

Abstract
Elevated levels of insulin-like growth factor-I (IGF-I) are associated with ovarian carcinogenesis and progression. However, the molecular mechanisms by which IGF-I contributes to ovarian cancer development remain to be elucidated. Cyclooxygenase-2 (COX-2) is a crucial player in the pathogenesis of human malignancies. Herein we showed that IGF-I efficiently induced COX-2 expression and PGE2 biosynthesis at physiologically relevant concentrations in human ovarian cancer cells. IGF-I treatment significantly increased COX-2 transcriptional activation. IGF-I also stabilized COX-2 mRNA through the COX-2 3′-untranslated region (3′-UTR), which appeared independent of the conserved AU-rich elements. We next investigated the signaling pathways involved in IGF-I-induced COX-2 expression. We found that PI3K inhibitor wortmannin or LY294002 blocked COX-2 expression induced by IGF-I. Wortmannin treatment or a dominant negative PI3K mutant significantly inhibited IGF-I-induced COX-2 mRNA stabilization, but only slightly decreased COX-2 transcriptional activation. We showed that ERK1/2 and p38 MAPKs were required for IGF-I-induced COX-2 expression and that activation of both pathways by IGF-I increased COX-2 transcriptional activation and its mRNA stability. IGF-I stimulated PKC activation in the cells and pretreatment with PKC inhibitor bisindolylmaleimide prevented IGF-I-induced COX-2 transcriptional activation and mRNA stabilization, and inhibited COX-2 mRNA and protein expression. Taken together, our data demonstrate that IGF-I induces COX-2 expression in human ovarian cancer cells, which is mediated by three parallel signaling cascades — PI3K, MAPK, and PKC pathways that differentially regulate COX-2 expression at transcriptional and post-transcriptional levels.
Keywords: insulin-like growth factor-I (IGF-I), cyclooxygenase-2 (COX-2), prostaglandins, ovarian cancer
1. Introduction
The type I insulin-like growth factor receptor (IGF-IR) and its ligand IGF-I have been implicated in the development and progression of human cancer (1–3). An accumulating body of epidemiological evidence suggests that high levels of circulating IGF-I are associated with the risk of a number of human malignancies, such as breast, prostate, colorectal, pancreas, lung, and ovarian cancer (3). Insulin-like growth factors (IGF-I and IGF-2) are small polypeptides structurally related to insulin and exert their mitogenic effects mainly through IGF-IR. IGF-2 is considered as the major IGF isoform in fetal development, whereas IGF-I plays more important role in adults (3). The bioactivities of IGFs are regulated by a family of IGF binding proteins (i.e., from IGFBP-1 to IGFBP-6) and a group of IGFBP proteases (2–4). Activation of IGF-IR by ligand binding initiates the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways, which are the major signaling cascades that mediate the mitogenic and antiapoptotic actions of IGFs (1–3). The IGF-IR is frequently overexpressed in several human cancers. The combination of the mitogenic and antiapoptotic effects of IGF-IR activation has a profound impact on tumor growth. In addition, IGF-IR is involved not only in the induction of cell transformation, but also in the maintenance of the transformed phenotype (5). Furthermore, IGF-I has been implicated in tumor neo-vascularization by increasing the expression of hypoxia-inducible factor 1α (HIF-1α) and vascular endothelial growth factor (VEGF) (6–9). Despite these findings, however, the molecular mechanisms by which IGF-I contributes to cancer progression remain to be elucidated.
The conversion of arachidonic acid to prostaglandins (PGs) is catalyzed by two isoforms of cyclooxygenases (COX), COX-1 and COX-2. COX-1 is constitutively expressed in most human tissues, and appears to be responsible for the production of PGs that modulate physiological functions. In contrast, COX-2 is expressed at low or undetectable levels in most normal tissues. COX-2 can be rapidly induced by inflammatory stimuli, and is frequently overexpressed in many human cancers (10–13). Genetic studies showed that transgenic mice overexpressing COX-2 in various types of tissues developed malignant tumors (10,11). Conversely, the development and growth of tumors were markedly retarded in COX-2 knockout mice (10,11). The role of COX-2 in carcinogenesis is further supported by experimental and clinical studies which demonstrate the effectiveness of selective COX-2 inhibitors in the prevention and treatment of human cancers (10,11). The biological effects of COX-2 are mediated by its prostanoid products that affect multiple mechanisms implicated in carcinogenesis. For example, prostaglandin E2 (PGE2) can stimulate cell proliferation and motility while inhibiting apoptosis and immune surveillance (11). Importantly, COX-2-derived prostanoids are involved in multiple key points of tumor angiogenesis, including enhancing VEGF production (12).
Expression of COX-2 in human cancer is regulated at both transcriptional and posttranscriptional levels by multiple signaling pathways (14). COX-2 expression can be induced by inflammatory cytokines and by growth factors (15). COX-2 expression can also be induced by activation of oncogenes and inactivation of tumor suppressor genes (15). However, little is known about the precise factors and molecular mechanisms regulating COX-2 gene expression in human ovarian cancer. The novel findings of this study are: (a) IGF-I potently upregulates COX-2, but not COX-1 expression in human ovarian cancer cells; (b) IGF-I activates PI3K, MAPK as well as PKC signaling cascades in the cells, and all these pathways are required for IGF-I-induced COX-2 expression; (c) PI3K, MAPK and PKC pathways are differentially involved in transcriptional and post-transcriptional regulation of IGF-I-induced COX-2 expression in the cells; (d) PKC is independent of Erk1/2 MAPK in the mediation of IGF-I-induced COX-2 expression. Our data suggest that increased COX-2 expression may be important for IGF-I involvement in ovarian cancer development and progression.
2. Materials and Methods
2.1. Cell Culture and Reagents
A2780 and OVCAR-3 human ovarian cancer cells were cultured in RPMI Medium 1640 (Life Technologies, Grand Island, NY), supplemented with 10% fetal bovine serum, 50 nM insulin (Sigma), 100 units/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained at 37°C and 5% CO2 in a humid environment. Actinomycin D, PD98059, bisindolylmaleimide, and recombinant human IGF-I were purchased from Sigma (St louis, MO). LY294002, wortmannin and SB203580 were purchased from Calbiochem (La Jolla, CA). Monoclonal antibodies against COX-2 and COX-1, and selective COX-2 inhibitor NS398 were obtained from Cayman Chemical Company (Ann Arbor, MI). Antibodies against phosphorylated AKT (Ser473) or total AKT, phosphorylated ERK1/2 (Thr202/Tyr204), and phosphorylated MAPKAPK2 (Thr334) were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against total p44/p42 MAPK were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibody against β-actin was from Sigma.
2.2. Immunoblotting
After appropriate treatments, cells were washed with ice-cold PBS, and collected by centrifugation. Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors (100 mM Tris, PH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1% deoxycholate acid, 0.1% SDS, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 2 mM DTT, 20 μg/ml leupeptin, 20 μg/ml pepstatin). Aliquots of protein lysates (50 μg) were fractionated by SDS-PAGE, transferred to a nitrocellulose membrane (Schleicher & Schuell Biosciences, Keene, NH), and subjected to immunoblotting according to the manufacturer’s instructions. Immunoreactivity was visualized with chemiluminescence detection reagent (Pierce Biotechnology, Rockford, IL).
