

Abstract
It has been proposed that opioid agonist efficacy may play a role in tolerance and the regulation of opioid receptor density. To address this issue, the present studies estimated the in vivo efficacy of three opioid agonists and then examined changes in spinal μ-opioid receptor density following chronic treatment in the mouse. In addition, tolerance and regulation of the trafficking protein dynamin-2 were determined. To evaluate efficacy, the method of irreversible receptor alkylation was employed and the efficacy parameter τ estimated. Mice were injected with the irreversible μ-opioid receptor antagonist clocinnamox (0.32–25.6mg/kg, i.p), and 24h later, the analgesic potency of s.c. morphine, oxycodone and etorphine were determined. Clocinnamox dose-dependently antagonized the analgesic effects of morphine, etorphine and oxycodone. The shift to the right of the dose-response curves was greater for morphine and oxycodone compared to etorphine and the highest dose of clocinnamox reduced the maximal effect of morphine and oxycodone, but not etorphine. The order of efficacy calculated from these results was etorphine > morphine > oxycodone. Other mice were infused for 7 days with oxycodone (10–150 mg/kg/day, s.c.) or etorphine (50–250ug/kg/day, s.c.) and the analgesic potency of s.c. morphine determined. The low efficacy agonist (oxycodone) produced more tolerance than the high efficacy agonist (etorphine) at equi-effective infusion doses. In saturation binding experiments, the low efficacy opioid agonists (morphine, oxycodone) did not regulate the density of spinal μ-opioid receptors, while etorphine produced ≈ 40% reduction in μ-opioid receptor density. Furthermore, etorphine increased spinal dynamin-2 abundance, while oxycodone did not produce any significant change in dynamin-2 abundance. Overall, these data indicate that high efficacy agonists produce less tolerance at equi-effective doses. Furthermore, increased efficacy was associated with μ-opioid receptor downregulation and dynamin-2 upregulation. Conversely, lower efficacy agonists produced more tolerance at equi-effective doses, but did not regulate μ-opioid receptor density or dynamin-2 abundance. Taken together, these studies indicate that agonist efficacy plays an important role in tolerance and regulation of receptors and trafficking proteins.
Index Terms: Etorphine, Oxycodone, Morphine, Clocinnamox, Efficacy, Tolerance, Analgesia
1. Introduction
Although tolerance to opioid agonists has been well-documented, studies indicate that the magnitude of tolerance is dependent on the particular opioid agonist that is used (e.g., Duttaroy et al., 1995; Paronis and Holztman, 1992; Stevens and Yaksh, 1989; Walker and Young, 2001). For example, if equi-effective doses (e.g., 100 times the ED50) of an opioid agonist are continuously infused in the intact animal, some agonists, such as etorphine, produce substantially less tolerance than other agonists such as morphine (Duttaroy et al., 1995). In addition, the ability of an opioid agonist to induce analgesic tolerance appears to be inversely related to its effectiveness to internalize and downregulate μ-opioid receptors. Specifically, morphine does not induce μ-opioid receptor internalization and downregulation, either in vitro or in vivo, while etorphine is very effective (Keith et al., 1996, 1998; Patel et al., 2002b; Stafford et al.,2001; Yoburn et al., 2004; however see Haberstock et al., 2005). Similarly, etorphine, but not morphine, upregulates the trafficking protein dynamin-2; and this may be an important mechanism in accelerating internalization and downregulation (Patel et al., 2002b; Yoburn et al., 2004).
Efficacy is a property of a ligand that may contribute to tolerance and regulation of μ-opioid receptors and trafficking proteins. Efficacy has been defined as “… the property of a molecule that causes the receptor to change its behavior toward the host cell“ (Kenakin, 2002a). Efficacy is generally interpreted to be a function of the receptor and its cellular environment (Kenakin, 2002b). Thus, efficacy is not simply a receptor based parameter of drug action; it depends upon activity at the receptor as well as the chain of events that are activated. In contrast, intrinsic efficacy was a term proposed by Furchgott (1966) that was intended to provide “… a method to compare the relative ability of agonists to produce a response for a given receptor occupancy … “ (Kenakin, 2002b). Intrinsic efficacy is a property solely of the drug-receptor complex and, theoretically, is independent of the cellular environment and the assay system.
Recently, it has been proposed that the Operational Model of Drug Agonism (Black and Leff; 1983; Black et al., 1985; Leff et al., 1990) is a preferred approach for characterizing agonist efficacy (e.g., Kenakin, 2004). A key feature of the operational model is the term τ. τ is a dimensionless proportionality factor that can relate receptor occupancy and receptor stimulus. τ is a transducer constant and characterizes the propensity of a system and an agonist to produce a response (Kenakin 2004). τ is analogous to the efficacy term, since it is a property of both the tissue, assay and the receptor system (Motulsky and Christopoulos, 2004). Estimation of τ using the operational model requires depletion of receptors, typically using an antagonist that will irreversibly bind to the receptor. In the present study, we employed the irreversible μ-opioid receptor antagonist clocinnamox and the data analysis method of Zernig et al (1995 Zernig et al (1996) to calculate τ values for 3 opioid agonists following clocinnamox treatment. Clocinnamox has been shown to dose-dependently decrease μ-opioid receptor BMAX in the intact animal (Burke et al., 1994; Chan et al., 1995; Paronis and Woods, 1997 ). Furthermore, clocinnamox produces a dose-dependent shift to the right of opioid agonist dose-response functions (Barrett et al., 2003; Burke et al., 1994; Chan et al., 1995; Comer et al., 1992; Zernig et al., 1995).
We have proposed that ligand efficacy is a determinant of the regulation of μ-opioid receptors and trafficking proteins, as well as changes in opioid agonist potency following treatment (Duttaroy et al., 1995; Patel et al., 2002b; Yoburn et al., 1993;). This suggestion is based on results from experiments using chronic treatment with opioid agonists and antagonists in the intact animal. High efficacy opioid agonists (e.g., etorphine) can reliably decrease μ-opioid receptor density, regulate μ-opioid receptor mRNA levels and upregulate dynamin-2 (e.g. Yoburn et al., 1993; Sehba et al.,1997; Duttaroy and Yoburn, 2000; Patel et al., 2002b). Furthermore, at equi-effective doses, high efficacy opioid agonists produce less tolerance. Conversely, opioid antagonists reliably increase μ-opioid receptor density, decrease dynamin-2 abundance, but do not alter μ-opioid receptor mRNA levels in vivo (e.g. Rajashekhara et al., 2003; Duttaroy et al., 1999; Unterwald et al., 1995). In addition, chronic opioid antagonist treatment produces a significant shift to the left of the agonist dose-response function for several outcome measures including analgesia and lethality (i.e., supersensitivity; Tempel et al., 1982; Paronis et al., 1991; Patel et al., 2002a; Yoburn, 1988). Therefore, from a broad perspective, the direction of receptor regulation (upregulation, downregulation) covaries with efficacy for agonists and antagonists. Similarly, following chronic agonist or antagonist treatment, the change in potency of agonists (tolerance, supersensitivity) also covaries with efficacy. Given these data, we hypothesized that, following chronic opioid treatment, the efficacy of a ligand can be used to predict changes in potency of agonists, as well as regulation of μ-opioid receptor density and dynamin-2. In this study, we determined the efficacy of oxycodone, morphine and etorphine and then evaluated tolerance and regulation of μ-opioid receptor density and dynamin-2 in the intact mouse. Overall, we report that efficacy does predict tolerance and regulation of μ-opioid receptors and dynamin-2.
2. Material and Methods
2.1 Subjects
Male, Swiss-Webster mice (20–25g) were obtained from Taconic Farms (Germantown, NY, USA). Mice were housed (5–10/cage) under standard colony conditions with food and water available ad libitum. All animal treatment protocols were approved by the St. John’s University Institutional Animal Care and Use Committee.
