

Abstract
Hepatocytes are resistant to tumor necrosis factor-α- (TNF) induced killing/apoptosis under normal circumstances, but primary hepatocytes from rats chronically fed alcohol have increased TNF cytotoxicity. Therefore, there must be mechanism(s) by which alcohol exposure “sensitizes” to TNF hepatotoxicity. Abnormal metabolism of methionine and Sadenosylmethionine (SAM) are well documented acquired metabolic abnormalities in ALD. Sadenosylhomocysteine (SAH) is the product of SAM in hepatic transmethylation reactions, and SAH hydrolase (SAHH) is the only enzyme to metabolize SAH to homocysteine and adenosine. Our previous studies demonstrated that chronic intracellular accumulation of SAH sensitized hepatocytes to TNF cytotoxicity in vitro. In the current study, we extended our previous observations by further characterizing the effects of chronic alcohol intake on mitochondrial SAM levels in liver and examining its possible involvement in SAH sensitization to TNF hepatotoxicity. Chronic alcohol consumption in mice not only increased cytosolic SAH levels, but also decreased mitochondrial SAM concentration, leading to decreased mitochondrial SAM to SAH ratio. Moreover, accumulation of hepatic SAH induced by administration of 3-deazaadenosine (DZA-a potent inhibitor of SAHH) enhanced lipopolysaccharide (LPS) /TNF hepatotoxicity in mice in vivo. Inhibition of SAHH by DZA resulted not only in accumulation of cytoplasmic SAH, but also in depletion of the mitochondrial SAM pool. Further studies using mitochondrial SAM transporter inhibitors showed that inhibition of SAM transport into mitochondria sensitized HepG2 cells to TNF cytotoxicity. In conclusion, our results demonstrate that depletion of the mitochondrial SAM pool by SAH, which is elevated during chronic alcohol consumption, plays a critical role in SAH induced sensitization to TNF hepatotoxicity.
Keywords: Liver, S-adenosylhomocysteine, S-adenosylmethionine, Tumor necrosis factor, Mitochondria
1. Introduction
Alcoholic liver disease (ALD) continues to be an important health problem in the United States. Compelling evidence generated over the past 1-2 decades demonstrates that tumor necrosis factor-α (TNF) is a critical effector molecule in ALD [1], and abnormal hepatic methionine metabolism is a major feature of ALD [2, 3].
Although the fact that TNF plays an etiologic role in ALD is now widely accepted, the exact molecular mechanism(s) involved in its hepatotoxicity are not well defined. TNF can induce both proliferative and cytotoxic responses in hepatocytes [4], and under normal conditions hepatocytes are resistant to TNF induced killing. However, primary hepatocytes isolated from rats chronically fed alcohol have increased TNF cytotoxicity [5], therefore a “sensitization” process must be induced by alcohol through which hepatocytes become vulnerable to TNF-induced cytotoxicity. Several factors have been demonstrated to sensitize hepatocytes to TNF cytotoxicity. Some of these factors include agents inhibiting protein synthesis or inducing transcriptional arrest [6]; agents causing total GSH depletion or selective mitochondrial GSH depletion [7, 8]; and agents inhibiting proteosome function [9].
S-adenosylhomocysteine (SAH) is the product of methionine in the hepatic transmethylation pathway whereby methyl groups from S-adenosylmethionine (SAM) are transferred to a vast number of molecules including DNA, RNA, biogenic amines, phospholipids, histones, and other proteins via specific methyltransferases. SAH is a potent competitive inhibitor of most methyltransferases. Either a decrease in SAM levels or an increase in SAH levels, or both, results in a decrease in the SAM:SAH ratio which is associated with inhibition of transmethylation reactions [10, 11]. Abnormal hepatic methionine metabolism is an acquired metabolic abnormality in ALD. Previous studies, including ours, reported that chronic alcohol in experimental animals not only caused SAM deficiency, but also elevation of SAH [3, 12, 13]. These observations are consistent with the study by Lieber et al. showing that alcohol consumption decreased hepatic phosphatidylethanolamine N-methyltransferase activity in a baboon model of alcohol-induced hepatic fibrosis [14]. Furthermore, a recent study by Lu's group documented decreased mRNA for multiple methyltransferases in liver biopsies from patients with alcoholic hepatitis [15]. More importantly, our recent studies have shown for the first time that elevation of intracellular SAH levels can sensitize to TNF hepatotoxicity [12]; however, the mechanism for this is still not fully understood.