2.3. RT-PCR
Total cellular RNAs were prepared using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Aliquots of total RNAs (1 μg) were used as templates to synthesize the first strand cDNA using reverse transcriptase (Promega). The following primers were used for PCR amplification: human COX-2: sense 5′-TTCAAATG AGATTGTGGGAAAATTGCT-3′, antisense 5′-AGATCATCTCTGCCTGAGTATCTT-3′; GAPDH: sense 5′-ACCACAGTCCATGCCATCAC-3′, antisense 5′-TCCACCACCCTGTTGCTGTA-3′. PCR was performed for 30 cycles, with each cycle at 95°C for 1 min, 56°C for 1 min, and 72°C for 1 min. PCR products were separated on a 2% agarose gel and visualized with ethidium bromide and photographed using an EagleEye II system (Stratagene, La Jolla, CA).
2.4. PGE2 ELISA assay
Cells were seeded at 2× 105 cells/well in 12-well plates, and cultured to 80% confluence in normal growth medium. The cells were cultured in serum-free medium overnight, then changed to fresh basal medium containing various concentrations of IGF-I in the presence or absence of 25 μM NS398 for 24 h. Levels of PGE2 released by the cells were measured using an ELISA kit (R&D systems, Minneapolis, MN) according to the manufacturer’s instructions.
2.5. Transient transfection and luciferase reporter assay
A reporter construct containing the 5′-flanking region of the human COX-2 gene (phPES2-1432/+59) was used to study transcriptional activation of the COX-2 promoter (16). The COX-2 3′-UTR reporter luciferase constructs, Luc-3′UTR containing the full-length human COX-2 3′-UTR region; Luc-3′UTRΔARE containing the 3′-UTR region with the deletion of a conserved 116-nucleotide AU-rich sequence element as described previously (17). Cells were seeded at a density of 2 × 105 cells/well in 12-well plates. The next day, 1 μg of plasmid DNA was transfected into cells using 2 μl of Lipofectamine (Invitrogen) as per manufacturer’s instructions. After the transfection, the cells were cultured overnight, changed to fresh basal medium with different treatments for 16 h. Luciferase and β–galactosidase activities were assayed in the cell lysates, and relative luciferase activity was the ratio of luciferase/β–galactosidase activity which was normalized to the control.
2.6. PKC Kinase Activity Assay
The activity of PKC was measured as described previously with modifications (18). Briefly, cells were lysed in a PKC extraction buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Tween 20, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol) supplemented with protease inhibitors (10 μg/ml of aprotinin, 10 μg/ml of leupeptin, 0.1 mM phenylmethylsulfonyl fluoride) and phosphatase inhibitors (1 mM NaF, 0.1 mM Na3VO4). PKC proteins were immunoprecipitated from 200 μg cellular of extracts with 2 μg of the anti-phospho-PKC (pan) antibodies (beta II, Ser660) (Cell Signaling Technology, Beverly, MA). The kinase assay was performed in 40 μl of PKC reaction buffer (20 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 2.5 mM EGTA, 1 mM NaF, 0.1 mM Na3VO4, 10 μM ATP) containing 1 μg of the substrate myelin basic protein and 5 μCi of [γ-32P]ATP. The reactions were performed at 30°C for 20 min, and terminated by adding SDS-PAGE sample buffer. Reaction products were analyzed by SDS-PAGE and visualized by autoradiography. The radioactive bands were excised and quantified by liquid scintillation counting. The activity of PKC is expressed as counts per min with each μg of protein.
2.7. Statistics
Comparisons between treatments were made by the Student’s t test. A difference between the treatments was considered significant at p < 0.05.
3. Results
3.1. IGF-I stimulates COX-2 mRNA and protein expression, and induces PGE2 biosynthesis in human ovarian cancer cells
To examine whether IGF-I was able to induce COX-2 expression, human ovarian cancer cells A2780 were incubated with various concentrations of IGF-I, and COX-2 mRNA levels were analyzed by RT-PCR. At the range of physiologically relevant concentrations, IGF-I induced COX-2 mRNA expression in a dose-dependent manner (Fig. 1A). Similar results were obtained using another human ovarian cancer cell line, OVCAR-3 (Fig. 1A). We next examined the time-dependent effect of IGF-I on COX-2 expression in the cells. A2780 and OVCAR-3 cells were incubated with IGF-I for 0, 0.5, 1, 2, 4, 6, 12, or 24 h; and COX-2 mRNA expression was examined. IGF-I increased COX-2 expression within 1 h, and the induction reached the maximum at 6 h (Fig. 1B). COX-2 protein levels were examined to determine whether the elevated COX-2 mRNA levels correlated with increased COX-2 protein expression. As shown in Fig. 2A, IGF-I greatly upregulated COX-2 protein expression in a dose-dependent fashion in both cell lines. COX-2 protein expression was gradually increased and peaked by 6–8 h, and remained elevated for up to 24 h (Fig. 2B). By contrast, both cell lines express constitutive levels of COX-1 that were not increased by IGF-I treatment (Figs. 2A and 2B). We further examined whether IGF-I-induced COX-2 protein expression was associated with induction of PGE2 biosynthesis. As shown in Fig. 2C, treatment of the cells with IGF-I increased PGE2 biosynthesis, and pretreatment of the cells with the COX-2 selective inhibitor NS398 completely inhibited the induction. These data show that COX-2 expression and activity are upregulated by IGF-I in human ovarian cancer cells.
Fig. 1. IGF-I upregulates COX-2 mRNA expression.
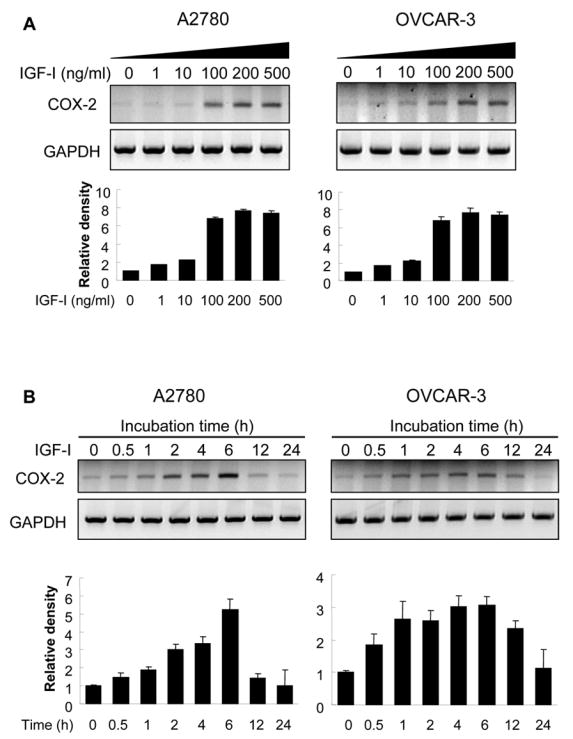
(A) A2780 and OVCAR-3 cells were cultured in serum-free medium for 16 h, then treated with the indicated concentrations of IGF-I for 6 h. COX-2 and GAPDH mRNA levels in the cells were detected by RT-PCR. (B) The cells were treated with 200 ng/ml of IGF-I for 0 to 24 h as indicated. COX-2 and GAPDH mRNA expression was analyzed by RT-PCR. The levels of COX-2 mRNA were quantified using NIH Image J software, and normalized to those of GAPDH levels. The mean ± SD of the corresponding densitometry data from replicate experiments were showed in lower panels of each figure.
Fig. 2. IGF-I induces COX-2 protein expression and PGE2 biosynthesis.