2.2 General Procedure
Initially, analgesic (tailflick, see below) ED50’s were determined for the opioid agonists oxycodone and etorphine. Other groups of mice (N=8–10/group) were injected i.p. with saline or clocinnamox (0.32–25.6 mg/kg). Twenty-four hours later, cumulative dose response studies were conducted with oxycodone, morphine or etorphine. For other studies, mice (N=8–12/group) were implanted s.c. with ALZET osmotic pumps (Model 2001; 24ul/day) infusing oxycodone (10–150mg/kg/day) or etorphine (50–250ug/kg/day). Controls were implanted with placebo pellets. At the end of 7days, pumps and pellets were removed and 16h later, either cumulative dose response studies were conducted using morphine or mice were sacrificed and spinal cords collected for radioligand binding and western blotting assays (see below). The 16h delay between opioid agonist treatment and sacrifice insures that residual etorphine does not interfere with the radioligand binding assay (see Yoburn et al., 1993). For consistency, this interval was also used for all drugs and assays following opioid agonist treatment. Pumps and pellets were implanted and removed while mice were briefly anesthetized with oxygen:halothane (96:4).
2.3 Analgesia assay
Antinociception was determined using the tailflick (Model TF6, Emdie instrument Co, Maidens, VA). A beam of light was focused on the dorsal tail surface approximately 2cm from tail tip and the latency for the mouse to remove its tail from the heat source was recorded. The intensity of the light stimulus was adjusted so that baseline latencies were 2–4s. The maximum latency (cutoff) was set at 10s and the test was terminated if a mouse did not flick by the cutoff. Mice with tail flick latencies of 10s were defined as analgesic. All testing was conducted in a blind manner.
2.4 Dose Response Protocols
Dose response studies were conducted using two procedures: standard and cumulative dosing protocols. In the standard dose response protocol, groups of mice (5–10/group) were injected s.c. with different doses of oxycodone or etorphine and tested for analgesia. For the standard dosing protocol, mice were injected with one dose of drug and tested once at the time of peak effect (15min; determined previously). Doses for infusion studies were based on the ED50 calculated from standard dosing.
For the cumulative dose response protocol, mice (8–10/group) were injected s.c. with a starting dose of morphine (1.5mg/kg), etorphine (0.25ug/kg) or oxycodone (0.4mg/kg) and tested for analgesia. Testing time was 30min following morphine and 15 min following etorphine and oxycodone. If the mouse had a tailflick latency of 10s, it was defined as analgesic and not tested further. Otherwise, the mouse was immediately injected s.c. with a dose of the same drug and retested. This procedure was continued until all mice were analgesic or cumulative dosing results indicated that no further changes in latencies were likely. The doses used for cumulative dosing protocols for etorphine, morphine and oxycodone are presented in Table 1. The cumulative dosing protocol was used in most cases to reduce the number of mice and the cost of supplies (e.g. osmotic pumps).
TABLE 1.
Cumulative Dosing Protocol
| Morphine |
| Individual dose sequence (mg/kg) 1.5, 1.5, 2.0, 3.0, 4.0, 4.0, 4.0, 4.0, 4.0, 8.0, 8.0, 8.0, 8.0, 16.0, 16.0, 16.0, 32.0 |
| Cumulative dose (mg/kg) 1.5, 3.0, 5.0, 8.0, 12.0, 16.0, 20.0, 24.0, 28.0 36.0 44.0, 52.0, 60.0, 76.0, 92.0, 108.0, 140.0 |
| Etorphine |
| Individual dose sequence (ug/kg) 0.25, 0.5, 1.0, 1.5, 2.0, 2.0, 2.0, 2.0, 2.0, 4.0, 4.0, 4.0, 4.0, 8.0, 8.0, 8.0, 8.0 |
| Cumulative dose (ug/kg) 0.25, 0.75, 1.75, 3.25, 5.25, 7.25, 9.25, 11.25, 13.25, 17.25, 21.25, 25.25, 29.25, 37.25, 45.25, 53.25 |
| Oxycodone |
| Individual dose sequence (mg/kg) 0.4, 0.6, 0.8, 1.6, 3.2, 6.4, 11.4, 16.4, 21.4, 26.4, 36.4 |
| Cumulative dose (mg/kg) 0.4, 1.0, 1.8, 3.4, 6.6, 13.0, 24.4, 40.8, 62.2, 88.6, 125.0 |
Cumulative dosing was used in most cases to reduce the number of mice and the cost of supplies (e.g. osmotic pumps). For the cumulative dose response protocol, mice were injected s.c. with a starting dose of morphine (1.5mg/kg), etorphine (0.25ug/kg) or oxycodone (0.4mg/kg) and tested for analgesia (tailflick). Testing time was 30 min following morphine and 15 min following etorphine and oxycodone. If a mouse did not flick by 10sec, a latency of 10sec was recorded and it was defined as analgesic and not tested further. Otherwise, the mouse was immediately injected s.c. with another dose of the same drug according to the dose sequence shown above and retested. This procedure was continued until all mice were analgesic or cumulative dosing results indicated that no further changes in latencies were likely. The individual dose sequence and corresponding cumulative dose are shown for each drug.
2.5 Radioligand Binding Studies
Following treatment, mice (N=10–12mice/group) were sacrificed and whole spinal cords were immediately removed and placed in 15ml (10–12 cords/tube) of ice cold 50mM Tris buffer (pH 7.4) and homogenized on ice (Brinkmann Polytron PT3000 Homogenizer 20,000rpm, 45s). Homogenates were centrifuged at 15,000rpm (≈ 26,000g) (2–4° C) for 15min, supernatants were discarded, and pellets were resuspended in 35ml of ice-cold 50mM Tris buffer and incubated in a shaking water bath for 30min at 25°C. Homogenates were centrifuged again at 15,000rpm (2–4° C) for 15min and pellets were resuspended in 18–20ml 50mM Phosphate buffer (pH 7.2). An aliquot of homogenate was assayed in triplicate in saturation binding assays using [3H] DAMGO (μ-opioid receptor ligand; range: 0.02–5nM; PerkinElmer Life Sciences, Boston, MA). Non-specific binding was determined in triplicate in the presence of levorphanol (1000nM). Tubes were incubated for 90min at 25° C. Incubation was terminated by addition of ice cold phosphate buffer and filtering the samples over GF/B filters (Brandel, Gaithersburg, MD). Tubes were washed three times with phosphate buffer, and filters were placed in vials containing scintillation cocktail and counted. Counts per minute (cpm) were converted into disintegration per minute (dpm) using the external standard method. Protein was assayed by the Bradford method (Bradford, 1976) using reagent from Bio-Rad (Richmond, CA). Binding studies were repeated 1–4 times.
