

Abstract
Age-related macular degeneration (AMD) is the major cause of blindness for people over 60. In the “wet” form of AMD compounds targeting growth factor signaling pathways such as VEGF have been a major focus for therapeutic interventions. In a previously developed rat model of CNV, we utilized two receptor tyrosine kinase inhibitors (RTKi) to block VEGFR-1, VEGFR-2 and PDGFR signaling following the establishment of CNV. AAV-VEGF 165 was injected into the subretinal space of rats at postnatal days 15-17. Six weeks later, a suspension of RTK inhibitors, AG013764 or AG013711, was injected intraperitoneally (IP, twice daily) or intravitreally (every five days) over a two week period. FITC-dextran whole-mounts of RPE-choroid-sclera were prepared after the animals were sacrificed. CNV area was quantified using Neurolucida to measure the hyperfluorescence on FITC-dextran whole-mounts. Histology and immunohistochemistry were performed as described previously. VEGF expression in control and treated eyes was confirmed by immunohistochemistry and histological sections indicated recovery of retinal morphology and CNV reduction in treated eyes. In the animals IP injected with AG013764 or AG013711 the mean CNV level was reduced by 25 to 33% compared to control, but this effect did not achieve statistical significance. Intravitreal injections of AG013764 or AG013711 reduced the level of CNV by approximately 60% compared to control (p< 0.005 or p< 0.05, respectively). These data show that two RTK inhibitors, AG013764 or AG013711, delivered intravitreally, significantly reduce blood vessel proliferation in this AAV-VEGF 165 model of CNV.
Keywords: Age-related macular degeneration, choroidal neovascularization, gene transfer, adeno-associated viral vector, anti-angiogenesis, receptor tyrosine kinase, VEGF, PDGF, combination therapy
INTRODUCTION
Ocular neovascularization leads to severe vision loss in many diseases, including age-related macular degeneration (AMD), diabetic retinopathy, and retinopathy of prematurity (Bird 2003; Ferris 2004; Grossniklaus and Green 2004). AMD is the leading cause of severe loss of vision in people 55 years of age and older in the developed countries (Congdon et al., 2004). Neovascular (wet) AMD represents approximately 10 percent of the overall disease prevalence, but it is responsible for 90 percent of the severe vision loss in AMD (Ferris et al., 1984). Neovascular AMD occurs in as many as 200,000 to 300,000 patients each year in the United States, and without treatment most patients progress to a visual acuity of 20/200 or worse in less than 2 years (Macular et al., 1993).
Neovascular AMD is characterized by choroidal neovascularization in which new blood vessels penetrate through Bruch’s membrane and extend into the subretinal pigment epithelium or subretinal space, often leading to exudation and hemorrhage, RPE or retina detachment, photoreceptor degeneration, loss of central vision and the formation of disciform scars (Green 1999; Grossniklaus and Green 2004). In AMD, a significant angiogenic role for VEGF was suggested by preferential localization of VEGF immunoreactivity in the cytoplasm of RPE from highly vascularized regions of surgically excised choroidal neovascular membranes (Lopez et al., 1996; Baffi et al., 2000; Spilsbury et al., 2000; Wang et al., 2003; Lebherz et al., 2005).
The importance of VEGF for the development of AMD-related CNV has led to multiple strategies to block its effects including anti-VEGF aptamer (pegaptanib sodium, Macugen™) (Eyetech et al., 2002; Eyetech et al., 2003; Gragoudas et al., 2004), anti-VEGF antibodies (Krzystolik et al., 2002), soluble VEGF receptor (Saishin et al., 2003; Liu and Regillo 2004), gene therapy, protein kinase C inhibitors, and siRNA for VEGF and its receptors (Tolentino et al., 2004). Anti-VEGF strategies with promise as potential therapeutic agents for the treatment of CNV are currently undergoing or have completed clinical trials (pegaptanib sodium, Macugen; ranibizumab, Lucentis; VEGF-Trap) (Barouch and Miller 2004). Macugen has been approved by the FDA for use in the treatment of neovascular AMD. Avastin, the parent molecule of Lucentis, is a fully humanized antibody against VEGF and has recently been shown to be clinically effective against CNV (Avery et al., 2006). The results of randomized, placebo-controlled phase III studies of Lucentis (MARINA and ANCHOR) have been reported (Rosenfeld et al., 2005; Brown et al., 2006; Heier et al., 2006) and Lucentis has being approved by FDA for treating wet AMD.
Recent work in tumor biology has focused on multi-kinase inhibitors, such as Sutent, PTK787 and AG013736 to block angiogenesis (Motzer et al., 2006; Nakahara et al., 2006; Schoffski et al., 2006). The present experiments utilized two similar compounds, AG013711 and AG013764, which are small molecule inhibitors of VEGFR-1, VEGFR-2 and related receptor tyrosine kinases (RTK) such as PDGFR (Niesman et al., 2000; Rewolinski et al., 2001). Combined activity against VEGF and PDGF signaling transduction is beneficial since both growth factors are involved in angiogenesis. VEGF induces endothelial cell proliferation, migration, tube formation, increases vascular permeability, leukocyte trafficking, and is critical in ensuring the survival of proliferating endothelial cells. PDGF induces pericyte recruitment and supports vascular maturation. Inhibitors of PDGFRs have been shown to produce pericyte loosening or detachment from endothelial cells of tumor vessels (Betsholtz 2004; Inai et al., 2004; Kinose et al., 2005). In addition, blood vessels lacking pericytes are more susceptible to VEGF deprivation. Therefore, inhibitors targeting both VEGFR and PDGFR can contribute to the regression of actively proliferating endothelial cells in tumors (Bergers et al., 2003; Nakahara, Norberg et al. 2006) and in multiple models of ocular neovascularization (Jo et al., 2006). Analogues of the compounds used in the present study, AG013736 and AG013958, are in phase II clinical trials for renal cell carcinoma (Schoffski, Dumez et al. 2006) and for wet AMD (Hoyng et al., 2005; Rewolinski et al., 2005), respectively.
The compounds used in this study target both VEGF and PDGF pathways and have the potential to induce regression of choroidal neovascularization beyond what Macugen or Lucentis can accomplish individually. Previously, we developed a rat model of choroidal neovascularization using adeno-associated virus (AAV)-VEGF. Our previous work showed that CNV is well established in this model by six weeks (Wang, Rendahl et al. 2003). In the present experiments, two small molecule inhibitors of VEGFR and PDGFR were administered after establishment of CNV. Our experiments test the notion that inhibitors against multiple receptor tyrosine kinases are more effective in causing CNV regression.
MATERIALS AND METHODS
Animals
All animal experiments were done according to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Subretinal Injection of AAV
Sprague Dawley (albino) rats were injected with AAV-VEGF or AAV-GFP at the ages of P15 - P17 as previously described (Green et al., 2001). Briefly, wild-type animals were anesthetized by ketamine/xylazine injection, and a local anesthetic (proparacain HCl) was applied topically to the cornea. The pupils were dilated with 1% atropine sulfate and 2.5% phenylephrine hydrochloride. Under the microscope, a small hole was made in the medial sclera, just behind the ora serrata, with a 28-gauge needle. The subretinal injections of 2 μl of AAV-VEGF were made by inserting a blunt 32-gauge needle (Hamilton, Reno, NV) through the sclera, vitreous, and retina and delivering the rAAV suspension into the subretinal space (SRS) in the posterior retina. The contralateral eye was either injected with AAV-VEGF or AAV-GFP. 1.4×1010 particles of each virus were injected. Only one viral preparation was used for all of AAV-VEGF experiments; this same preparation was previously used in the CNV model paper (Wang, Rendahl et al. 2003). Subretinal blebs were formed in the SRS and disappeared over 24 hours as the fluid was absorbed. No choroidal bleeding was observed during and after subretinal injection, indicating that the integrity of Bruch’s membrane was not compromised.