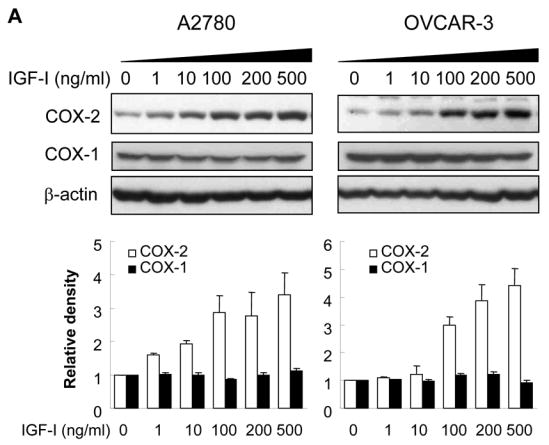
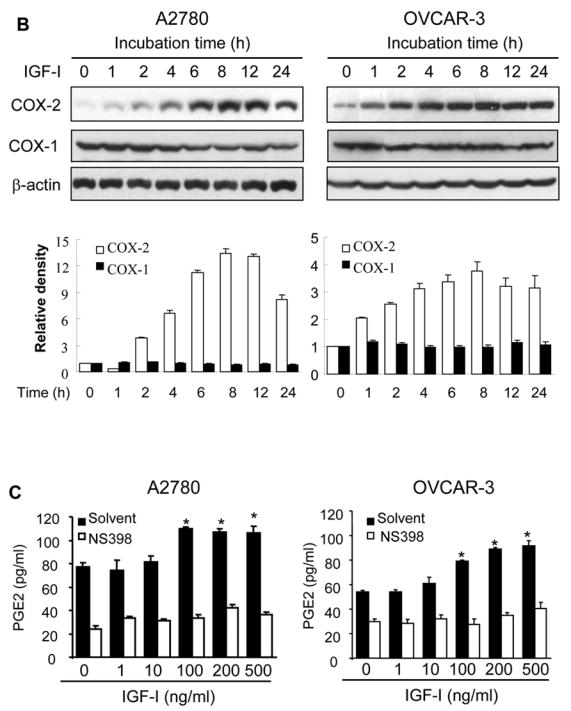
(A) The serum-starved cells were treated with the indicated concentrations of IGF-I for 8 h. Total cellular lysates were subjected to immunoblotting using antibodies against COX-2, COX-1, or β-actin. (B) The serum-starved cells were treated with 200 ng/ml IGF-I for 0 to 24 h as indicated. COX-2, COX-1, and β-actin protein levels were analyzed by immunoblotting. The COX-2 and COX-1 protein signals were quantified and normalized to those of β-actin levels. The mean ± SD of the densitometry data from triplicate experiments were showed in the lower panels of Fig. 2A and 2B. (C) The cells were incubated in basal medium containing the indicated concentrations of IGF-I in the presence or absence of 25 μM NS398 for 24 h. PGE2 levels in the medium were determined by ELISA assay. * indicates that the value is significantly different when compared to that of the control (p<0.05).
3.2. IGF-I increases COX-2 transcriptional activation and its mRNA stability
To study the mechanism underlying the induction of COX-2 by IGF-I, A2780 cells were transfected with a COX-2 promoter luciferase reporter phPHES2(−1432/+59), which carries the full-length promoter region of human COX-2 gene (16). Treatment of the cells with IGF-I significantly increased COX-2 promoter activity (Fig. 3A), suggesting that IGF-I was able to stimulate COX-2 transcriptional activation.
Fig. 3. IGF-I treatment induces COX-2 transcriptional activation and mRNA stability.

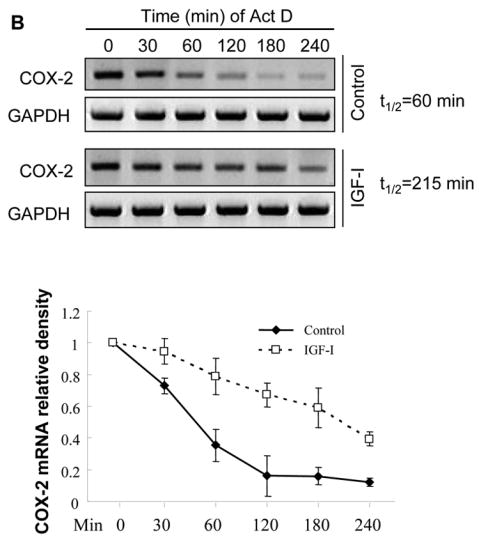

(A) A2780 cells were co-transfected with a COX-2 promoter reporter construct phPHES2(−1432/+59) and pCMV-β-gal, and incubated overnight. The cells were treated with the indicated concentrations of IGF-I for 16 h, and relative luciferase activity was obtained by the ratio of luciferase to β-gal activity, and normalized to that of the control. (B) The cells were treated with solvent (control) or 200 ng/ml IGF-I for 1 h, then treated with 5 μg/ml actinomycin D (Act D) to stop the transcription. The cells were harvested at the indicated time after the addition of Act D, and the levels of COX-2 mRNA were detected by RT-PCR. The mean ± SD of densitometry COX-2 mRNA signals from triplicate experiments were shown in the lower panel. The half-life (t1/2) of COX-2 mRNA was calculated using the regression program of Microsoft Excel 2000. (C) The cells were transfected with a luciferase reporter construct containing full-length COX-2 3′-UTR region (Luc-3′UTR). Relative luciferase activity was analyzed in the cells as described above after the cells were treated for 16 h with the indicated concentrations of IGF-I. (D) The cells were transfected with Luc-3′UTR or Luc-3′UTRΔARE (the deletion of AU-rich sequence element from 3′-UTR region). Luciferase activity was assayed after the treatment of the cells with 200 ng/ml IGF-I for 16 h. The data were presented as mean ±SD. All transient transfection assays were performed in triplicate and repeated at least three times. *indicates that the value is significantly different when compared to that of the control (p<0.05).
Post-transcriptional stabilization of COX-2 mRNA may play an important role in the regulation of COX-2 expression. To determine whether IGF-I affects the stability of COX-2 mRNA, the rate of COX-2 mRNA decay was examined in A2780 cells. In the absence of IGF-I treatment, the half-life (t1/2) of COX-2 mRNA in the cells was 60 min. IGF-I treatment increased the stability of COX-2 mRNA with the t1/2 of 215 min (Fig. 3B). It is known that the 3′-untranslated region (3′-UTR) of COX-2 mRNA is involved in promoting its mRNA decay (17). We therefore asked whether IGF-I treatment affected COX-2 3′-UTR activity. We found that treatment of cells with IGF-I caused up to 2-fold increase of COX-2 3′-UTR reporter activity, suggesting that IGF-I increased COX-2 mRNA stability (Fig. 3C). Within the proximal end of COX-2 3′-UTR, there is a highly conserved AU-rich mRNA decay sequence element (ARE) (17). To determine whether the stabilization of COX-2 mRNA by IGF-I was dependent on the conserved ARE sequence, cells were transfected with the luciferase reporter constructs Luc-3′UTR and Luc-3′UTR ARE, respectively, as was described (17). We found that the luciferase activity was dramatically increased when the ARE region was deleted from the full-length COX-2 3′-UTR (Fig. 3D). This result suggests that in human ovarian cancer cells, the COX-2 ARE may facilitate COX-2 mRNA decay. Interestingly, IGF-I treatment similarly increased luciferase reporter activity when the ARE was deleted (Fig. 3D), implying that IGF-I promotes COX-2 mRNA stabilization through other regions of the 3′UTR. Taken together, these results show that IGF-I upregulates COX-2 expression through both transcriptional activation and post-transcriptional mechanism.