2.6 Western Blotting Assay
Following treatment, mice (N=5/treatment) were sacrificed, spinal cords removed and each spinal cord placed in 500μl ice cold western buffer A [2% sodium dodecyl sulfate (SDS), 1.5mM sodium orthovanadate, 12.5mM Tris buffer (pH 7.4)], homogenized on ice (Brinkmann Polytron Homogenizer, at 20,000 rpm for 30sec) and centrifuged at 10,000 rpm (≈ 20,000g), at 15° C for 10min. The supernatant was aliquoted into fresh tubes. An aliquot (20μl) was removed to determine protein concentration (Bradford, 1976). Remaining sample was diluted 1:1 using Western Buffer B (4% SDS, 2% β-mercaptoethanol, 20% glycerol, 112.5mM Tris base, 0.01% bromophenol blue) and then boiled for 5–7min. Aliquots of each sample (8ul; 0.125–1μg protein) were loaded on polyacrylamide gels (PAGEr Gold Precast Gels 10% Tris Glycine, Cambrex Bio Sciences, Rockland, ME) and separated by electrophoresis (150V for 60 min). A sample from one spinal cord was loaded on each lane. Proteins were transferred to PVDF membrane (Immobilon-P, Millipore Corporation, Billerica, MA) at 100V for 85–95 min. Membranes were incubated in blocking buffer [0.2% Aurora blocking reagent, MP Biomedicals, Irvine, CA; 1X Phosphate buffered saline (75mM)] overnight at 4°C to block nonspecific binding sites, followed by incubation (1h, 24° C) with primary dynamin-2 antibody [Goat Polyclonal IgG (1:300), Santa Cruz Biotechnology, Santa Cruz, CA] in blocking buffer. Membranes were washed with blocking buffer and incubated in secondary antibody [Donkey Anti- Goat IgG-AP (1:5000), Santa Cruz Biotechnology] in blocking buffer. Membranes were washed with blocking buffer and incubated in alkaline phosophatase chemiluminescence substrate (CDP Star Substrate, Novagen, Madison, WI) for 5min. Target protein was visualized and digitized (KODAK Image Station R, Eastman Kodak Company, New Haven, CT). The images were analyzed for density using Gel Pro image analysis software (Version 3.1, Media Cybernetics, Silver Spring, MD). A standard curve using increasing amounts (0.125–1μg) of untreated spinal cord was included in each assay to convert the optical densities into valid estimates of percent change in protein. Western blot data were analyzed as percent of control values.
2.7 Drugs
Etorphine hydrochloride, oxycodone hydrochloride, morphine sulphate, morphine pellets (containing 25mg morphine base) and placebo pellets were obtained from Research Triangle Institute (Research Triangle Park, NC) through the Research Technology Branch of the National Institute on Drug Abuse. All drugs were dissolved in 0.9% normal saline and doses expressed as the free base.
2.8 Data Analysis
Cumulative and standard dose-response studies were analyzed as quantal (percent analgesic) or graded (tailflick latency) data. The estimation of agonist ED50 requires quantal data, however the calculation of agonist efficacy following receptor depletion is typically based on graded data. In this report, both quantal and graded outcome measures were used, although potency changes determined using either dependent measure are similar (e.g., see Fig 4). Quantal dose response data were analyzed with the computer program BLISS-21 (Department of Statistics, University of Edinburgh). This program uses probit analysis (Finney, 1973) to calculate ED50 values, standard errors and 95% confidence intervals. Graded data were analyzed using nonlinear regression using Prism (version 4.03, GraphPad software, San Diego, CA); which estimates EC50 values, standard errors and 95% confidence intervals. Potency changes are based on the ratio of the ED50 or EC50 value in the treated groups relative to control. This change is referred to as the shift in the ED50 or EC50 value.
Fig 4.
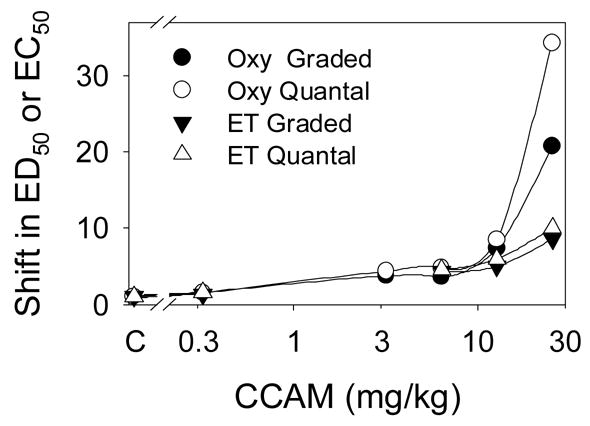
The effect of clocinnamox (CCAM) on the potency of oxycodone (Oxy) and etorphine (ET). Mice were pretreated with clocinnamox as described (see Fig 3). ED50’s (quantal; percent analgesic) and EC50’s (graded, mean tailflick latency) were determined using probit analysis and nonlinear regression, respectively. The shift in the ED50’s and EC50’s was calculated as the value following clocinnamox divided by the value for the saline treated control group (C). In order to statistically compare the results, each data set was fit with the equation:
where y is the shift in the ED50 or the EC50; a is the y-intercept; and k is the rate constant. There was no significant difference among the fitted a values, but the k values for oxycodone were greater than for etorphine for both graded data (2.9±.24, 1.16±.49) and quantal data (3.93±.13, 1.27±.48), respectively, (p<0.05).
The data from clocinnamox studies were analyzed according to the model proposed by Black and Leff (1983) and applied to behavioral data using the methods described extensively by Zernig et al. (1996). In this method, τ is an operational definition of efficacy (Zernig et al., 1996; Walker et al., 1998). τ is the reciprocal of the fraction of receptors necessary to give a half-maximum response. An estimate of the fraction of receptors available for agonist interaction after blockade with a given dose of clocinnamox corresponds to q in Furchott’s model (1966). The q value is calculated by dividing the τ value obtained after inactivation of the receptors with the insurmountable antagonist by the τ value obtained under control conditions (see Zernig et al., 1996). For non-rectangular hyperbolic E/[A] curves, Black et al. (1985) propose a function of the form:
where E is the effect (mean tail-flick latency), Em is the maximum attainable response (10 sec), [A] is the agonist dose, KA is the estimate for the agonist apparent in vivo dissociation constant, and n is the slope factor of the transducer function. The semilogarithmic form of the above equation is extended by c, the baseline response, with tau represented as (q*taucontrol):
All dose-response curves obtained with a given agonist in the presence or absence of clocinnamox pretreatment were simultaneously fit to the above equation. The efficacy estimates were based on simultaneous fitting of 6, 5 and 3 dose-response functions for oxycodone, etorphine and morphine (see Fig 3), respectively, using a non-linear fitting program (Zernig et al., 1995) and the general mathematical software package Mathematica Version 4.2.1 (Wolfram Research, Champaign, IL, USA).
Fig 3.
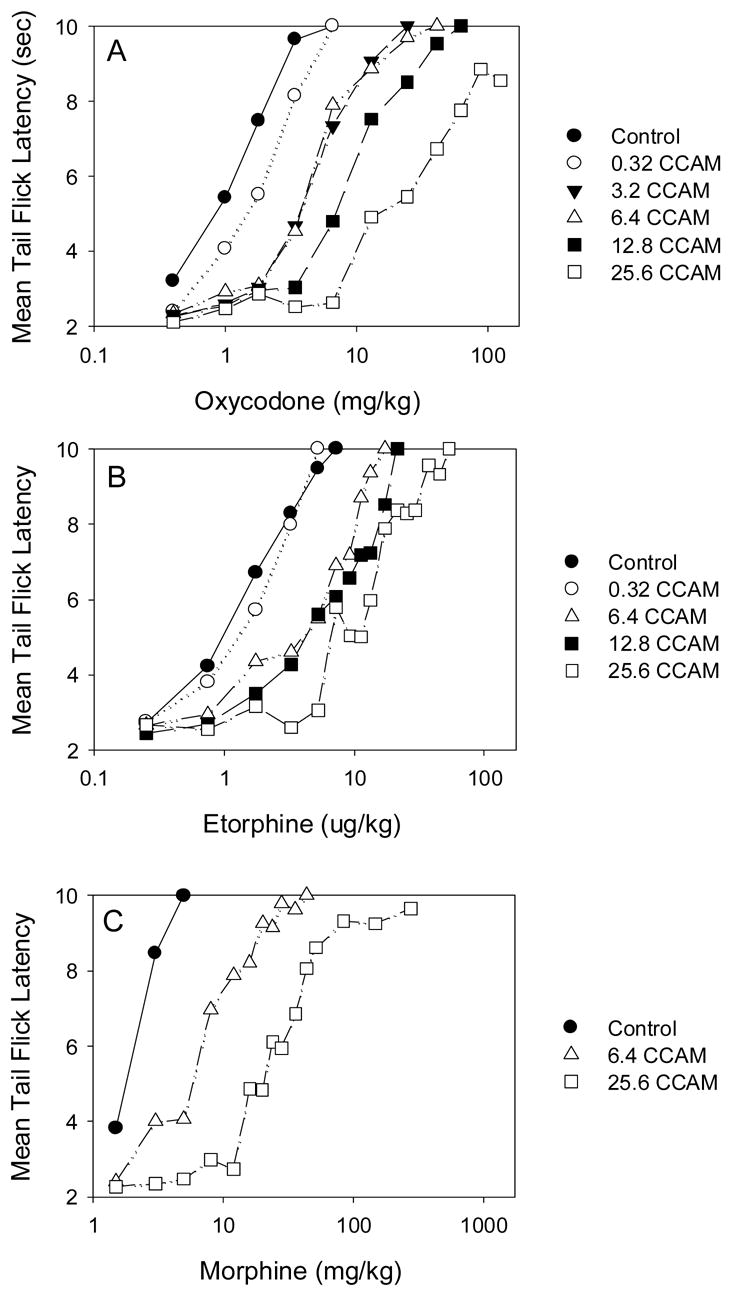
The effect of clocinnamox (CCAM) on the analgesic potency of oxycodone (A), etorphine (B) and morphine (C). Mice were injected with saline (N=8–10) or 0.32–25.6 mg/kg clocinnamox i.p., (N=6–10/CCAM dose); and 24 h later, cumulative dose response studies were conducted for each opioid agonist. Each clocinnamox dose for each drug was evaluated with a corresponding saline treated group (control) and results for each clocinnamox dose were determined 1–5 times for a given opioid agonist. Each graph presents the cumulated mean data from all experiments.