Drug Delivery by Intraperitoneal (IP) or Intravitreal injection
The two compounds shown below, AG013764 and AG013711, were delivered via intraperitoneal or intravitreal injections using 5-8 rats per group. Rats were treated starting at 6 weeks after subretinal injection of AAV-VEGF in both eyes. Suspensions of compounds were made in sterile 0.1% carboxymethylcellulose (CMC) –PBS. For experiments with IP injection, animals previously injected AAV-VEGF in both eyes were randomly divided into three groups. Each group of animals was injected IP twice daily with 1.25 ml of vehicle (0.1% CMC-PBS), AG013764 (10 mg/Kg) or AG013711 (25 mg/Kg).
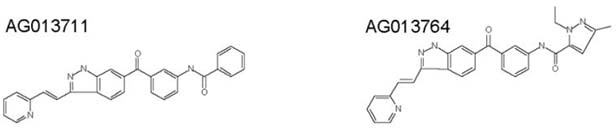
For experiments with intravitreal injection, animals previously injected AAV-VEGF in both eyes were randomly divided into two groups. In each animal, one eye was intravitreally injected with vehicle (0.1% CMC–PBS) and the contralateral eye was injected with 5 μl of AG013764 (4 mg/ml) or AG013711 (10mg/ml) every five days (at 42, 47 and 52 days after the initial AAV-VEGF injection; three doses over two weeks).
Immunohistochemistry
Rats were euthanized by overdose of carbon dioxide inhalation and their eyes were enucleated. After the cornea and lens were removed, the posterior eyecups (retina-RPE-choroid-sclera) were fixed in 4% formaldehyde for 1 hour. After three 5-minute washes in PBS, eyecups were cryoprotected in 30% sucrose overnight at 4°C, and embedded in OCT at -20°C. 15 μm sections were cut on a cryostat, placed on glass slides, and allowed to dry overnight. GFP fluorescence image was obtained using an Axiophot microscope (Zeiss, Thornwood, NY) equipped with a CCD camera and processed with Adobe Photoshop 6.0 (San Jose, CA). For VEGF immunohistochemical staining, sections were permeabilized with 0.3% Triton X-100 (PBS) for 10 min and processed with a customized Zymed Histostain-SP kit (South San Francisco, CA) according to the instructions. Briefly, sections were treated with Peroxo-Block to eliminate endogenous peroxidase activity and blocked with 10% non-immune rabbit serum to reduce non-specific binding. Following three washes with 0.3% Triton X-100 PBS, sections were incubated in 10 μg/ml goat anti-human VEGF primary antibodies (R & D systems, Minneapolis, MN) diluted in blocker for 1 hr at room temperature. After three washes, sections were incubated with biotinylated rabbit anti-goat secondary antibodies for 10 min, followed by streptavidin-peroxidase conjugate. Sections were incubated with substate-chromogen solution (hydrogen peroxide-DAB), counterstained with hematoxylin, dehydrated with graded series of alcohol, cleared in xylene, and mounted with Histomount.
Histology
As previously described (Lau et al., 2000), rats were euthanized by overdose of carbon dioxide inhalation and immediately perfused intracardially with 2% formaldehyde and 2.5% glutaraldehyde in PBS, pH7.4. Heads were removed and immersed in same fixative overnight at 4°C. Eyes were then enucleated and bisected along the vertical meridian through the optic nerve head. Eye hemispheres were osmicated, dehydrated, and embedded in Epon-Araldite resin. Serial sections (1-1.5 μm) were cut along the vertical meridian of the eye and stained with 1% toluidine blue in TBS. Each epoxy embedded eye was sectioned completely, from one side of the eye all the way through to the other side. This allowed us to identify the complete boundary of the CNV membrane. Images were obtained with the same microscope as above.
Fluorescein Angiography and FITC-Dextran Flat-mount of RPE-Choroid-Sclera
After two weeks of treatment by intraperitoneal and intravitreal injection, rats were euthanized with 100% CO2 (eight weeks after initial AAV-VEGF injection). Using a method similar to that previously described (Edelman and Castro 2000), each animal was perfused intracardially with 50 ml PBS to wash out the blood and followed immediately by 20 ml of 10% gelatin and 2.5% FITC-dextran (MW: 2×106 Sigma, St. Louis, MO, USA). FITC-dextran perfused rats were kept in the refrigerator at 4°C for two to four hours to solidify the gelatin. Eyes were enucleated and stored in a cold 4% neutralized formaldehyde solution.
In the experimental groups receiving intraperitoneal injection, anterior segments were first removed, and then the eye cups were kept in the cold 10% neutralized formalin in a Petri dish with the eye cup facing up. Angiograms were taken with digital camera mounted to a fluorescence dissection microscope using FITC filters. Angiograms were graded for fluorescence area and intensity from low to high (grade 1 to 5) by a reader in a masked manner. Six-eight radial cuts were made in the eye cups and then the retina was carefully peeled away. The RPE-choroid-sclera complexes with CNV were flattened on a glass slide with RPE facing up and a cover slip placed on the RPE with mounting medium (VectorShield, Vector Laboratory, CA).
In the experimental groups receiving intravitreal injection, anterior segments were removed and the eye cups were cut and flat-mounted using the methods described above. Angiograms could not be obtained because some of the injected compound was still deposited on the retina blocking the flourescence signal from FITC-dextran filled blood vessels.
Imaging and quantifying Choroidal Neovascularization
Whole mounts of RPE-choroid-sclera were viewed under a Nikon Eclipse 600 fluorescence microscope. Contours of whole mount, optic nerve, and choroidal neovascularization area were drawn on a computer using the Neurolucida system (MicrobrightField Inc.) coupled to a Nikon E600 microscope (200× magnification). The Neurolucida system (MBF Bioscience; http://www.mbfbioscience.com) can be used to automatically trace a boundary that is much larger than the field of view under the microscope. The computer mouse cursor is projected onto this view field. The motorized stage shifts automatically as the cursor moves outside the box in the middle of the view field (Amorapanth et al., 2000). This system allows one to view the RPE-choroid-sclera whole mounts at high magnification and thereby clearly visualize and mark the boundary between hyperfluorescence area (CNV) and the normal (choriocapillaris) florescence area. The system was calibrated using a slide with a grid of known size. Area of choroidal neovascularization was calculated by the Neurolucida software. A series of photographs (30 per eye) of whole mounts of RPE-choroid-sclera were taken with a Zeiss Axioplan 2 fluorescence microscope at 200× magnification. The photographs were then stitched together in Adobe Photoshop.
