonseca RC, Tempel BL, Noben-Trauth K. Mutations in a plasma membrane Ca2+-ATPase gene cause deafness in deafwaddler mice. Nat Genet. 1998;19:390–4. doi: 10.1038/1284. [DOI] [PubMed] [Google Scholar]
- Wood JD, Muchinsky SJ, Filoteo AG, Penniston JT, Tempel BL. Low endolymph calcium concentrations in deafwaddler2J mice suggest that PMCA2 contributes to endolymph calcium maintenance. J Assoc Res Otolaryngol. 2004;5:99–110. doi: 10.1007/s10162-003-4022-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, Wiegand SJ, Sleeman MW. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci U S A. 2004;101:8227–32. doi: 10.1073/pnas.0402763101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamoah EN, Lumpkin EA, Dumont RA, Smith PJ, Hudspeth AJ, Gillespie PG. Plasma membrane Ca2+-ATPase extrudes Ca2+ from hair cell stereocilia. J Neurosci. 1998;18:610–24. doi: 10.1523/JNEUROSCI.18-02-00610.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]