Binding data from saturation studies were analyzed by Prism using nonlinear regression analysis. All binding data were best fit by a one-site model. Significant differences (p<0.05) among the data were analyzed using t-tests or ANOVA with appropriate post-hoc comparisons.
3. Results
The quantal ED50’s (percent analgesic) were determined for oxycodone and etorphine using both the standard and cumulative dose-response protocols (Fig 1 and 2). The ED50 for oxycodone using the standard dosing protocol was 0.81±0.10 mg/kg and using the cumulative dosing protocol was 1.74±0.46 mg/kg. The ED50 for etorphine using the standard dosing protocol was 0.85±0.17 ug/kg and using the cumulative dosing protocol was 1.12±0.07 ug/kg. Similar results were observed when graded data (mean tailflick latencies) were examined (not shown).
Fig 1.
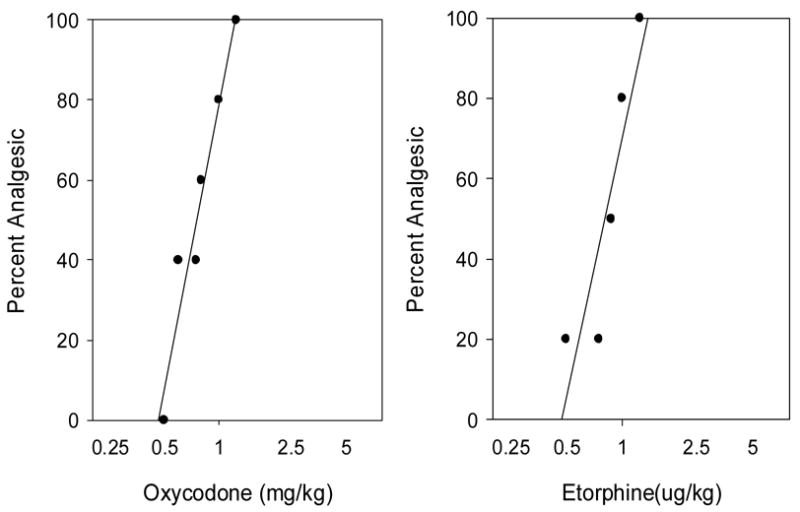
Dose response functions for oxycodone (left) and etorphine (right) using the standard dosing protocol (see methods). Mice (N=5–10/dose/drug) were injected s.c. with oxycodone or etorphine and tested using the tail flick 15min later. The data shown are the combined results from 3–4 experiments per drug. The mean ED50’s ± sd for oxycodone and etorphine based on all experiments were 0.81±0.10 mg/kg and 0.85±0.17 ug/kg. Linear regression fits for the data are plotted.
Fig 2.
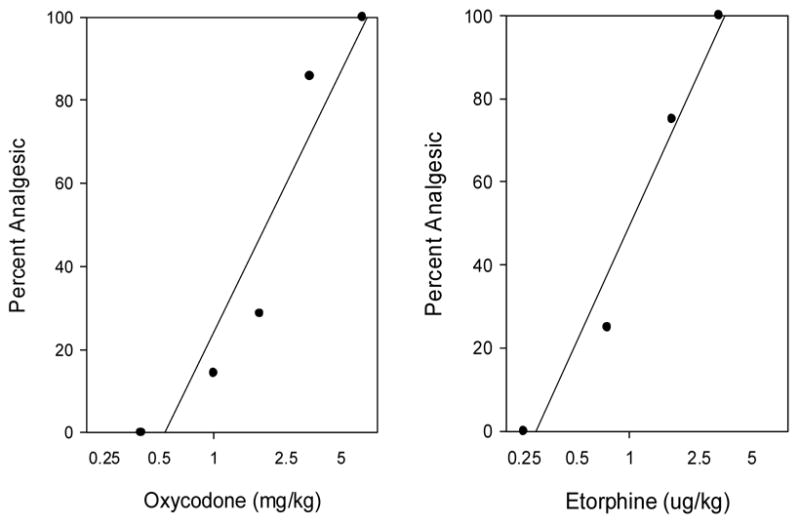
Dose response curves for oxycodone (left) and etorphine (right) determined using the cumulative dosing protocol (see methods). Mice (N=8–10/drug) were injected s.c. with oxycodone or etorphine and tested with the tail flick 15min later. Mice that were not analgesic were injected again and retested until all mice were analgesic. The data shown are the combined results of 2–6 experiments per drug. The mean ED50‘s ± sd for oxycodone and etorphine based on all experiments were 1.74±0.46 mg/kg and 1.12±0.07 ug/kg. Linear regression fits for the data are plotted.
To determine the efficacy of oxycodone and etorphine, mice were injected with clocinnamox and then cumulative dose-response studies were conducted 24h later (Fig 3a, b). Clocinnamox dose dependently increased the ED50 for both oxycodone and etorphine. The shift in the dose-response function at the highest dose of clocinnamox (25.6 mg/kg) was greater for oxycodone than for etorphine regardless of whether quantal or graded data were evaluated (Fig 4). In addition, the maximum effect for oxycodone was reduced following treatment with the highest clocinnamox dose (Fig 3a). Calculation of τ indicated that oxycodone had significantly lower efficacy than etorphine (Table 2). For comparison, we also evaluated the effect of 2 doses of clocinnamox on morphine potency (Fig 3c); which was sufficient to determine that the efficacy of morphine was significantly less than that for etorphine (Table 2).
Table 2.
Apparent efficacy (τ) estimates for etorphine, oxycodone and morphine
The efficacy (τ) for each was determined using the irreversible alkylation method. The values for τ derived by fitting the data from Fig 3 to the operational model of drug agonism (Black and Leff, 1983) as described by Zernig et al. (1996); see methods for complete description of fitting methods.
= significantly different from etorphine p<0.05.