Biochemical Activity Assays for Human VEGF-R2, VEGF-R1, PDGFRs
The enzyme phosphorylation (activation) assay for VEGFR2 activity was performed using a R2 construct previously described (McTigue et al., 1999). For this assay, VEGF-R1 and PDGFRβ coupled assay components (2 mM PEP, 300 μM NADH, 5 mM DTT, 15 U/mL PK, 15 U/mL LDH, 200 mM HEPES, pH 7.5) were added to the protein. Poly(Glu, Tyr) (PGT) was the peptide substrate. Incubation conditions were: VEGF-R2: 3mM ATP, 60 mM Mg, 2 mM Mn, 20 mM PGT; VEGF-R1: 3mM ATP, 40 mM Mg, 2 mM Mn, 20 mM PGT; PDGFRβ: 3mM ATP, 40 mM Mg, 20 mM PGT. After incubating for 10 minutes at 37°C, the reaction was measured spectrophotometrically at 340 nm. In a final reaction volume of 25 μL, PDGFRα (5-10 mU) was incubated with 8 mM MOPS pH 7.0, 0.2 mM EDTA, 0.1 mg/mL poly(Glu, Tyr) 4:1, 10 mM MnCl2, 10 mM MgAcetate and [γ-33P-ATP] (specific activity approximately 500 cpm/pmol, concentration as required). The reaction was initiated by the addition of the MgATP mix. After incubation for 40 minutes at room temperature, the reaction was stopped by the addition of 5 μL of a 3% phosphoric acid solution. Ten microliters of the reaction was then spotted onto a Filtermat A and washed three times for 5 minutes in 75 mM phosphoric acid and once in methanol prior to drying and scintillation counting. Ki or IC50 was calculated from a 9-point dose-response curve. The % inhibition assay is run as a single point assay in duplicate. Data summarized in Table 1.
Table 1.
| AG013711 | AG013764 | |
|---|---|---|
| Biochemistry Assays | ||
| VEGFR-2 (KDR) Ki | 0.046 nM | 0.057 nM |
| VEGFR-1 (Flt-1) Ki | NA | 0.242 nM |
| VEGFR-1 % inhibition @ 50 nM | 93% | 96% |
| PDGFR alpha IC 50 | 31 nM | 79 nM |
| PDGFR beta IC 50 | 89 nM | 186 nM |
| Cell-Based Assays IC50 nM | ||
|
| ||
| VEGF stimulated HUVEC survival | 0.273 nM | 0.295 nM |
| VEGF stimulated HUVEC survival + 2.3% BSA | 6.7 nM | 7.1 nM |
The enzyme phosphorylation (activation) assay for VEGFRs and PDGFRs activity was performed using purified protein. Ki or IC50 was calculated from a 9-point dose-response curve. The % inhibition assay is run as a single point assay in duplicate. The compounds were also examined in their activity for inhibiting VEGF-stimulated human umbilical vein endothelial cells survival. Serial dilutions of AG013711 or AG013764 were then added to drug-treatment wells in triplicates and three independent assays were performed.
VEGF stimulated HUVEC Survival Assay
Human umbilical vein endothelial cells (HUVEC) were purchased from Clonetics (# CC2517). Cells were initially thawed and grown in T75 tissue culture flasks in EGM2 media (Clonetics # CC3162). Cell media was then changed to F12K HUVEC growth medium consisting of F12K, 10% fetal bovine serum (FBS), 60 μg/mL ECGS (Sigma, # E-0760), 10 μg/mL heparin (Sigma, # H-3149), and 10 mM HEPES. Cells were removed from tissue culture flasks and seeded in 96-well cell culture plates with 100 μL of F12K HUVEC growth medium for several days. After removing medium from each well, starvation medium (F12K with 1% FBS, 10 μg/mL heparin, and 10 mM HEPES) was added to each well. Plates were incubated at 37°C, 5% CO2 for 18 hours. Fifteen microliters of 10 nM inhibitor in starvation medium was then added to each well to inhibit background stimulation by exogenous or endogenous VEGF or other mitogen in control and starved wells. Serial dilutions of AG013764 or AG013711 were then added to drug-treatment wells in triplicate and 15 μL of vehicle (1% DMSO in starvation media) was added to blank, starvation and control wells. After a one hour incubation, 20 μL of growth factor is added to drug treatment and control wells. Twenty microliters of starvation media was added to blank and starvation wells. Cells were then stimulated with recombinant human VEGF 165 (R&D Systems). Fifteen microliters of MTT dye solution was added to each well and incubated for 4.5 hours at 37°C, 5% CO2. Stop solution was then added and the plates incubated overnight at 37°C, 5% CO2. MTT and MTT Stop solutions were purchased from Promega (# G4100). Three independent assays were performed. Data summarized in Table 1.
Statistics
Results are expressed as means ± SEM unless otherwise indicated. Statistical significance was calculated using the two-tailed, paired Student’s t-test for the data from intravitreal injections. To evaluate the intraperitoneal injection data, we used a two-sided, mixed model analysis of variance that takes into account the correlation between paired eyes (Littell et al., 1996). P ≤ 0.05 is regarded as statistically significant in all comparisons.
RESULTS
VEGF165 and GFP were overexpressed in RPE at 8 weeks after AAV-VEGF (Fig. 1C) and AAV-GFP (Fig. 1A) injection, respectively. There was little or no VEGF expression in RPE cells overexpressing GFP (Fig. 1B). Since the antibody for human VEGF cross reacts with rat VEGF, the weak staining that we did observe is probably due to endogenous rat VEGF This pattern of expression is consistent with our previously published data (Wang, Rendahl et al. 2003). Increased VEGF staining was also seen in the choroid of the VEGF injected eye (Fig. 1C) compared to control (Fig. 1B), possibly due to secretion of VEGF165 from the basolateral side of the RPE.
Figure 1. VEGF and GFP expression in rat eyes 8 weeks after AAV-VEGF or AAV-GFP injection.
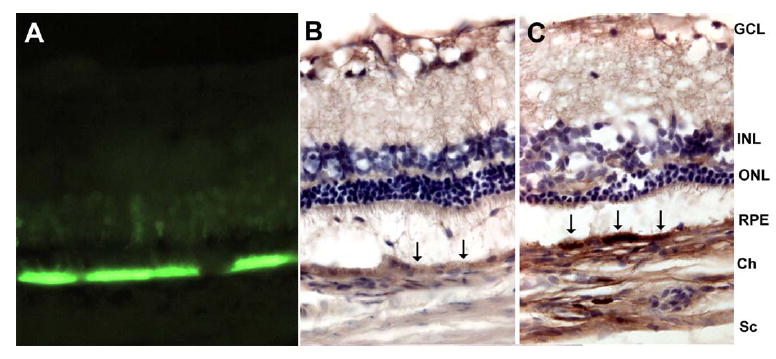
Eyes were fixed, embedded in OCT medium, sectioned (10μm) and stained. A. GFP strongly expressed in RPE and faintly expressed in photoreceptors from an eye injected with AAV-GFP. B. In AAV-GFP injected eye, there is relatively little DAB staining in RPE (black arrows). C. In AAV-VEGF -injected eye, there is intense DAB staining at the RPE (black arrows), indicating VEGF overexpression. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium; Ch, choroid; Sc, sclera.
Several hundred histological sections (1.5 μm) were obtained from each eye of four animals. These sections were cut along the inferior-superior axis and traversed the entire area of CNV. In each animal, one eye was injected with control (METHODS) and the other eye was injected with one of two anti-angiogenic compounds, either AG013764 (n = 2) or AG013711 (n = 2). These two compounds are extremely potent inhibitors of both KDR and Flt-1 and also inhibit PDGF receptors α and β at sub-micromolar concentrations (Table 1). This in vitro data helps provide an estimate for the highest in vivo dose (25 mg/Kg body wt or 56 nM, assuming the drug is evenly distributed over the entire body). The HCl salt of AG013764 and related compounds have demonstrated reduction in tumor blood vessel growth and blood flow (Kim et al., 2005; Mancuso et al., 2006) and would be expected to have a similar efficacy in CNV. We qualitatively assessed the efficacy of this treatment by comparing sections chosen from comparable locations within the area of CNV. As in a previous analysis (Wang et al., 2002; Wang et al., 2003), we compared the linear distance of retinal detachment along Bruch’s membrane, retinal morphology and choroidal neovascularization in hundreds of sections from control and treated eyes.