SAM is the source for methyl groups in almost all biological methylation reactions [16]. Methylation reactions occur in both cytosol and mitochondria. Normal intra-mitochondrial SAM concentrations play a pivotal role in mitochondrial functions because methylation reactions are required for the methylation of RNA and proteins, and they function as intermediates in the biosynthesis of lipoic acid, ubiquinone and biotin [17-20]. Since mitochondria have a relatively large pool of SAM [21], and the enzyme required for SAM synthesis (methionine adenosyltransferase, MAT) is present only in cytosol and not in the mitochondria [22], a specific SAM transporter is needed to maintain normal mitochondrial SAM levels. A human mitochondrial SAM transporter has been recently identified [23]. Moreover, it has been reported that increased cytosolic SAH caused a decrease in SAM concentration in the mitochondria in rat hepatocytes [24]. Furthermore, studies by Colell et al. [25] demonstrated that exogenous SAM supplementation prevented the depletion of mitochondrial GSH pool induced by alcohol consumption and protected hepatocytes from alcohol-consuming rats from TNF-α induced hepatotoxicity. A very recent study by Bailey et al. [26] showed that SAM treatment prevented mitochondrial dysfunction induced by chronic alcohol consumption in rats. It is our hypothesis that SAH accumulation caused by chronic alcohol consumption inhibits mitochondrial SAM transport in hepatocytes, and the subsequent depletion of the mitochondrial SAM pool contributes to the well-documented sensitization to TNF hepatotoxicity observed in ALD.
In this study, we first examined the effects of chronic alcohol consumption on hepatic SAM and SAH levels in both cytosol and mitochondria, and documented that mitochondrial SAM was indeed significantly decreased in a mouse model of ALD. We next evaluated the effect of endogenously elevated SAH levels by 3-deaza-adenosine (DZA), a potent inhibitor of SAH hydrolase (SAHH), on mitochondrial SAM levels, as well as on the sensitization to TNF-induced hepatotoxicity both in vivo and in vitro. Based on these studies, we then performed in vitro studies to evaluate the effects of inhibition of SAM transport into mitochondria (using inhibitors of the mitochondrial SAM transporter) on sensitization to TNF induced hepatotoxicity.
2. Materials and Methods
2.1. Materials
Lipopolysaccharide (LPS) (Escherichia coli O111:B4) was purchased from Difco Laboratories (Detroit, MI). Before use, LPS was dissolved in sterile, pyrogen-free water, sonicated, and diluted with sterilized saline. Penicillin, streptomycin, Dulbecco's modified Eagle's medium (DMEM), trypsin, and fetal bovine serum were purchased from Invitrogen (Grand Island, NY); cell culture plates were from Corning (Corning, NY). Both human and rat recombinant TNF-α were from R&D Systems (Minneapolis, MN). DNA fragmentation ELISA kit was from Roche (Indianapolis, IN). All other reagents were of the highest purity available and, unless indicated otherwise, were obtained from Sigma (St. Louis, MO).