Based on the estimated efficacies of etorphine and oxycodone, we predicted that tolerance induced by oxycodone would be greater than that for etorphine. Mice were infused with etorphine (50–250ug/kg/day) or oxycodone (10–150mg/kg/day) for 7 days. These doses represent ≈ 60–300 times the ED50 for etorphine and ≈ 12–185 times the ED50 for oxycodone determined using the standard dosing protocol. Higher doses could not be used due to solubility issues (oxycodone) and toxicity (etorphine). Following drug infusions, tolerance to morphine was examined. Morphine was used to estimate tolerance so that the magnitude of potency shifts induced by oxycodone and etorphine could be directly compared. Infusion of oxycodone and etorphine produced dose-dependent increases in the morphine ED50 determined using a cumulative dosing protocol (Fig 5). The magnitude of the shift in morphine’s ED50 was greater following equi-effective infusion doses of oxycodone compared to etorphine (Fig 6).
Fig 5.
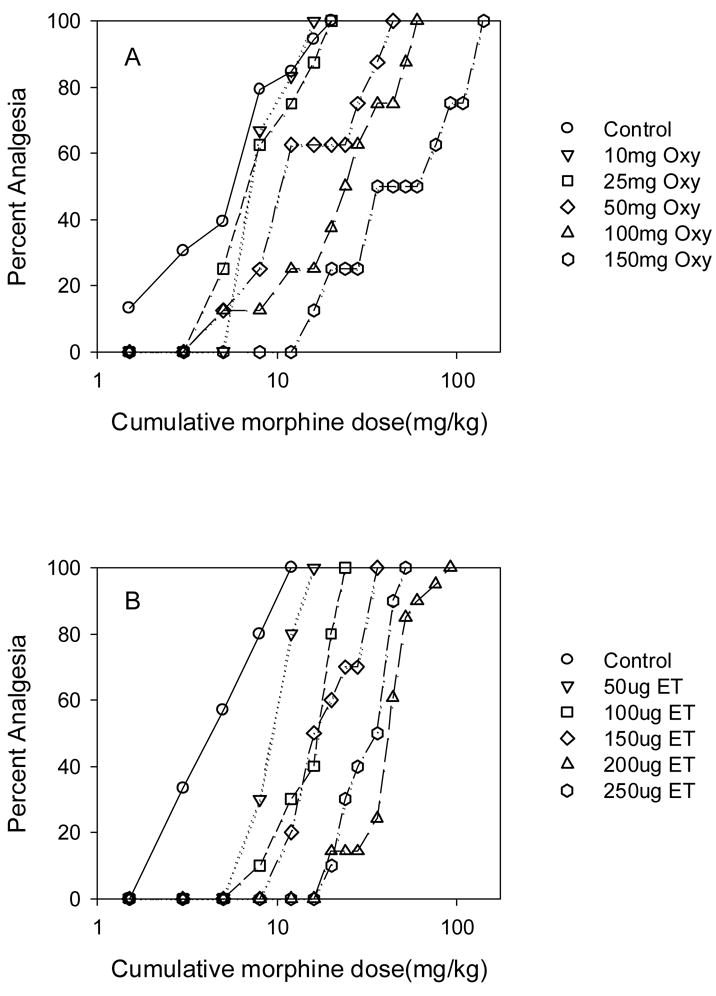
The effect of continuous oxycodone (A; Oxy) or etorphine (B; ET) infusion on morphine’s quantal analgesic potency. Mice were implanted with placebo pellets (control) or infused using osmotic pumps with oxycodone (10–150mg/kg/day) or etorphine (50–250ug/kg/day) for 7days (N=8–10/drug/infusion dose). On the 7th day, pumps and placebo pellets were removed and 16h later, morphine cumulative dose response studies were conducted. The data shown are representative of one of 1–2 experiments for each infusion dose.
Fig 6.
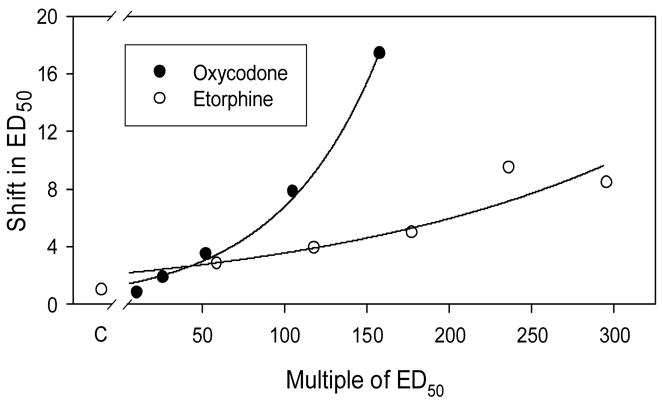
The effect of continuous oxycodone or etorphine infusion on the shift in morphine’s ED50. The shift in the ED50’s was calculated as the value following continuous agonist infusion divided by the value for the placebo treated control group (C). In order to statistically compare the results, each data set was fit with the equation:
where y is the shift in the ED50; a is the y-intercept; and k is the rate constant. There was no significant difference between the fitted a values, but the k value for oxycodone was greater than for etorphine (0.016±0.001; 0.005±0.001), respectively, (p<0.05).
Using the same 7 day infusion approach, we next determined the consequences on μ-opioid receptor density and dynamin-2 abundance in mouse spinal cord. We anticipated that the lower efficacy agonist oxycodone would be ineffective in regulating μ-receptors or the trafficking protein dynamin-2. Conversely, etorphine has been previously shown to produce μ-opioid receptor downregulation and increase dynamin-2 abundance (Patel et al., 2002b; Yoburn et al., 2004; Zhang et al., 2005). Oxycodone did not alter μ-opioid receptor density or affinity at 50 or 150mg/kg/day (Fig 7); although both these dosing regimens produced tolerance (Fig 6). Etorphine (250ug/kg/day) significantly reduced μ-opioid receptor density in spinal cord by ≅ 40%, without altering affinity (Fig 7). Similarly, etorphine (200ug/kg/day), but not oxycodone, regulated dynamin-2 abundance (Fig 8). We have previously reported this dose of etorphine (200ug/kg/day) also produces μ-opioid receptor downregulation (Patel et al., 2002b; Yoburn et al., 2004). Finally, for comparison, we infused mice with a dose of morphine that has been shown to induce substantial tolerance (Stafford et al., 2001). Morphine did not regulate μ-opioid receptor density or affinity in mouse spinal cord (Fig 9).
Fig 7.
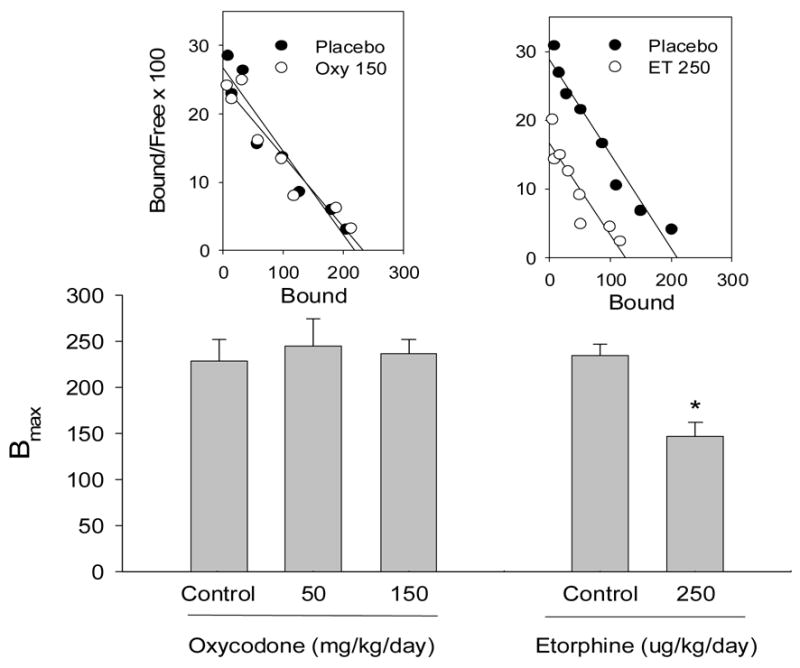
The effect of continuous oxycodone (Oxy) or etorphine (ET) infusion on μ-opioid receptor density in mouse spinal cord. Separate groups of mice (N=8–10/dose/drug) were infused s.c. for 7 days with oxycodone (50, 150mg/kg/day) or etorphine (250ug/kg/day). Controls were implanted with placebo pellets. The pumps and pellets were removed at the end of treatment and 16h later, mice were sacrificed and spinal cords collected for saturation binding studies ([3H] DAMGO). The histograms for oxycodone are the combined mean (± SEM) BMAX data from 4 experiments. A representative scatchard plot for oxycodone is shown above the histogram. The histogram (BMAX ± SE) and scatchard plot for etorphine are the results from a single study. We have reported similar results for etorphine, previously (e.g., Patel et al., 2002b; Yoburn et al., 2004; Stafford et al., 2001). * = significantly different from control p<0.05
Fig 8.