3.3. PI3K signaling pathway is involved in IGF-I-induced COX-2 expression
We next investigated the signaling pathways that are required for IGF-I-induced COX-2 expression. PI3K signaling is a major pathway initiated by growth factor receptors including IGF-IR. To determine whether PI3K regulates IGF-I-induced COX-2 expression, the cells were treated with PI3K inhibitors. As shown in Figs. 4A and 4B, PI3K inhibitors wortmannin and LY294002 inhibited IGF-I-induced COX-2 protein and mRNA expression, indicating that PI3K activation plays an important role in IGF-I-mediated COX-2 expression. We next examined whether PI3K is involved in IGF-I-induced COX-2 transcriptional activation or post-transcriptional mRNA stability. As shown in Fig. 4C, wortmannin treatment slightly reduced COX-2 promoter activity (up to 15% reduction) in IGF-I-treated cells. In contrast, wortmannin completely inhibited IGF-I-induced increase of the COX-2 3′-UTR activity (Fig. 4D). Similar results were observed by using a dominant-negative PI3K construct (Figs. 4E and 4F). These data indicate that PI3K activation regulates IGF-I-induced COX-2 expression in human ovarian cancer cells primarily through increasing stabilization of COX-2 mRNA.
Fig. 4. PI3K signaling is required for IGF-I-induced COX-2 expression.
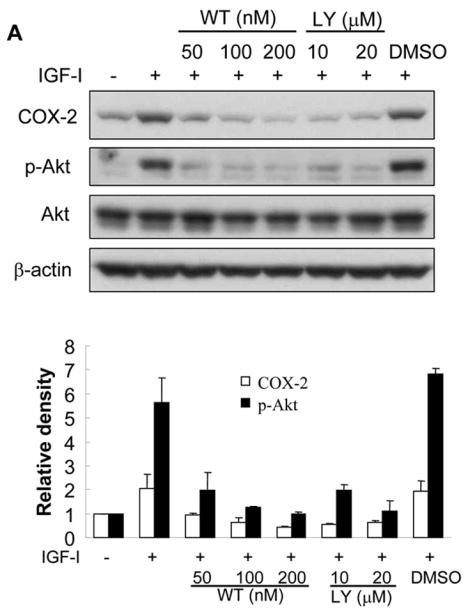
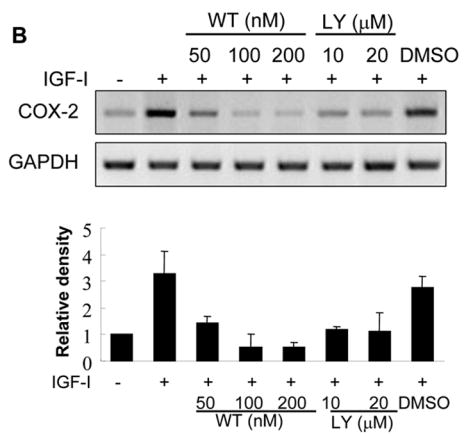

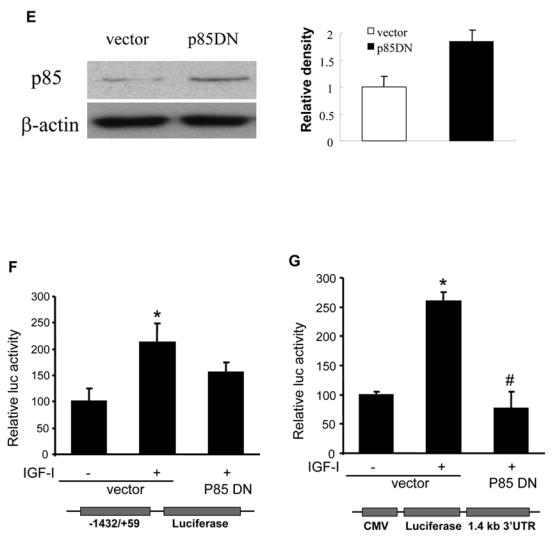
(A) Serum-starved A2780 cells were pretreated with wortmannin (WT), LY294002 (LY) or the solvent DMSO followed by the incubation with 200 ng/ml IGF-I for 8 h. COX-2, phospho-Akt (p-Akt, Ser473), total Akt, and β–actin protein levels were analyzed by immunoblotting. The COX-2 protein signals were normalized to those of β-actin levels, and p-Akt signals were normalized to those of total Akt levels. The mean ± SD of the corresponding densitometry data from duplicate experiments were shown in the lower panel. (B) The cells were pretreated with wortmannin (WT), LY294002 (LY) or solvent (DMSO) followed by incubation with 200 ng/ml IGF-I for 6 h. COX-2 and GAPDH mRNA levels were detected by RT-PCR. The COX-2 mRNA signals were normalized to those of GAPDH levels with the mean ± SD of the corresponding densitometry data from duplicate experiments shown in the lower panel. (C and D) The cells were transfected with phPHES2(−1432/+59) and Luc-3′UTR, respectively; and cultured overnight. The cells were switched to serum-free medium in the absence or presence of wortmannin or 200 ng/ml IGF-I for 16 h. Relative luciferase activity was the ratio of luciferase/β-gal activity, and normalized to that of the control cells. Data are presented as mean ± SD from three independent experiments. * indicates that the value is significantly different when compared to that of the control (p<0.05); # indicates that the value is significantly different when compared to that of IGF-I treatment alone (p<0.05). (E) A2780 cells were transfected with empty vector or a vector expressing dominant-negative PI3K construct (p85DN). After 24 h, the cells were collected and lysed in RIPA buffer. Total proteins were analyzed by immunoblotting using antibodies against p85 and β-actin. The right panel shows the relative densitometry data of p85 protein signals normalized to those of β-actin levels. (F) A2780 cells were co-transfected with phPHES2(−1432/+59), pCMV-βgal, empty vector, or a vector expressing dominant-negative PI3K construct (p85DN); and cultured overnight. The cells were switched to serum-free medium, and incubated in the absence or presence of 200 ng/ml IGF-I for16 h. (G) The cells were co-transfected with Luc-3′UTR, pCMV-βgal, empty vector, or a vector expressing p85DN. The cells were switched to serum-free medium and incubated in the absence or presence of 200 ng/ml IGF-I for 16 h. Relative luciferase activity was analyzed as described above. *indicates that the value is significantly different when compared to that of the control (p<0.05); # indicates that the value is significantly different when compared to that of the vector-transfected cells treated with IGF-I alone (p<0.05).
3.4. ERK1/2 and p38 MAP kinases are important for COX-2 induction by IGF-I at both transcriptional and post-transcriptional levels
IGF-I treatment induced the activation of Erk1/2 and p38 MAPKs in human ovarian cancer cells, and treatment with MEK1 inhibitor PD98059 or p38 inhibitor SB203580 inhibited IGF-I-induced COX-2 expression, which correlated with the inhibition of Erk1/2 and p38 kinase activation (Figs. 5A and 5B). Consistent with their effects on COX-2 protein expression, the MAPK inhibitors completely prevented IGF-I-induced COX-2 mRNA expression (Fig. 5C). To test whether ERK1/2 and p38 regulate COX-2 transcriptional activation, COX-2 promoter reporter activity was analyzed in the cells treated by PD98509 and SB203580. As shown in Figs. 5D and 5E, PD98509 and SB203580 treatment inhibited IGF-I-induced COX-2 promoter reporter activity in a dose-dependent manner, indicating that ERK1/2 and p38 activities are required for COX-2 transcriptional activation. PD98509 and SB203580 similarly inhibited the COX-2 3′-UTR reporter activity, suggesting that ERK1/2 and p38 also regulate IGF-I-induced COX-2 expression by increasing COX-2 mRNA stabilization.
Fig. 5. IGF-I-induced COX-2 expression is dependent on MAPK activation.