Figure 2 compares representative sections from the eyes of one animal. The control section (panels A & C) and the section from the contralateral eye injected with AG013764 (panels B & D) illustrate the AAV-VEGF –induced development of choroidal neovascularization at eight weeks after injection (Wang, Rendahl et al. 2003). Compared to control (panels A & C), the AG013764 injected eye (panel B, inset D), shows fewer blood vessels in the subretinal space (black arrows). The AG013764 treated eyes also have less retinal neovascularization, edema, and photoreceptor disorganization than the control eyes. The length of RPE and retinal detachment along Bruch’s membrane is shorter in the section from the AG013764 treated eye (indicated by Ltin Fig. 2B) than in the section from the control eye (Lc in Fig. 2A). This probably indicates a smaller total neovascular area in the AG013764 treated eye. These observations, characteristic of all sections, were quantified in subsequent experiments using FITC-dextran whole mounts.
Figure 2. AG013764 reduces VEGF-induced CNV in SD rats.
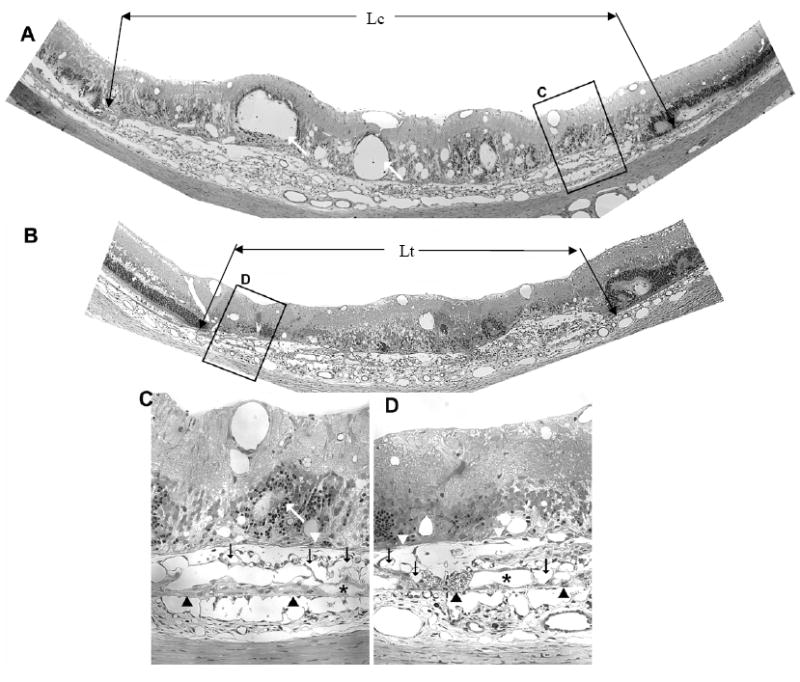
Panels A and B are 1.5 μm sections from eyes injected intravitreally with CMC carrier (control) or AG013764 (three 20 μg injections at 5 day intervals). Compared with the control eye, sections from the AG013764 treated eye show less blood vessel proliferation in the SRS (black arrows), areas of disorganized photoreceptors (white arrows), RPE proliferation (white arrowheads), and RPE “lakes” (asterisks). Bruch’s membrane indicated by black arrowheads. Panels C and D are sections in A and B at higher magnification.
The efficacy of compounds delivered systemically was qualitatively evaluated using fluorescein angiography on fifteen animals (five control and five treated with AG013764 and AG013711, respectively). AG013764 (1.25ml, 2 mg/ml), AG013711(1.25ml, 2 mg/ml) or control (1.25 ml) was injected twice daily for two weeks (IP), starting at six weeks after AAV-VEGF injection (METHODS). The animals were killed and the angiograms obtained at 8 weeks after subretinal injection of AAV-VEGF. In 7 out of 10 eyes treated with AG013764 and in 6 out of 10 eyes treated with AG013711, the angiograms showed less hyperfluorescence than the control eyes (represented in Figs 3A, B, C). This qualitative data suggests that both compounds may be capable of reducing neovascularization after two weeks of systemic treatment.
Figure 3. Inhibition of CNV by intraperitoneal injection of AG013764 and AG013711.
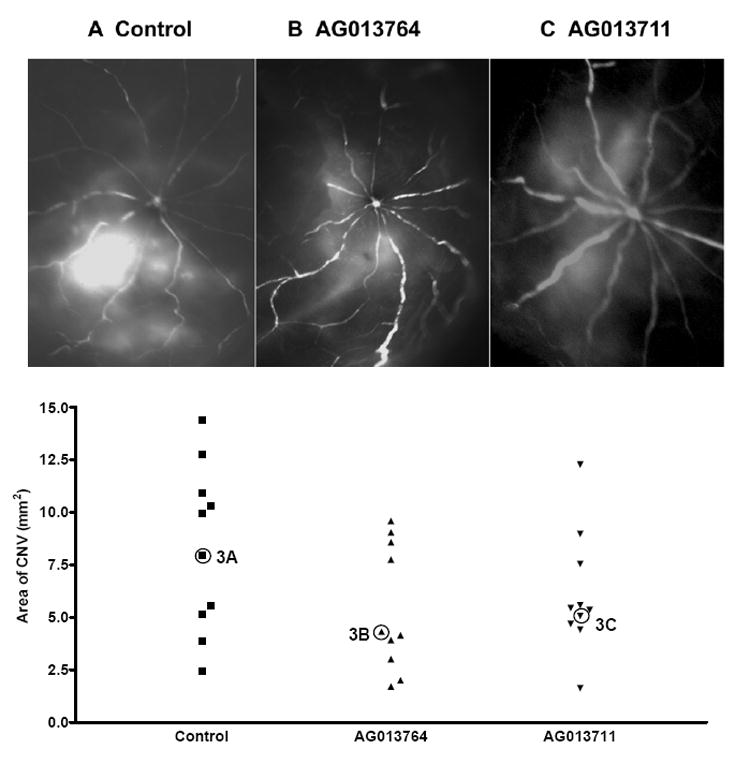
Animals(n=15) intraperitoneally were injected with control, AG013764 or AG013711 solutions twice daily for two weeks. Rats were first cardiac perfused with FITC-dextran, the eyes then removed and dissected free of anterior segments, and angiograms were taken. A. Angiogram from control animal injected with CMC carrier solution shows extensive area of hyperfluorescence. Angiograms from animals injected with AG013764 ( B ) or AG013711 ( C ) show a lower level of hyperfluorescence in a smaller area compared to control ( A ). D. RPE-choroid-sclera whole mount was prepared and the area of CNV was measured using the Neurolucida system. The mean area of CNV in the AG013764 treated eyes (n = 10, triangles) was reduced by ≈ 33% compared to control (squares). The mean area of CNV in the AG013711 treated eyes (n = 10, inverted triangle) was reduced by 25% compared to control. The angiogram for each eye labeled as 3A, 3B, and 3C is shown in panels A,B,& C respectively.