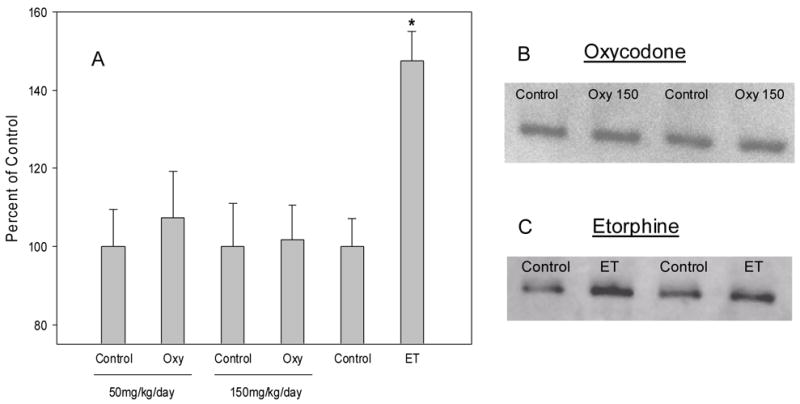
Effect of continuous oxycodone (Oxy) or etorphine (ET) infusion on dynamin-2 protein abundance. Mice were infused with oxycodone (50, 150mg/kg/day) or etorphine (200ug/kg/day) for 7 days, and 16h later, mice were sacrificed and spinal cords removed for western blot assays. Controls were implanted with a placebo pellet. Panel A presents the percent of control results (mean ± sd) from 3 oxycodone experiments, and 2 etorphine experiments. B and C are representative blots from one experiment. * = significantly different from control p<0.05.
Fig 9.
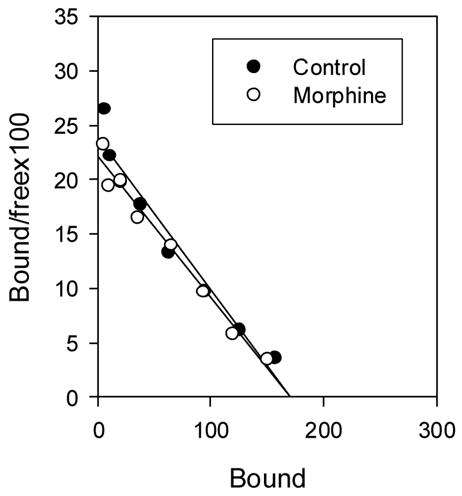
The effect of continuous morphine treatment on μ-opioid receptor density in mouse spinal cord. Mice were implanted with a 25mg morphine pellet and an osmotic pump infusing morphine (40mg/kg/day) for 7 days. Controls were implanted with placebo pellets. The pumps and pellets were removed at the end of treatment and 16h later, mice were sacrificed and spinal cords collected for saturation binding studies ([3H] DAMGO). The data are from a single experiment. We have previously reported that the same treatment protocol does not alter μ-opioid receptor binding in spinal cord (e.g., Yoburn et al., 2003; Patel et al., 2002).
4. Discussion
Opioid agonists vary in the magnitude of tolerance produced following chronic treatment. Several studies suggest that opioid agonists with lower efficacy will induce more tolerance than higher efficacy agonists (Duttaroy et al., 1995; Paronis and Holtzman, 1992; Stevens and Yaksh, 1989; Walker and Young, 2001). Previously, we have used an equi-effective dosing approach to directly determine the magnitude of tolerance produced by different opioid agonists (Duttaroy and Yoburn, 1995). This procedure involves determining the ED50 for some criterion effect (e.g., analgesia) and then using this information to examine tolerance. The chronic treatment dose is converted to a multiple of the acute ED50. This tactic serves to equate the chronically administered opioid drugs on a common measure (acute ED50) and allow an equi-effective dosing protocol. In the present study, we utilized this procedure to examine the role of efficacy of opioid agonists in producing tolerance and in regulating μ-opioid receptor density and the abundance of the trafficking protein dynamin-2. It was hypothesized that tolerance would be inversely related to efficacy; while downregulation of μ-opioid receptors and upregulation of dynamin-2 would be directly related to efficacy.
The current results demonstrate that morphine and oxycodone are of significantly lower efficacy than etorphine. The operational model of agonism (Black and Leff, 1983) was employed to estimate efficacy. This model requires receptor depletion, usually by irreversible receptor occupancy by an antagonist in order to calculate efficacy (τ). The antagonist clocinnamox was used to produce irreversible blockade of the μ-opioid receptor. Clocinnamox has been shown previously to dose-dependently decrease BMAX in radioligand binding studies. (Burke et al., 1994; Chan et al. 1995; Paronis and Woods, 1997;). In the present study, clocinnamox dose-dependently increased the ED50 for morphine, etorphine and oxycodone. The shift to the right in the dose-response functions was greatest for oxycodone and morphine, compared to etorphine (Figs 3 and 4). Furthermore, at the higher clocinnamox dose, the maximal effect for these 2 drugs was reduced (see Fig 3). Overall, inspection of the dose-response functions for oxycodone, morphine and etorphine (see Fig 3 and 4) confirms the efficacy estimates (Table 2) resulting from analysis of these data using the operational model of agonism and supports previous findings that etorphine is a higher efficacy agonist than morphine (Walker et al., 1998). The fact that oxycodone was of lower efficacy than etorphine led us to predict that this drug would not regulate μ-opioid receptors or dynamin-2, but would produce greater tolerance following an equi-effective infusion.
The tolerance induced by oxycodone was substantially greater than that produced by etorphine. The increase in tolerance to morphine following oxycodone is most apparent when comparing multiples of the ED50 between 50 – 200 (see Fig 6). We did not evaluate tolerance following morphine treatment using this equi-effective dosing approach, since the solubility of morphine is insufficient to allow adequate doses to be loaded into the osmotic pumps. However, morphine has been established to produce dose-dependent tolerance using an alternate tactic (e.g., Stafford et al., 2001).
While etorphine produced robust μ-opioid receptor downregulation in spinal cord, neither oxycodone nor morphine was effective. Similarly, etorphine regulated dynamin-2 in mouse spinal cord, but oxycodone did not. We have previously shown that the same chronic dosing protocol for morphine (25mg pellet + osmotic pump infusing 40mg/kg/day for 7 days) induces substantial tolerance, but does not regulate dynamin-2 or μ-opioid receptors (Patel et al., 2002b; Yoburn et al., 2004; Stafford et al., 2001). Taken together, these data indicate that opioid agonist efficacy is predictive of the magnitude of changes in potency (tolerance), as well as regulation of μ-opioid receptor density and dynamin-2 abundance. This conclusion extends to opioid antagonists, which are by definition of lower (or negative) efficacy. Chronic opioid antagonist treatment increases μ-opioid receptor density and decreases dynamin-2 (Rajshekhara et al., 2003) while at the same time inducing functional supersensitivity to agonists (Temple et al., 1982; Paronis et al., 1991). Thus, over a broad range of ligands, efficacy is related to regulation of receptors, a trafficking protein (dynamin-2) and modulation of agonist potency.