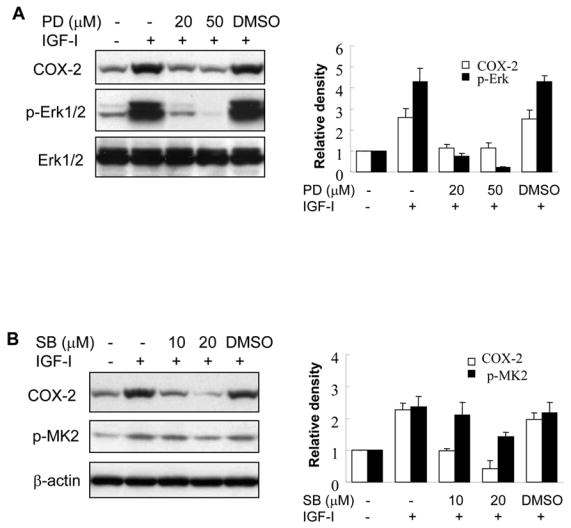
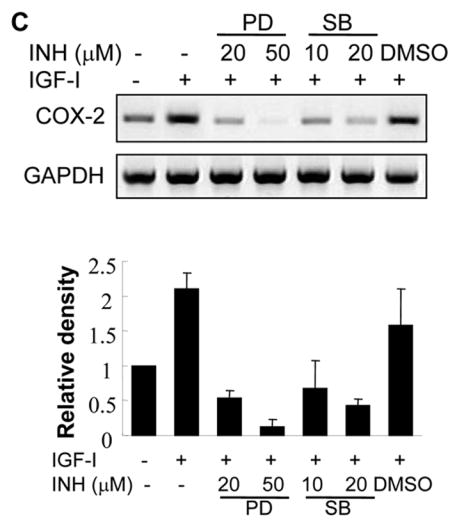

(A and B) A2780 cells were pretreated with the solvent (-), PD98059 (PD) or SB203580 (SB) for 1 h, then treated with 200 ng/ml IGF-I for 8 h. Specific protein levels were analyzed by immunoblotting using antibodies against COX-2, phospho-Erk1/2 (p-Erk1/2, Thr202/Tyr204), total Erk1/2, phospho-MK2 (p-MK2, Thr33), and β–actin, respectively. The right panels indicated the mean ± SD of the corresponding densitometry data from duplicate experiments. (C) The cells were pretreated with PD, SB, or DMSO for 1 h, followed by incubation with 200 ng/ml IGF-I for 6 h. COX-2 and GAPDH mRNA levels were analyzed by RT-PCR. The lower panel indicated the mean ± SD of the corresponding densitometry data from duplicate experiments. (D and E) The cells were transfected with phPHES2(−1432/+59) and Luc-3′UTR plasmids, respectively; and cultured overnight. The cells were switched to serum-free mdium, and incubated in the absence or presence of DMSO, PD, or SB with 200 ng/ml IGF-I as indicated for 16 h. Relative luciferase activity was analyzed as described in Fig. 3. Data are mean ± SD from five independent experiments. * indicates that the value is significantly different when compared to that of the control (p<0.05); # indicates that the value is significantly different when compared to that of IGF-I treatment alone (p<0.05).
3.5. PKC activation is required for IGF-I-mediated COX-2 expression
To determine whether IGF-I stimulates PKC activation, PKC kinase activity was analyzed in human ovarian cancer cells. We found that IGF-I treatment significantly increased PKC activity which was inhibited by PKC inhibitor bisindolylmaleimide (Fig. 6A). We next determined whether IGF-I-induced PKC activation contributed to COX-2 induction. Treatment of the cells with bisindolylmaleimide inhibited COX-2 protein and mRNA expression induced by IGF-I (Figs. 6B and 6C). Blockade of PKC activity also significantly inhibited IGF-I-induced COX-2 promoter and 3′-UTR reporter activities (Figs. 6D and 6E), indicating that PKC activation was important for IGF-I-mediated COX-2 transcriptional activation and mRNA stabilization. PKC was shown to regulate IL-1β-induced COX-2 expression through ERK1/2 activation in astrocytes (19). However, we found that pretreatment with the PKC inhibitor did not alter IGF-I-induced ERK1/2 activation in the cells (Fig. 6F), suggesting that regulation of IGF-I-induced COX-2 expression by PKC is independent of ERK1/2 activation in human ovarian cancer cells.
Fig. 6. PKC activity is important for IGF-I-induced COX-2 expression.
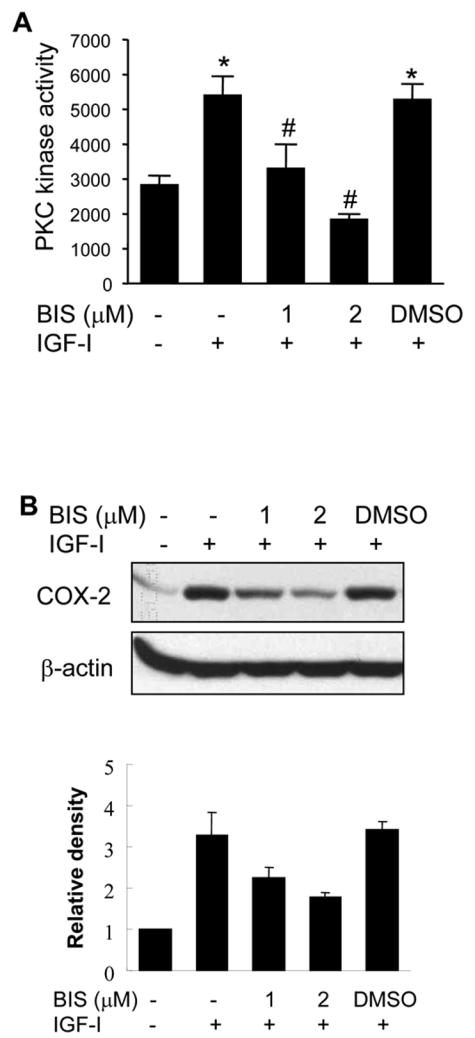
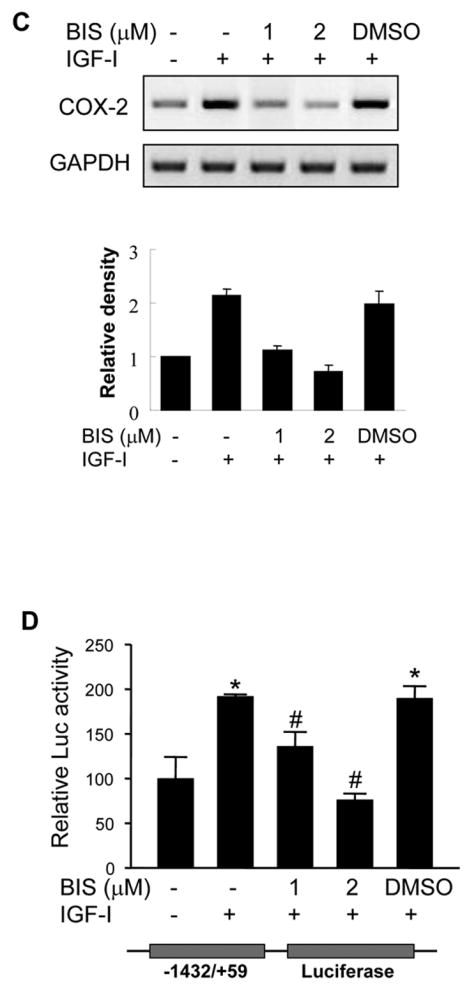
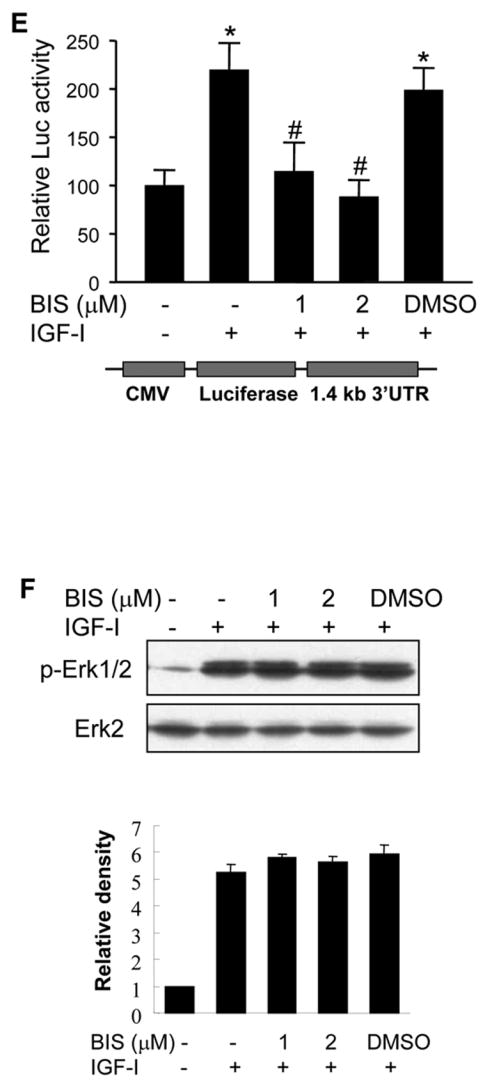
(A) Serum-starved A2780 cells were pretreated with PKC inhibitor bisindolylmaleimide (BIS) for 30 min, followed by incubation with 200 ng/ml IGF-I for 6 h. Total PKC activity was measured using myelin basic protein as a substrate as described in Experimental Procedures. (B) The serum-starved cells were pretreated with BIS and treated with 200 ng/ml IGF-I for 6 h. COX-2 and β–actin protein levels were analyzed by immunoblotting. The lower panel indicated the mean ± SD of the relative densitometry data of COX-2 protein levels from duplicate experiments. (C) The cells were treated as above. COX-2 and GAPDH mRNA levels in the cells were analyzed by RT-PCR. The lower panel indicated the mean ± SD of the relative densitometry data of COX-2 mRNA levels from duplicate experiments. (D and E) The cells were transfected with phPHES2 (−1432/+59) and Luc-3′UTR reporter constructs, respectively. The cells were cultured overnight after the transfection, then switched to serum-free medium in the absence or presence of BIS and 200 ng/ml IGF-I for 16 h. Relative luciferase activity was analyzed in the cells. Data represent mean ± SD from five independent experiments. *indicates that the value is significantly different when compared to that of the control (p<0.05); #indicates that the value is significantly different when compared to that of IGF-I treatment alone (p<0.05). (F) The serum-starved cells were pretreated with BIS, followed by incubation with 200 ng/ml IGF-I for 4 h. Specific protein levels were analyzed by immunoblotting using antibodies against phospho-Erk1/2 (p-Erk1/2, Thr202/Tyr204), and total Erk1/2, respectively. The lower panel indicated the mean ± SD of the relative p-Erk1/2 levels from duplicate experiments.
4. Discussion