The Neurolucida system was used to quantify the area of CNV (Fig. 3D) and assess the ability of these two putative anti-angiogenic compounds to reduce VEGF induced choroidal neovascularization (see METHODS). The mean areas of CNV from animals treated with AG013764 or AG013711 (n=10 in each group) were approximately two-thirds and three-quarters of the CNV area (9 mm2) of control animals (n=10), but this trend did not achieve significance (p > 0.05 for both comparisons of control vs AG013764 and vs AG013711).
The possible efficacy of these compounds was further evaluated by intravitreal injections of AG013764 and AG013711 at six weeks after AAV-VEGF injection (METHODS). Whole mounts were examined using a fluorescence microscope to identify the areas of CNV, which were quantified using the Neurolucida system.
Figure 4 shows fluorescence images from whole mounts of contralateral control (panels A, B, C) and AG013764 – treated eyes (panels D, E, F). Panels A and D are an integrated composite of 30 images taken at high magification. Panels B and E show the areas enclosed by three contours: (1) the entire whole mount area in brown; (2) the area of neovascularization in purple; and (3) the optic nerve head in blue. The Neurolucida system was used to measure the area within each contour. The whole mount of RPE-choroid-sclera is approximately 50 mm2 for both control and AG013764 treated eyes. Panels A & B and D & E contain a green rectangular enclosed area that identifies the boundary separating an area of CNV-induced hyperfluorescence and a non-CNV area of choroidal blood vessels with much less fluorescence. Panels C and F show this boundary (yellow dotted line) at a higher magnification. The area of CNV in A, shown schematically in B as the area in purple, is approximately 14.3 mm2. In the AG013764 - treated eye (D & E) the area of CNV is approximately 5.6 mm2.
Figure 4. FITC-dextran whole mount of eyes intravitreally injected with AG013764 or control.
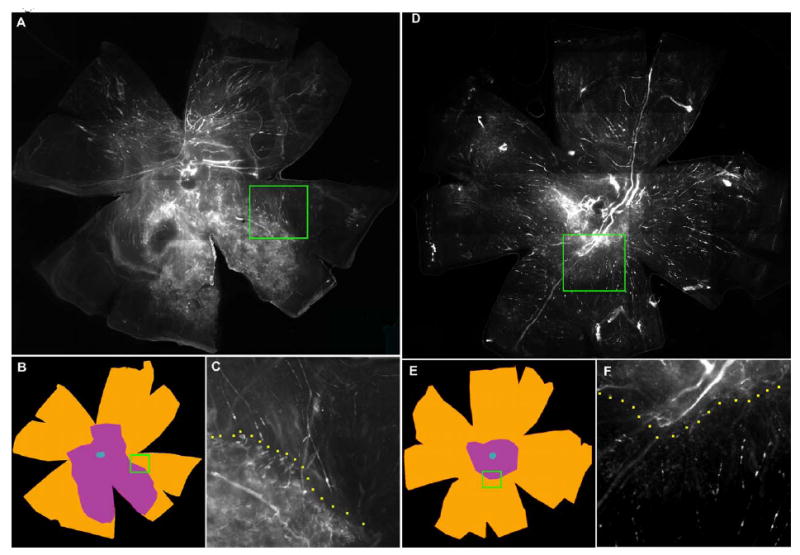
Animals injected every five days for two weeks starting at six weeks after AAV-VEGF injection. Panels A, B, C from a control eye injected with carrier and panels D, E, F from AG013764 injected eye. Panels A and D, photographs of whole eye flat mount. Panels B and E, typical sets of contours drawn using the Neurolucida system. The total flat mount area (≈ 50 mm2) is colored brown and the blue center denotes the optic nerve. Area of CNV shown in purple. Panels C and F, insets from A and D at higher magnification, show a typical boundary between areas of proliferation (CNV) and non-proliferation.
Figure 5 is a typical set of whole mount images from AG013711- treated eyes (panels D, E, F) and its contralateral control (panels A, B, C). The six panels were arranged as in Figure 4. The area of CNV in A and D is marked by hyperfluorescence compared to the surround and is approximately 3.2 mm2 in control panels 5A & 5B and approximately 1.4 mm2 in the AG013711 - treated eye (panels 5D & 5E).
Figure 5. FITC-dextran whole mount of eyes intravitreally injected with AG013711 or control.

Animals injected every five days for two weeks starting at six weeks after AAV-VEGF injection. Panels A, B, C from a control eye injected with carrier and panels D, E, F from AG013711 injected eye. Panels A and D, photographs of whole eye flat mount. Panels B and E, typical sets of contours drawn using the Neurolucida system. The total flat mount area (≈ 50 mm2) is colored brown and the blue center denotes the optic nerve. Area of CNV shown in purple. Panels C and F, insets from A and D at higher magnification, show a typical boundary between areas of proliferation (CNV) and non-proliferation.
Figure 6 summarizes the results from twenty-five animals using the protocol illustrated in Figures 4 & 5. In 12 of 13 animals, the area of CNV was reduced by 16% to 100% following intravitreal injections of AG013764. The mean CNV area (2.5 ± 1.1 mm2, mean ± SEM) for the AG013764 injected eyes (n=13) is 57% smaller than control (5.8 ± 1.4 mm2, p< 0.005, t-test). In 11 of 12 animals, the CNV area was reduced by 16% to 100% following intravitreal injection of AG013711 and the mean (1.8 ± 0.8 mm2, n=12) is 60% smaller (4.5 ± 1.5 mm2, p< 0.05, t-test).
Figure 6. CNV reversal by intravitreal injection of AG013764 and AG013711.
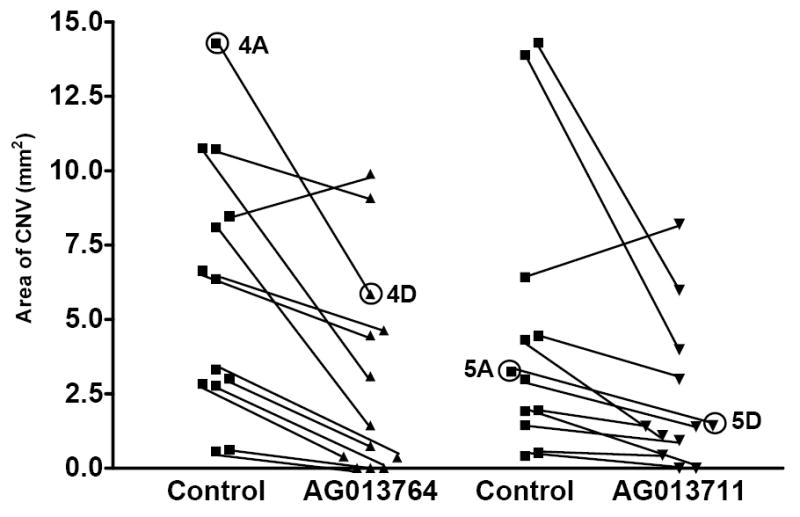
Four groups of animals (n=25) were injected either with AG013764, AG013711 or control carrier solution for two weeks starting six weeks after AAV-VEGF injection. Methods as in Fig. 3D legend. Data from each pair of eyes are connected with a black line. The mean area of CNV in the AG013764 treated eyes (n = 13) was reduced by 57% (p < 0.005). The mean area of CNV in the AG013711 treated eyes (n = 12) was reduced by 60% (p < 0.05). The data points labeled 4A & 5A are from control eyes and 4D & 5D are from treated eyes. These four data points were obtained from the whole mount images shown in Figs 4 & 5.