The role of efficacy in producing tolerance has been previously examined in the intact animal (Duttaroy et al., 1995; Paronis et al., 1997; Walker and Young, 2001). These studies concluded that low efficacy agonists tend to produce more tolerance. However, none of these reports included quantitative estimates of efficacy. Thus, one advantage of the present study is that we have determined efficacy and tolerance in the same set of experiments. In the present study, tolerance to morphine following chronic treatment was determined for both etorphine and oxycodone. Since morphine is primarily a μ-active agonist and lacks activity in knockout mice (e.g., Kieffer, 1999), our results allow us to compare tolerance at the μ receptor induced by both oxycodone and etorphine.
It is possible that etorphine and oxycodone produce some part of their in vivo analgesic effects via non-μ opioid receptor mechanisms. There is evidence that δ-opioid receptors may modulate the function of μ-opioid receptors in tolerance (see Zhang et al., 2006; Ananthan, 2006) However, etorphine has been shown to have somewhat higher affinity for the μ-opioid receptor; while μ antagonists, but not κ or δ antagonists, inhibit etorphine analgesia in the mouse (Yoburn et al., 1995; Aceto et al., 1997). With regard to oxycodone, it has been suggested that it may activate κ receptors and these receptors may mediate analgesic effects (e.g., Ross and Smith, 1997). However, data indicate that oxycodone has little affinity for the κ (and δ) receptor (Lalovic et al., 2006; Yoburn et al., 1995). Furthermore, the analgesic effects of oxycodone are only marginally altered by κ and δ antagonists, but blocked by a μ antagonist (see Nozaki et al., 2006). It is notable that, while it has been proposed that oxycodone metabolites may be important in mediating its effects, recent findings suggest that this is unlikely (Lalovic et al, 2006). Taken together, these data suggest that the pharmacodynamics of oxycodone and etorphine in the present study were mediated by the μ-opioid receptors. In addition, since both oxycodone and etorphine-induced tolerance were determined using morphine which lacks efficacy in μ knockouts (Keiffer, 1999), it is likely that the magnitude of tolerance represents changes in μ-opioid receptor signaling. Finally, it should be noted that in this series of studies tolerance was determined after the termination of treatment and that differential recovery from the tolerant state following oxycodone and etorphine may play a role in these findings.
The mechanism by which lower efficacy results in more tolerance is not entirely clear. However, it is possible that chronic treatment with lower efficacy agonists desensitizes more receptors since, by definition, a low efficacy agonist must occupy more receptors to produce an equivalent effect to a higher efficacy agonist (Zimmerman et al., 1987; Adams et al., 1990; Comer et al., 1992). Furthermore, the inability of lower efficacy agonists to induce internalization may reduce the recycling of the resensitized receptor that has been proposed to occur with higher efficacy agonists (Koch et al.,1998). In either case, drug efficacy is an important parameter that can be used to predict the biochemical and functional effects of chronic treatment with opioid ligands.
Acknowledgments
These data represent a portion of a thesis presented by the first author (MP) to the faculty of the College of Pharmacy and Allied Health Professions, St. John’s University, in partial fulfillment of the requirements for the M.S. degree in Pharmaceutical Sciences. We thank Dr. M.T. Turnock, Qiuyu Zhang, Pinky Sharma for advice and technical assistance. We also thank Dr. Terry Kenakin for several discussions regarding the nature of efficacy. This work was supported in part by DA 19959 and a seed grant from St. John’s University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JU, Paronis CA, Holtzman SG. Assessment of relative intrinsic activity of μ-opioid analgesic in vivo by using β–funaltrexamine. J Pharmacol Exp Ther. 1990;255:1027–1032. [PubMed] [Google Scholar]
- Ananthan S. Opioid ligands with mixed mu/delta opioid receptor interactions: an emerging approach to novel analgesics. AAPS J. 2006;8:E118–25. doi: 10.1208/aapsj080114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AC, Smith ES, Picker MJ. Use of irreversible antagonists to determine the relative efficacy of mu-opioids in a pigeon drug discrimination procedure: comparison of beta-funaltrexamine and clocinnamox. J Pharmacol Exp Ther. 2003;305:1061–70. doi: 10.1124/jpet.102.047068. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proceed Royal Soc London. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Black JW, Leff P, Shankley NP, Wood J. An operational model of pharmacological agonism: The effect of E/[A] curve shape on agonism on agonist dissociation constant estimation. British J Pharmacol. 1985;84:561–571. doi: 10.1111/j.1476-5381.1985.tb12941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TF, Woods JH, Lewis JW, Medzihradsky F. Irreversible opioid antagonist effects of clocinnamox on opioid analgesia and μ receptor binding in mice. J Pharmacol Exp Ther. 1994;271:715–721. [PubMed] [Google Scholar]
- Chan KW, Brodsky M, Davis T, Franklin S, Inturrisi CE, Yoburn BC. The effect of irreversible μ-opioid receptor antagonist clocinnamox on morphine potency, receptor binding and receptor mRNA. Eur J Pharmacol. 1995;287:135–143. doi: 10.1016/0014-2999(95)00488-2. [DOI] [PubMed] [Google Scholar]
- Comer SD, Burke TF, Lewis JW, Woods JH. Clocinnamox: A novel, systemically-active, irreversible opioid antagonist. J Pharmacol Exp Ther. 1992;262:1051–1056. [PubMed] [Google Scholar]
- Duttaroy A, Yoburn BC. In vivo regulation of μ-opioid receptor density and gene expression in Cxbk and outbred swiss webster mice. Synapse. 2000;37:118–124. doi: 10.1002/1098-2396(200008)37:2<118::AID-SYN6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Duttaroy A, Shen J, Shah S, Chen B, Sehba F, Carroll J, Yoburn BC. Opioid receptor upregulation in μ-opioid receptor deficient Cxbk and outbred swiss webster mice. Life Sciences. 1999;65:113–123. doi: 10.1016/s0024-3205(99)00228-3. [DOI] [PubMed] [Google Scholar]
- Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82:1226–1236. doi: 10.1097/00000542-199505000-00018. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. Advances Drug Res. 1966;3:21–55. [Google Scholar]
- Heberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. Morphine promotes rapid, arrestin-dependent endocytosis of μ opioid receptors in striatal neurons. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–84. [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–4. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2002a;42:349–379. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Efficacy at G protein-coupled receptors. Nat Rev Drug Discov. 2002b;1:103–109. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Principles: Receptor theory in pharmacology. Trends Pharmacol Sci. 2004;25:186–192. doi: 10.1016/j.tips.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Kieffer BL. Opioids: first lessons from knockout mice. Trends PharmSci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Schroder H, Wolf R, Raulf E, Hollt V. Carboxyl-terminal splicing of rat μ-opioid receptor modulates agonists-mediated internalization and receptor resensitization. J Biol Chem. 1998;273:13652–13657. doi: 10.1074/jbc.273.22.13652. [DOI] [PubMed] [Google Scholar]