The exact roles of IGF-I in regulating COX-2 expression are not well understood. The findings in this study demonstrated that IGF-I induced COX-2 expression and prostaglandin synthesis in human ovarian cancer cells. A recent study showed that enhanced autocrine expression of IGF-II up-regulated COX-2 mRNA expression in Caco-2 human colon cancer cells (20). In human keratinocytes, COX-2 mRNA and protein expression was stimulated by exogenous IGF-II (21). It was recently reported that local IGF-I expression in colorectal cancer tissue was associated with COX-2 mRNA levels (22). IL-1β–induced COX-2 expression was enhanced by IGF-I treatment in rat renal mesangial cells (23). However, other recent studies showed that IGF-I failed to induce COX-2 expression in HT-29 colon cancer cells, hepatic stellate cells, and human vein vascular endothelial cells (13,24,25), and IGF-I treatment even decreased COX-2 expression in osteoblasts (26). Thus, the effects of IGF-I on COX-2 expression appear to be cell-type specific, which may reflect its ability to promote organ-specific tumorigenesis (2–4).
Recent studies showed that COX-2 was overexpressed in ovarian cancer, and that COX-2 overexpression correlated with a more aggressive phenotype and a worse clinical outcome (27,28). In addition, some studies showed that COX-1 was overexpressed in ovarian cancer (29,30). The discrepancy of COX isoform expression in ovarian cancer may be due to the differences in patient population and detection methodology. In this study, we found that both COX-1 and COX-2 are expressed in A2780 and OVCAR-3 ovarian cancer cells, and that basal levels of COX-1 are relatively high in the cells. IGF-1 treatment dramatically increases COX-2 expression and PGE2 levels, but not COX-1 expression in the cells. Recent studies show that high levels of COX-2 and PGE2 expression are associated with cancer progression and angiogenesis in other human cancers such as colon, gastric, and breast cancers (12,15,31,32). Since IGF-I and its receptor IGF-IR are frequently overexpressed in human ovarian cancer (2–4), the induction of COX-2 expression by IGF-I may play an important role in the angiogenesis switch during ovarian cancer development and progression. In addition, PEG2 is a signaling molecule which may be involved in tumor development and angiogenesis through autocrine effect by binding to its cognate E-prostanoid receptors or paracrine effect by acting on the neighboring cells through the induction of growth factors including VEGF and bFGF (33,34). Thus, IGF-I-induced expression of COX-2 and PGE2 may have profound impacts on ovarian cancer development, progression, and angiogenesis.
Overexpression of COX-2 in human cancer is due to deregulation at transcriptional and post-transcriptional levels (10,11,14). Here we have shown that IGF-I was able to stimulate both COX-2 transcriptional activation and mRNA stability, which correlated with elevated COX-2 mRNA and protein expression. Increasing evidence indicates that post-transcriptional regulation via the stabilization of COX-2 mRNA plays an important role in the COX-2 expression (11,14). We found that the conserved ARE sequence in COX-2 3′-UTR attenuates COX-2 expression in human ovarian cancer cells. The data are consistent with several recent studies showing that the 3′-UTR of COX-2 mRNA and its ARE sequence are important for regulating COX-2 mRNA stability (17,35,36). Interestingly, the stabilization of COX-2 mRNA induced by IGF-I was independent of the ARE region in the COX-2 3′-UTR. Recently other cis-acting regions were identified within the COX-2 3′-UTR that are important for the post-transcriptional regulation of COX-2 (37,38). It would be of interest to identify these mRNA regions that are regulated by IGF-I to induce COX-2 expression in human ovarian cancer cells in the future study.
We found that treatment of the cells with PI3K inhibitors blocked both COX-2 protein and mRNA expression induced by IGF-I, indicating that PI3K signaling plays an important role in IGF-I-induced COX-2 expression. Our results are in agreement with several recent reports that PI3K positively regulates COX-2 expression induced by other factors. PI3K/Akt activation was shown to be required for K-Ras-induced COX-2 expression in rat intestinal epithelial cells (39,40). PI3K activity also mediates COX-2 expression in response to LPS, interferon-γ, UVB treatment, and high glucose stimulation (41–44). Our data indicate that PI3K regulates IGF-I-induced COX-2 expression predominantly by modulating the stability of COX-2 mRNA. In addition, we recently showed that IGF-I induced PI3K-dependent phosphorylation of several key components of protein translational apparatus, including p70S6K1, S6 ribosomal protein, 4E-BP1, and eIF4E in human ovarian cancer cells (6). Thus, it is possible that PI3K/Akt pathway may also regulate COX-2 protein translation in response to IGF-I treatment.