DISCUSSION
The hypothesis that a specific antagonist of an angiogenesis factor can be used to treat neovascular diseases was first proposed more than 30 years ago (Folkman 1971). In particular, VEGF has been identified as one of the most important mediators of physiologic and pathologic angiogenesis. Many strategies for blocking VEGF signaling in neovascular diseases have been developed, including anti-VEGF antibodies, anti-VEGF aptamer, soluble VEGF receptors or chimeric VEGF receptors, protein kinase C inhibition and VEGF receptor kinase inhibitors (Takeda et al., 2003; Barouch and Miller 2004; Liu and Regillo 2004; Kinose, Roscilli et al. 2005). Several clinical trials (Gragoudas, Adamis et al. 2004; Rosenfeld, Schwartz et al. 2005) have demonstrated therapeutic efficacy and recently Macugen (anti-VEGF aptamer) has been approved for treating wet AMD. Macugen is injected intravitreally every six weeks and there are reports of increased complications associated with repeated injections (eight times per year) for one year (Gragoudas et al. 2004), but the long term effects of repeated intravitreal injections are still unknown. In this study, we have compared the efficacy of intravitreal versus intraperitoneal injections, but other delivery routes are also possible (Okada et al., 2004; Augustin et al., 2005; Kato et al., 2005).
The present experiments demonstrate that AG013764 and AG013711, significantly inhibited choroidal neovascularization when the compounds were delivered via intravitreal injection, six weeks after inducing CNV (Fig. 6). We confirmed the expression of VEGF in these animals at eight weeks after AAV-VEGF injection (Fig. 1). Histological sections (Fig. 2) from the eye injected with AG013764 showed less blood vessel proliferation in the SRS, smaller areas of disorganized photoreceptors, and less RPE proliferation. In FITC-dextran flat mounts, the mean area of CNV from IP-injected animals (Fig. 3D) treated with RTK inhibitors was reduced compared to control animals, but based on a analysis that accounted for possible interactions between paired eyes (Littell et al. 1996), this trend did not reach statistical significance.
VEGF signaling occurs mainly through the KDR receptor while Flt-1 is thought to regulate the amount of free VEGF in the extracellular space (Ferrara 2001). As shown in Table 1, AG013764 and AG013711 are extremely potent inhibitors of both KDR and Flt-1 but have a much higher affinity for KDR. Other growth factors such as platelet derived growth factor have also been implicated in ocular neovascular diseases (Jain 2003). This suggests that inhibitors such as AG013764 and AG013711, which target both VEGFRs and PDGFRs, could potentially induce regression of proliferating blood vessels (Bergers, Song et al. 2003). Table 1 also shows that AG013764 and AG013711 inhibit PDGF receptors α and β at sub-micromolar concentrations. Electron microscopic examination of CNV in AMD patients and laser-induced CNV in monkey revealed that pericytes associated with endothelial cells undergo phenotypic changes with different phases of neovascularization (Archer and Gardiner 1981; Killingsworth 1995). PDGF is required for recruiting pericytes and smooth muscle cells to endothelial cells. Anti-PDGF antibody disrupted endothelial-pericyte association and destabilized the developing retinal vasculature. Inhibition of both VEGF and PDGF signaling pathways is more effective at preventing and regressing CNV in the mouse laser model and neovascularization in other ocular models (Jo et al. 2006).
Previously we showed that CNV is well developed six weeks after AAV-VEGF injection (Wang, Rendahl et al. 2003). AAV-VEGF -induced CNV was significantly reduced in this model by the concomitant addition of anti-angiogenic molecules, sFlt1 and PEDF, also delivered by AAV (Wang et al. 2003; Wang et al., 2004). The present results suggest that AG013764 and AG013711 can inhibit the further development of CNV and potentially could be used to inhibit CNV associated with the “wet” form of AMD thus providing additional possible pathways of therapeutic intervention.
In these experiments, FITC-dextran whole mounts or angiograms of the posterior segment showed more intense subretinal hyper-fluorescence over a larger area in control compared to treated eyes (Figs 3-5). Angiograms from animals receiving intravitreal injections of the compounds could not be obtained because the compounds were deposited on the surface of the retina and blocked the fluorescence signal. In those eyes, the retina was removed and CNV quantified by Neurolucida analysis of whole mount preparations (Figs 4-6). It could be that AG013764 or AG013711 reduced the amount of AAV-VEGF induced CNV, or reduced the permeability of CNV for dextran, or both (Edelman and Castro 2000; Ferris 2004). In laser induced CNV, high molecular weight fluorescein labeled dextran (2×106 molecular weight) is thought to be confined within the neo-vascular blood vessels (Edelman and Castro 2000). However, overexpression of VEGF may make these blood vessels more permeable to dextran, as evidenced by an area of hyper-fluorescence that has well-demarcated boundaries and a uniform intensity (Fig. 3A, control).
The area of hyperfluorescence on the FITC-dextran flat-mount was used as a quantitative measure of CNV. The measured area of the flat mount (50 mm2) is probably smaller than our estimate of the posterior section geometrical area for two reasons: (1) because of possible tissue shrinkage after fixation; and (2) because the tissues are not perfectly flat when mounted on the glass slide. The present results could be explained in part by assuming that the tissues treated with AG013764 or AG013711 had greater shrinkage or were less flat on the glass slide compared to the control tissues. We think that possibility unlikely since the total area of the flat mount, measured from the intravitreal injection group, was unchanged (p=0.58) between control (54.4 ± 1mm2; n = 10) and treated animals (55.3 ± 1.3mm2; n = 10). The observed reduction in CNV is probably an underestimate since it does not reflect changes in vascular volume. We are currently developing a method to measure the volume of CNV using optical coherence tomography (OCT). This will allow us to determine the volume of neovascular complex in the subretinal space and to evaluate the treatment induced changes in CNV in real time.
A noteworthy aspect of the present model is that it allows us to generate relatively large areas of CNV (50-100 times the area of a laser-induced CNV; ˜ 0.03 - 0.08 mm2 (Edelman and Castro 2000; Hangai et al., 2001; Mori et al., 2001; Dejneka et al., 2004) ). In the present study, the intervention started six weeks after the initial injection of AAV-VEGF and at least 1-2 weeks after the development of CNV. The ability of AG013764 or AG013711 to significantly reduce a large area of well developed CNV suggests that these compounds may provide a clinically effective treatment for CNV.
Acknowledgments
It is our pleasure to thank Arvydas Maminishkis for his help on the image analysis, Katherine Rendahl for help in packaging AAV virus, and George Reed for statistical advice.