- Leff P, Prentice DJ, Giles H, Martin GR, Wood J. Estimation of agonist affinity and efficacy by direct, operational model-fitting. J Pharmacol Methods. 1990;23:225–237. doi: 10.1016/0160-5402(90)90066-t. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Christopoulos A. A practical guide to curve fitting. Oxford University Press; New York: 2004. Fitting models to biological data using linear and nonlinear regression. [Google Scholar]
- Nozaki C, Saitoh A, Kamei J. Characterization of the antinociceptive effects of oxycodone in diabetic mice. Eur J Pharmacol. 2006;535:145–51. doi: 10.1016/j.ejphar.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Paronis CA, Holtzman SG. Increased analgesic potency of μ agonists after continuous naloxone infusion in rats. J Pharmacol Exp Ther. 1991;259:582–589. [PubMed] [Google Scholar]
- Paronis CA, Woods JH. Clocinnamox dose dependently antagonizes morphine analgesia and [3H] DAMGO binding in rats. Eur J Pharmacol. 1997;337:27–34. doi: 10.1016/s0014-2999(97)01296-x. [DOI] [PubMed] [Google Scholar]
- Paronis CA, Holtzman SG. Development of tolerance to the analgesic activity of μ agonists following continuous infusion of morphine, mepridine, or fentanyl in rats. J Pharmacol Exp Ther. 1992;262:1–9. [PubMed] [Google Scholar]
- Paronis CA, Woods JH. Clocinnamox dose-dependently antagonizes morphine-analgesia and [3H]DAMGO binding in rats. Eur J Pharmacol. 1997;337:27–34. doi: 10.1016/s0014-2999(97)01296-x. [DOI] [PubMed] [Google Scholar]
- Patel M, Gomes B, Patel C, Yoburn BC. Antagonist-induced μ-opioid receptor upregulation decreases G-protein receptor kinase-2 and dynamin-2 abundance in mouse spinal cord. Eur J Pharmacol. 2002a;446:37–42. doi: 10.1016/s0014-2999(02)01823-x. [DOI] [PubMed] [Google Scholar]
- Patel MB, Patel CN, Rajashekara V, Yoburn BC. Opioid agonists differentially regulate μ-opioid receptors and trafficking proteins in vivo. Molec Pharmacol. 2002b;62:1464–1470. doi: 10.1124/mol.62.6.1464. [DOI] [PubMed] [Google Scholar]
- Rajashekara V, Patel CN, Patel K, Purohit V, Yoburn BC. Chronic opioid antagonist treatment dose-dependently regulates μ-opioid receptors and trafficking proteins in vivo. Pharmacol Biochem Behav. 2003;75:909–913. doi: 10.1016/s0091-3057(03)00166-7. [DOI] [PubMed] [Google Scholar]
- Ross FB, Smith MT. The intrinsic antinociceptive effects of oxycodone appear to be kappa-opioid receptor mediated. Pain. 1997;73:151–7. doi: 10.1016/S0304-3959(97)00093-6. [DOI] [PubMed] [Google Scholar]
- Sehba F, Duttaroy A, Shah S, Chen B, Carroll J, Yoburn BC. In vivo homologous regulation of μ-opioid receptor gene expression in the mouse. Eur J of Pharmacol. 1997;339:33–41. doi: 10.1016/s0014-2999(97)01360-5. [DOI] [PubMed] [Google Scholar]
- Stafford K, Gomes AB, Shen J, Yoburn BC. Pharmacol Biochem Behav. Vol. 69. 2001. μ-Opioid receptor downregulation contributes to opioid tolerance in vivo; pp. 233–237. [DOI] [PubMed] [Google Scholar]
- Stevens CW, Yaksh TL. Potency of infused spinal antinociceptive agents is inversely related to magnitude of tolerance after continuous infusion. J Pharmacol Exp Ther. 1989;250:1–8. [PubMed] [Google Scholar]
- Tempel A, Zukin RS, Gardner EL. Supersensitivity of brain opiate receptor subtype after chronic naltrexone treatment. Life Science. 1982;31:1401–1404. doi: 10.1016/0024-3205(82)90391-5. [DOI] [PubMed] [Google Scholar]
- Unterwald EM, Rubenfeld JM, Imai Y, Wang JB, Uhl GR, Kreek MJ. Chronic opioid antagonist administration upregulates μ-opioid receptor binding without altering μ-opioid receptor mRNA levels. Molec Brain Res. 1995;33:351–355. doi: 10.1016/0169-328x(95)00143-g. [DOI] [PubMed] [Google Scholar]
- Walker EA, Zernig G, Young AM. In vivo apparent affinity and efficacy estimates for μ opiates in a rat tail-withdrawal assay. Psychopharmacol. 1998;136:15–23. doi: 10.1007/s002130050534. [DOI] [PubMed] [Google Scholar]
- Walker EA, Young AM. Differential tolerance to antinociceptive effects of mu opioids during repeated treatment with etonitazene, morphine, or buprenorphine in rats. Psychopharmacol (Berl) 2001;154:131–42. doi: 10.1007/s002130000620. [DOI] [PubMed] [Google Scholar]
- Yoburn BC. Opioid antagonist-induced upregulation and functional supersensitivity. Rev Clin Basic Pharm. 1988;7 :109–28. doi: 10.1515/jbcpp.1988.7.1-4.109. [DOI] [PubMed] [Google Scholar]
- Yoburn BC, Shah S, Chan K, Duttaroy A, Davis T. Supersensitivity to opioid analgesics following chronic opioid antagonist treatment: relationship to receptor selectivity. Pharmacol Biochem Behav. 1995;51:535–9. doi: 10.1016/0091-3057(94)00375-s. [DOI] [PubMed] [Google Scholar]
- Yoburn BC, Billings B, Duttaroy A. Opioid receptor regulation in mice. J Pharmacol Exp Ther. 1993;265:314–320. [PubMed] [Google Scholar]
- Yoburn BC, Purohit V, Patel K, Zhang Q. Opioid agonist and antagonist treatment differentially regulates immunoreactive μ-Opioid receptors and dynamin-2 in vivo. Eur J Pharmacol. 2004;498:87–96. doi: 10.1016/j.ejphar.2004.07.052. [DOI] [PubMed] [Google Scholar]
- Zernig G, Butelman ER, Lewis JW, Walker EA, Woods JH. In vivo determination of μ opioid receptor turnover in rhesus monkeys after irreversible blockade with clocinnamox. J Pharmacol Exp Ther. 1994;269:57–65. [PubMed] [Google Scholar]
- Zernig G, Issaevitch T, Broadbear JH, Burke TF, Lewis JW, Brine GA, Woods JH. Receptor reserve and affinity of μ opioid agonists in mouse antinociception: Correlation with receptor binding. Life Science. 1995;57:2113–2125. doi: 10.1016/0024-3205(95)02204-v. [DOI] [PubMed] [Google Scholar]
- Zernig G, Issaevitch T, Woods JH. Calculation of agonist efficacy, apparent affinity, and receptor population changes after administration of insurmountable antagonists: comparison of different analytical approaches. J Pharmacol Tox Meth. 1996;35:223–237. doi: 10.1016/1056-8719(96)00053-6. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bao L, Guan JS. Role of delivery and trafficking of delta-opioid peptide receptors in opioid analgesia and tolerance. Trends Pharmacol Sci. 2006;27:324–9. doi: 10.1016/j.tips.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Purohit V, Yoburn BC. Continuous opioid agonist treatment dose-dependently regulates μ-opioid receptors and dynamin-2 in mouse spinal cord. Synapse. 2005;56:123–128. doi: 10.1002/syn.20137. [DOI] [PubMed] [Google Scholar]
- Zhiyi Z. Role of opioid receptor internalization and arrestin in development of opioid tolerance. Anesth Analg. 2005;101:728–734. doi: 10.1213/01.ANE.0000160588.32007.AD. [DOI] [PubMed] [Google Scholar]
- Zimmerman D, Leander JD, Reel J, Hynes M. Use Of β–funaltrexamine to determine μ opioid receptor involvement in the analgesic activity of various opioid ligands. J Pharmacol Exp Ther. 1987;241:374–378. [PubMed] [Google Scholar]