Many factors known to induce COX-2 expression also activate MAPK signaling pathway. Our findings indicate that ERK1/2 and p38 MAPKs are involved in the regulation of IGF-I-induced COX-2 expression in human ovarian cancer cells. We found that inhibition of MEK-1/ERK1/2 by PD98509 completely blocked IGF-I-induced COX-2 protein and mRNA expression, and that ERK1/2 activation was involved in both transcriptional and post-transcriptional regulation of COX-2 expression in response to IGF-I treatment. Similarly, IGF-I-induced activation of the p38 MAPK pathway was shown to regulate COX-2 expression on these levels. We found that the p38 selective inhibitor SB203580 suppressed IGF-I-induced activation of COX-2 promoter activity and mRNA stabilization. This is consistent with previous findings showing that p38 MAPK was involved in COX-2 transcription presumably through increasing AP-1 activity (41,45–47), and that p38 MAPK regulated COX-2 mRNA stabilization by its downstream kinase MK-2 (9,36,48).
PKC activity regulated COX-2 expression induced by inflammatory stimuli and stress signals (16,19,47,49,50). Activation of PKC promotes COX-2 gene transcription through the transcription factors NF-κB and C/EBP (16,50). PKC also regulates the stability of COX-2 mRNA (46). In this study, we showed that IGF-I was able to stimulate PKC activation in human ovarian cancer cells. We further demonstrated that PKC was involved in both transcriptional and post-transcriptional regulation of COX-2 expression. It was observed that MAPK could be a downstream signal of PKC for regulating COX-2 expression (19,47). However, we found that in ovarian cancer cells, blockade of PKC activity did not inhibit IGF-I-induced ERK1/2 activation, suggesting that induction of COX-2 expression by PKC is independent of MAPK in human ovarian cancer cells.
In summary, the present study provides first evidence that IGF-I upregulates COX-2 expression and the associated PGE2 biosynthesis in human ovarian cancer cells, and this effect is mediated by PI3K, MAPK, and PKC signaling cascades which are differentially involved in the transcriptional and posttranscriptional regulation for COX-2 expression (Fig. 7). Emerging evidence indicates that COX-2 is important in multiple steps of malignant transformation and tumor progression (10,11). Our data suggest that increased COX-2 expression may be important for ovarian cancer development, progression, and angiogenesis in the cancers associated with high levels of IGF-I expression.
Fig. 7. Schematic representation of the mechanism by which IGF-I induces COX-2 expression in human ovarian cancer cells.

Acknowledgments
We are grateful to Dr. Hiroyasu Inoue (Nara Women’s University, Japan) for providing phPHES2(−1432/+59) plasmid.
Footnotes
This work was supported by National Cancer Institute Grants CA109460 and CA123675 (B.H. Jiang), CA75911 and CA099925 (B. Chandran); and by American Cancer Society Research Scholar Grant 04-076-01-TBE (B.H. Jiang).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107:873–877. doi: 10.1002/ijc.11487. [DOI] [PubMed] [Google Scholar]
- 2.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 3.Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst. 2000;92:1472–1489. doi: 10.1093/jnci/92.18.1472. [DOI] [PubMed] [Google Scholar]
- 4.Kalli KR, Conover CA. The insulin-like growth factor/insulin system in epithelial ovarian cancer. Front Biosci. 2003;8:d714–d722. doi: 10.2741/1034. [DOI] [PubMed] [Google Scholar]
- 5.LeRoith D, Baserga R, Helman L, Roberts CT., Jr Insulin-like growth factors and cancer. Ann Intern Med. 1995;122:54–59. doi: 10.7326/0003-4819-122-1-199501010-00009. [DOI] [PubMed] [Google Scholar]
- 6.Cao Z, Fang J, Xia C, Shi X, Jiang BH. trans-3,4,5′-Trihydroxystibene inhibits hypoxia-inducible factor 1alpha and vascular endothelial growth factor expression in human ovarian cancer cells. Clin Cancer Res. 2004;10:5253–5263. doi: 10.1158/1078-0432.CCR-03-0588. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 8.Reinmuth N, Liu W, Fan F, Jung YD, Ahmad SA, Stoeltzing O, Bucana CD, Radinsky R, Ellis LM. Blockade of insulin-like growth factor I receptor function inhibits growth and angiogenesis of colon cancer. Clin Cancer Res. 2002;8:3259–3269. [PubMed] [Google Scholar]
- 9.Stoeltzing O, Liu W, Reinmuth N, Fan F, Parikh AA, Bucana CD, Evans DB, Semenza GL, Ellis LM. Regulation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor, and angiogenesis by an insulin-like growth factor-I receptor autocrine loop in human pancreatic cancer. Am J Pathol. 2003;163:1001–1011. doi: 10.1016/s0002-9440(10)63460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chun KS, Surh YJ. Signal transduction pathways regulating cyclooxygenase-2 expression: potential molecular targets for chemoprevention. Biochem Pharmacol. 2004;68:1089–1100. doi: 10.1016/j.bcp.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 11.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–436. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 12.Gately S, Li WW. Multiple roles of COX-2 in tumor angiogenesis: a target for antiangiogenic therapy. Semin Oncol. 2004;31:2–11. doi: 10.1053/j.seminoncol.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 13.Wu G, Mannam AP, Wu J, Kirbis S, Shie JL, Chen C, Laham RJ, Sellke FW, Li J. Hypoxia induces myocyte-dependent COX-2 regulation in endothelial cells: role of VEGF. Am J Physiol Heart Circ Physiol. 2003;285:H2420–H2429. doi: 10.1152/ajpheart.00187.2003. [DOI] [PubMed] [Google Scholar]
- 14.Dixon DA. Regulation of COX-2 expression in human cancers. Prog Exp Tumor Res. 2003;37:52–71. doi: 10.1159/000071363. [DOI] [PubMed] [Google Scholar]
- 15.Wendum D, Masliah J, Trugnan G, Flejou JF. Cyclooxygenase-2 and its role in colorectal cancer development. Virchows Arch. 2004;445:327–333. doi: 10.1007/s00428-004-1105-2. [DOI] [PubMed] [Google Scholar]
- 16.Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem. 1995;270:24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- 17.Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- 18.Soh JW, Lee EH, Prywes R, Weinstein IB. Novel roles of specific isoforms of protein kinase C in activation of the c-fos serum response element. Mol Cell Biol. 1999;19:1313–1324. doi: 10.1128/mcb.19.2.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131:152–159. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Popolo A, Memoli A, Apicella A, Tuccillo C, di Palma A, Ricchi P, Acquaviva AM, Zarrilli R. IGF-II/IGF-I receptor pathway up-regulates COX-2 mRNA expression and PGE2 synthesis in Caco-2 human colon carcinoma cells. Oncogene. 2000;19:5517–5524. doi: 10.1038/sj.onc.1203952. [DOI] [PubMed] [Google Scholar]
- 21.Kim HJ, Kim TY. IGF-II-mediated COX-2 gene expression in human keratinocytes through extracellular signal-regulated kinase pathway. J Invest Dermatol. 2004;123:547–555. doi: 10.1111/j.0022-202X.2004.23317.x. [DOI] [PubMed] [Google Scholar]