Grant Support: UC BioSTAR, Pfizer Inc., NEI Intramural Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amorapanth P, LeDoux JE, Nader K. Different lateral amygdala outputs mediate reactions and actions elicited by a fear-arousing stimulus. Nat Neurosci. 2000;3:74–79. doi: 10.1038/71145. [DOI] [PubMed] [Google Scholar]
- Archer DB, Gardiner TA. Electron microscopic features of experimental choroidal neovascularization. Am J Ophthalmol. 1981;91:433–457. doi: 10.1016/0002-9394(81)90230-0. [DOI] [PubMed] [Google Scholar]
- Augustin AJ, D’Amico DJ, Mieler WF, Schneebaum C, Beasley C. Safety of posterior juxtascleral depot administration of the angiostatic cortisene anecortave acetate for treatment of subfoveal choroidal neovascularization in patients with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2005;243:9–12. doi: 10.1007/s00417-004-0961-4. [DOI] [PubMed] [Google Scholar]
- Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology. 2006;113:363–372. e365. doi: 10.1016/j.ophtha.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Baffi J, Byrnes G, Chan CC, Csaky KG. Choroidal neovascularization in the rat induced by adenovirus mediated expression of vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 2000;41:3582–3589. [PubMed] [Google Scholar]
- Barouch FC, Miller JW. Anti-vascular endothelial growth factor strategies for the treatment of choroidal neovascularization from age-related macular degeneration. Int Ophthalmol Clin. 2004;44:23–32. doi: 10.1097/00004397-200404430-00005. [DOI] [PubMed] [Google Scholar]
- Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev. 2004;15:215–228. doi: 10.1016/j.cytogfr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Bird AC. The Bowman lecture. Towards an understanding of age-related macular disease. Eye. 2003;17:457–466. doi: 10.1038/sj.eye.6700562. [DOI] [PubMed] [Google Scholar]
- Brown DM, Shapiro H, Schneider S. Subgroup Analysis of First-Year Results of ANCHOR: A Phase III, Double-Masked, Randomized Comparison of Ranibizumab and Verteporfin Photodynamic Therapy for Predominantly Classic Choroidal Neovascularization Related to Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2006;47:S2963. [Google Scholar]
- Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Dejneka NS, Kuroki AM, Fosnot J, Tang W, Tolentino MJ, Bennett J. Systemic rapamycin inhibits retinal and choroidal neovascularization in mice. Mol Vis. 2004;10:964–972. [PubMed] [Google Scholar]
- Edelman JL, Castro MR. Quantitative image analysis of laser-induced choroidal neovascularization in rat. Exp Eye Res. 2000;71:523–533. doi: 10.1006/exer.2000.0907. [DOI] [PubMed] [Google Scholar]
- Eyetech, Study, Group. Preclinical and phase 1A clinical evaluation of an anti-VEGF pegylated aptamer (EYE001) for the treatment of exudative age-related macular degeneration. Retina. 2002;22:143–152. doi: 10.1097/00006982-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Eyetech, Study, Group. Anti-vascular endothelial growth factor therapy for subfoveal choroidal neovascularization secondary to age-related macular degeneration: phase II study results. Ophthalmology. 2003;110:979–986. doi: 10.1016/S0161-6420(03)00085-X. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001;280:C1358–1366. doi: 10.1152/ajpcell.2001.280.6.C1358. [DOI] [PubMed] [Google Scholar]
- Ferris FL., 3rd A new treatment for ocular neovascularization. N Engl J Med. 2004;351:2863–2865. doi: 10.1056/NEJMe048316. [DOI] [PubMed] [Google Scholar]
- Ferris FL, 3rd, Fine SL, Hyman L. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol. 1984;102:1640–1642. doi: 10.1001/archopht.1984.01040031330019. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- Green ES, Rendahl KG, Zhou S, Ladner M, Coyne M, Srivastava R, Manning WC, Flannery JG. Two animal models of retinal degeneration are rescued by recombinant adeno-associated virus-mediated production of fgf-5 and fgf-18. Mol Ther. 2001;3:507–515. doi: 10.1006/mthe.2001.0289. [DOI] [PubMed] [Google Scholar]
- Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27. [PubMed] [Google Scholar]
- Grossniklaus HE, Green WR. Choroidal neovascularization. Am J Ophthalmol. 2004;137:496–503. doi: 10.1016/j.ajo.2003.09.042. [DOI] [PubMed] [Google Scholar]
- Hangai M, Moon YS, Kitaya N, Chan CK, Wu DY, Peters KG, Ryan SJ, Hinton DR. Systemically expressed soluble Tie2 inhibits intraocular neovascularization. Hum Gene Ther. 2001;12:1311–1321. doi: 10.1089/104303401750270968. [DOI] [PubMed] [Google Scholar]
- Heier JS, Shapiro H, Singh AA. Randomized, Controlled Phase III Study of Ranibizumab (Lucentis) for Minimally Classic or Occult Neovascular Age-Related Macular Degeneration: Two-Year Efficacy Results of the MARINA Study. Invest Ophthalmol Vis Sci. 2006;47:S2959. [Google Scholar]
- Hoyng CB, Vingerling JR, Hooymans JMM, Klamerus KJ, Younis H, Khalil D, Scassellati-Sforzolini B, Russell S, Schlingemann RO. AG-013958 (VEGFR Inhibitor) Achieves Effective Choroidal Concentrations With Minimal Systemic Effects in Cynomolgus Monkeys and Humans With Age-Related Macular Disease. Invest Ophthalmol Vis Sci. 2005;46:2365. [Google Scholar]
- Inai T, Mancuso M, Hashizume H, Baffert F, Haskell A, Baluk P, Hu-Lowe DD, Shalinsky DR, Thurston G, Yancopoulos GD, McDonald DM. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol. 2004;165:35–52. doi: 10.1016/S0002-9440(10)63273-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Jo N, Mailhos C, Ju M, Cheung E, Bradley J, Nishijima K, Robinson GS, Adamis AP, Shima DT. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol. 2006;168:2036–2053. doi: 10.2353/ajpath.2006.050588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Kimura H, Okabe K, Okabe J, Kunou N, Nozaki M, Ogura Y. Suppression of laser-induced choroidal neovascularization by posterior sub-tenon administration of triamcinolone acetonide. Retina. 2005;25:503–509. doi: 10.1097/00006982-200506000-00017. [DOI] [PubMed] [Google Scholar]
- Killingsworth MC. Angiogenesis in early choroidal neovascularization secondary to age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 1995;233:313–323. doi: 10.1007/BF00200479. [DOI] [PubMed] [Google Scholar]
- Kim YR, Yudina A, Figueiredo J, Reichardt W, Hu-Lowe D, Petrovsky A, Kang HW, Torres D, Mahmood U, Weissleder R, Bogdanov AA., Jr Detection of early antiangiogenic effects in human colon adenocarcinoma xenografts: in vivo changes of tumor blood volume in response to experimental VEGFR tyrosine kinase inhibitor. Cancer Res. 2005;65:9253–9260. doi: 10.1158/0008-5472.CAN-03-2619. [DOI] [PubMed] [Google Scholar]
- Kinose F, Roscilli G, Lamartina S, Anderson KD, Bonelli F, Spence SG, Ciliberto G, Vogt TF, Holder DJ, Toniatti C, Thut CJ. Inhibition of retinal and choroidal neovascularization by a novel KDR kinase inhibitor. Mol Vis. 2005;11:366–373. [PubMed] [Google Scholar]
- Krzystolik MG, Afshari MA, Adamis AP, Gaudreault J, Gragoudas ES, Michaud NA, Li W, Connolly E, O’Neill CA, Miller JW. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch Ophthalmol. 2002;120:338–346. doi: 10.1001/archopht.120.3.338. [DOI] [PubMed] [Google Scholar]
- Lau D, McGee LH, Zhou S, Rendahl KG, Manning WC, Escobedo JA, Flannery JG. Retinal degeneration is slowed in transgenic rats by AAV-mediated delivery of FGF-2. Invest Ophthalmol Vis Sci. 2000;41:3622–3633. [PubMed] [Google Scholar]
- Lebherz C, Maguire AM, Auricchio A, Tang W, Aleman TS, Wei Z, Grant R, Cideciyan AV, Jacobson SG, Wilson JM, Bennett J. Nonhuman primate models for diabetic ocular neovascularization using AAV2-mediated overexpression of vascular endothelial growth factor. Diabetes. 2005;54:1141–1149. doi: 10.2337/diabetes.54.4.1141. [DOI] [PubMed] [Google Scholar]
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD. SAS System for Linear Models. Cary, NC: SAS Institute; 1996. [Google Scholar]
- Liu M, Regillo CD. A review of treatments for macular degeneration: a synopsis of currently approved treatments and ongoing clinical trials. Curr Opin Ophthalmol. 2004;15:221–226. doi: 10.1097/01.icu.0000122122.24016.f1. [DOI] [PubMed] [Google Scholar]
- Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age- related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- Macular, Photocoagulation, Study, Group. Macular Photocoagulation Study Group. Laser photocoagulation of subfoveal neovascular lesions of age-related macular degeneration. Updated findings from two clinical trials. Arch Ophthalmol. 1993;111:1200–1209. doi: 10.1001/archopht.1993.01090090052019. [DOI] [PubMed] [Google Scholar]
- Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, Yao VJ, Inai T, Brooks P, Freimark B, Shalinsky DR, Hu-Lowe DD, McDonald DM. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610–2621. doi: 10.1172/JCI24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue MA, Wickersham JA, Pinko C, Showalter RE, Parast CV, Tempczyk-Russell A, Gehring MR, Mroczkowski B, Kan CC, Villafranca JE, Appelt K. Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: a key enzyme in angiogenesis. Structure. 1999;7:319–330. doi: 10.1016/s0969-2126(99)80042-2. [DOI] [PubMed] [Google Scholar]
- Mori K, Ando A, Gehlbach P, Nesbitt D, Takahashi K, Goldsteen D, Penn M, Chen CT, Mori K, Melia M, Phipps S, Moffat D, Brazzell K, Liau G, Dixon KH, Campochiaro PA. Inhibition of choroidal neovascularization by intravenous injection of adenoviral vectors expressing secretable endostatin. Am J Pathol. 2001;159:313–320. doi: 10.1016/S0002-9440(10)61697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motzer RJ, Hoosen S, Bello CL, Christensen JG. Sunitinib malate for the treatment of solid tumours: a review of current clinical data. Expert Opin Investig Drugs. 2006;15:553–561. doi: 10.1517/13543784.15.5.553. [DOI] [PubMed] [Google Scholar]
- Nakahara T, Norberg SM, Shalinsky DR, Hu-Lowe DD, McDonald DM. Effect of inhibition of vascular endothelial growth factor signaling on distribution of extravasated antibodies in tumors. Cancer Res. 2006;66:1434–1445. doi: 10.1158/0008-5472.CAN-05-0923. [DOI] [PubMed] [Google Scholar]
- Niesman MR, Grove CG, Rewolinski DA, Cartwright CA, Bender SL, Zou HY, Heller DC, Misialek S, Parast C, Appelt K. Inhibitors of VEGF receptor-2 induce capillary regression in the retina of rats during vascular development and following exposure to cyclic hyperoxia/hypoxia. Invest Ophthalmol Vis Sci. 2000;41:S142–S142. [Google Scholar]
- Okada AA, Wakabayashi T, Kojima E, Asano Y, Hida T. Trans-Tenon’s retrobulbar triamcinolone infusion for small choroidal neovascularisation. Br J Ophthalmol. 2004;88:1097–1098. doi: 10.1136/bjo.2003.039719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rewolinski DA, Grove CG, Cobbs MN, Goldman JH, Niesman MR. A Potent and Selective Inhibitor of VEGFR and PDGFR Inhibits Ocular Angiogenesis. Invest Ophthalmol Vis Sci. 2005;46:469. [Google Scholar]
- Rewolinski DA, Jacobs SA, Grove CG, Cartwright CA, Scales LS, Goldman JH, Zou HY, Cripps SJ, Kania RS, Niesman MR. Analysis of angiogenic growth factors in rat retina following inhibition of VEGF-receptor 2. Invest Ophthalmol Vis Sci. 2001;42:S92–S92. [Google Scholar]
- Rosenfeld PJ, Schwartz SD, Blumenkranz MS, Miller JW, Haller JA, Reimann JD, Greene WL, Shams N. Maximum Tolerated Dose of a Humanized Anti-Vascular Endothelial Growth Factor Antibody Fragment for Treating Neovascular Age-Related Macular Degeneration. Ophthalmology. 2005 doi: 10.1016/j.ophtha.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Saishin Y, Saishin Y, Takahashi K, Lima e Silva R, Hylton D, Rudge JS, Wiegand SJ, Campochiaro PA. VEGF-TRAP(R1R2) suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 2003;195:241–248. doi: 10.1002/jcp.10246. [DOI] [PubMed] [Google Scholar]
- Schoffski P, Dumez H, Clement P, Hoeben A, Prenen H, Wolter P, Joniau S, Roskams T, Van Poppel H. Emerging role of tyrosine kinase inhibitors in the treatment of advanced renal cell cancer: a review. Ann Oncol. 2006 doi: 10.1093/annonc/mdj133. [DOI] [PubMed] [Google Scholar]
- Spilsbury K, Garrett KL, Shen WY, Constable IJ, Rakoczy PE. Overexpression of vascular endothelial growth factor (VEGF) in the retinal pigment epithelium leads to the development of choroidal neovascularization. Am J Pathol. 2000;157:135–144. doi: 10.1016/S0002-9440(10)64525-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Hata Y, Shiose S, Sassa Y, Honda M, Fujisawa K, Sakamoto T, Ishibashi T. Suppression of experimental choroidal neovascularization utilizing KDR selective receptor tyrosine kinase inhibitor. Graefes Arch Clin Exp Ophthalmol. 2003;241:765–772. doi: 10.1007/s00417-003-0688-7. [DOI] [PubMed] [Google Scholar]
- Tolentino MJ, Brucker AJ, Fosnot J, Ying GS, Wu IH, Malik G, Wan S, Reich SJ. Intravitreal injection of vascular endothelial growth factor small interfering RNA inhibits growth and leakage in a nonhuman primate, laser-induced model of choroidal neovascularization. Retina. 2004;24:132–138. doi: 10.1097/00006982-200402000-00018. [DOI] [PubMed] [Google Scholar]
- Wang F, Rendahl K, Manning WC, Quiroz D, Coyne M, Miller SS. Inhibition of choroidal neovascularization (CNV) by adeno-associated virus (AAV) mediated expression of sFLT1. Invest Ophthalmol Vis Sci. 2002;43:U1307–U1307. doi: 10.1167/iovs.02-0281. [DOI] [PubMed] [Google Scholar]
- Wang F, Rendahl KG, Manning WC, Quiroz D, Coyne M, Miller SS. AAV-mediated expression of vascular endothelial growth factor induces choroidal neovascularization in rat. Invest Ophthalmol Vis Sci. 2003;44:781–790. doi: 10.1167/iovs.02-0281. [DOI] [PubMed] [Google Scholar]
- Wang F, Shi G, Rendahl KG, Manning WC, Jafari A, Liu W, Coyne M, Miller SS. Inhibition of choroidal Neovascularization (CNV) by adeno-associated virus (AAV) mediated expression of PEDF. Invest Ophthalmol Vis Sci. 2003;44:U281–U281. doi: 10.1167/iovs.02-0281. [DOI] [PubMed] [Google Scholar]
- Wang FE, Jafari A, Shi G, Rendahl KG, Manning WC, Coyne M, Miller SS. Changes in mRNA expression and protein level of angiogenic and anti-angiogenic factors in cultured human fetal retinal pigment epithelium (hfRPE) Invest Ophthalmol Vis Sci. 2004;45:U198–U198. [Google Scholar]