- 22.Bustin SA, Dorudi S, Phillips SM, Feakins RM, Jenkins PJ. Local expression of insulin-like growth factor-I affects angiogenesis in colorectal cancer. Tumour Biol. 2002;23:130–138. doi: 10.1159/000064029. [DOI] [PubMed] [Google Scholar]
- 23.Guan Z, Buckman SY, Baier LD, Morrison AR. IGF-I and insulin amplify IL-1 beta-induced nitric oxide and prostaglandin biosynthesis. Am J Physiol. 1998;274:F673–F679. doi: 10.1152/ajprenal.1998.274.4.F673. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Reinmuth N, Stoeltzing O, Parikh AA, Tellez C, Williams S, Jung YD, Fan F, Takeda A, Akagi M, Bar-Eli M, Gallick GE, Ellis LM. Cyclooxygenase-2 is up-regulated by interleukin-1 beta in human colorectal cancer cells via multiple signaling pathways. Cancer Res. 2003;63:3632–3636. [PubMed] [Google Scholar]
- 25.Mallat A, Gallois C, Tao J, Habib A, Maclouf J, Mavier P, Preaux AM, Lotersztajn S. Platelet-derived growth factor-BB and thrombin generate positive and negative signals for human hepatic stellate cell proliferation. Role of a prostaglandin/cyclic AMP pathway and cross-talk with endothelin receptors. J Biol Chem. 1998;273:27300–27305. doi: 10.1074/jbc.273.42.27300. [DOI] [PubMed] [Google Scholar]
- 26.Rechler MM. Insulin-like growth factor binding proteins. Vitam Horm. 1993;47:1–114. doi: 10.1016/s0083-6729(08)60444-6. [DOI] [PubMed] [Google Scholar]
- 27.Munkarah A, Ali-Fehmi R. COX-2: a protein with an active role in gynecological cancers. Curr Opin Obstet Gynecol. 2005;17:49–53. doi: 10.1097/00001703-200502000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Nasi ML, Castiglione M. Cyclooxygenase-2 (COX-2) a new prognostic and predictive factor for ovarian cancer? Are all the criteria fulfilled? Ann Oncol. 2002;13:1169–1171. doi: 10.1093/annonc/mdf312. [DOI] [PubMed] [Google Scholar]
- 29.Dore M, Cote LC, Mitchell A, Sirois J. Expression of prostaglandin G/H synthase type 1, but not type 2, in human ovarian adenocarcinomas. J Histochem Cytochem. 1998;46:77–84. doi: 10.1177/002215549804600110. [DOI] [PubMed] [Google Scholar]
- 30.Gupta RA, Tejada LV, Tong BJ, Das SK, Morrow JD, Dey SK, DuBois RN. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003;63:906–911. [PubMed] [Google Scholar]
- 31.Chang SH, Liu CH, Conway R, Han DK, Nithipatikom K, Trifan OC, Lane TF, Hla T. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA. 2004;101:591–596. doi: 10.1073/pnas.2535911100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 expression is related to prostaglandin biosynthesis and angiogenesis in human gastric cancer. Clin Cancer Res. 2000;6:135–138. [PubMed] [Google Scholar]
- 33.Wang D, DuBois RN. Cyclooxygenase 2-derived prostaglandin E2 regulates the angiogenic switch. Proc Natl Acad Sci USA. 2004;101:415–416. doi: 10.1073/pnas.0307640100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhi YH, Liu RS, Song MM, Tian Y, Long J, Tu W, Guo RX. Cyclooxygenase-2 promotes angiogenesis by increasing vascular endothelial growth factor and predicts prognosis in gallbladder carcinoma. World J Gastroenterol. 2005;11:3724–3728. doi: 10.3748/wjg.v11.i24.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade. Mol Cell Biol. 2000;20:4265–4274. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cok SJ, Morrison AR. The 3′-untranslated region of murine cyclooxygenase-2 contains multiple regulatory elements that alter message stability and translational efficiency. J Biol Chem. 2001;276:23179–23185. doi: 10.1074/jbc.M008461200. [DOI] [PubMed] [Google Scholar]
- 38.Faour WH, He Y, He QW, de Ladurantaye M, Quintero M, Mancini A, Di Battista JA. Prostaglandin E(2) regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. J Biol Chem. 2001;276:31720–31731. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- 39.Sheng H, Shao J, Dixon DA, Williams CS, Prescott SM, DuBois RN, Beauchamp RD. Transforming growth factor-beta1 enhances Ha-ras-induced expression of cyclooxygenase-2 in intestinal epithelial cells via stabilization of mRNA. J Biol Chem. 2000;275:6628–6635. doi: 10.1074/jbc.275.9.6628. [DOI] [PubMed] [Google Scholar]
- 40.Sheng H, Shao J, DuBois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–2675. [PubMed] [Google Scholar]
- 41.Bachelor MA, Cooper SJ, Sikorski ET, Bowden GT. Inhibition of p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase decreases UVB-induced activator protein-1 and cyclooxygenase-2 in a SKH-1 hairless mouse model. Mol Cancer Res. 2005;3:90–99. doi: 10.1158/1541-7786.MCR-04-0065. [DOI] [PubMed] [Google Scholar]
- 42.Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- 43.Sheu ML, Chao KF, Sung YJ, Lin WW, Lin-Shiau SY, Liu SH. Activation of phosphoinositide 3-kinase in response to inflammation and nitric oxide leads to the up-regulation of cyclooxygenase-2 expression and subsequent cell proliferation in mesangial cells. Cell Signal. 2005;17:975–984. doi: 10.1016/j.cellsig.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 44.Sheu ML, Ho FM, Yang RS, Chao KF, Lin WW, Lin-Shiau SY, Liu SH. High glucose induces human endothelial cell apoptosis through a phosphoinositide 3-kinase-regulated cyclooxygenase-2 pathway. Arterioscler Thromb Vasc Biol. 2005;25:539–545. doi: 10.1161/01.ATV.0000155462.24263.e4. [DOI] [PubMed] [Google Scholar]
- 45.Subbaramaiah K, Hart JC, Norton L, Dannenberg AJ. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 AND p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275:14838–14845. doi: 10.1074/jbc.275.20.14838. [DOI] [PubMed] [Google Scholar]
- 46.Subbaramaiah K, Norton L, Gerald W, Dannenberg AJ. Cyclooxygenase-2 is overexpressed in HER-2/neu-positive breast cancer: evidence for involvement of AP-1 and PEA3. J Biol Chem. 2002;277:18649–18657. doi: 10.1074/jbc.M111415200. [DOI] [PubMed] [Google Scholar]
- 47.Subbaramaiah K, Marmo TP, Dixon DA, Dannenberg AJ. Regulation of cyclooxgenase-2 mRNA stability by taxanes: evidence for involvement of p38, MAPKAPK-2, and HuR. J Biol Chem. 2003;278:37637–37647. doi: 10.1074/jbc.M301481200. [DOI] [PubMed] [Google Scholar]
- 48.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cosentino F, Eto M, De Paolis P, van der LB, Bachschmid M, Ullrich V, Kouroedov A, delli GC, Joch H, Volpe M, Luscher TF. High glucose causes upregulation of cyclooxygenase-2 and alters prostanoid profile in human endothelial cells: role of protein kinase C and reactive oxygen species. Circulation. 2003;107:1017–1023. doi: 10.1161/01.cir.0000051367.92927.07. [DOI] [PubMed] [Google Scholar]
- 50.Huang WC, Chen JJ, Inoue H, Chen CC. Tyrosine phosphorylation of I-kappa B kinase alpha/beta by protein kinase C-dependent c-Src activation is involved in TNF-alpha-induced cyclooxygenase-2 expression. J Immunol. 2003;170:4767–4775. doi: 10.4049/jimmunol.170.9.4767. [DOI] [PubMed] [Google Scholar]