2.2. Primary Rat Hepatocyte Isolation and Culture
A two-step collagenase perfusion technique was used for primary hepatocyte isolation. Briefly, 6-8 week old Sprague-Dawley male rats were anesthetized and the portal vein was cannulated thereafter and perfused with Ca2+-free Hanks bicarbonate perfusion buffer. The perfusion was then switched to a re-circulating system with the perfusion medium (100 mL) as above but also containing CaCl2 (4 mM) and 0.05% collagenase (type IV) and continued for another 4-6 min. The digested liver was then chopped, filtered, and centrifuged at 50 × g and 4° C. The sediment, containing hepatocytes was washed 2-3 times with Ca2+-free Hanks, resuspended, counted, and evaluated for viability by Trypan blue. Isolated primary hepatocytes were seeded into collagen-pre-coated 6 or 24-well plates at a density of 5 × 105 cells/ml William E medium supplemented with 10% fetal calf serum, penicillin (100 U/ml), streptomycin (0.1 mg/ml), insulin (100 nM), and dexamethasone (100 nM). After an attachment period of 4 hours, the medium was replaced by fresh WME and further cultured at 37°C in an atmosphere of 5% CO2/95% O2. Experiments were started 24 hours later.
2.3. Cells and Culture Conditions
HepG2 cells, a human hepatoma cell line, and WRL68 cells, non-transformed human fetal hepatocytes, were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were cultured in DMEM containing 10% (v/v) fetal bovine serum, 2 mM glutamine, 5 U/ml penicillin, and 50 μg/ml streptomycin at 37 °C in a humidified O2/CO2 (19:1) atmosphere.
2.4. Animal Models and Experimental Protocol
In the alcohol feeding studies, male C57BL/6 mice weighing 20±0.5 g (mean ± SEM) were obtained from The Jackson Laboratory (Bar Harbor, ME). The mice were housed in the animal quarters at the University of Louisville Research Resources Center and the studies were approved by the Institutional Animal Care and Use Committee, which is certified by the American Association of Accreditation of Laboratory Animal Care. In the first week, sixteen mice were pair-fed liquid diets containing 18% of energy as protein, 35% as fat, 11% as carbohydrate and 30% as either ethanol (ethanol diet, 8 mice) or as an isocaloric maltose-dextrin mixture (control diet, 8 mice), according to Lieber and De Carli [27]. The energy from ethanol was increased by 2% (to replace carbohydrate) in each following week. Mice were kept on the treatments for 4 weeks before being humanely killed. The in vivo effect of SAH accumulation on LPS-induced liver injury was examined using C57 BL/6 male mice with 2 DZA injections, In this study, a total of 24 mice were divided into 4 groups (6 mice per group) consisting of control, DZA injections, LPS injection, and DZA+LPS injections. In DZA injection groups, mice received two i.p. injections of DZA (50mg/kg BW) within a 3-hour interval. Two hours after the last DZA injection, mice were i.p. injected either with LPS at a dose of 2mg/kg BW or an equal volume of vehicle (saline). Animals were humanely killed 24 hours later and blood and tissue samples were harvested for histology and biochemical assays. To measure time-course effect of DZA injections on SAM and SAH levels in both cytosol and mitochondria, DZA was injected as above-described and liver tissues were harvested at different time points after last DZA injection for the sub-cellular fractionation and HPLC analysis for SAM and SAH.
2.5. In Situ Apoptosis Detection
At the time of killing, the liver was harvested and small pieces of liver tissue were fixed immediately in 10% buffered formalin. After paraffin embedding, 5-μm sections were deparaffinized in xylene, and rehydrated through a series of decreasing concentrations of ethanol. Apoptotic hepatocytes were detected by the Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) assay with a TUNEL assay Kit (Intergen Company, Purchase, NY), and numbers were counted in 5 high-power fields. Images of liver sections were captured with a Nikon Eclipse E600 microscope equipped with a digital camera (SPOT; Diagnostic Instruments, Sterling Heights, MI).
2.6. Sub-cellular Fractionation of Liver Tissues and HepG2 Cells
Liver tissues or HepG2 cells were homogenized with cold isolation buffer (220 mM DMannitol, 70 mM sucrose, 2 mM K-HEPES, pH 7.4) and centrifuged at 4°C for 5 min at 600 × g. The supernatants were further centrifuged at 7000 × g for 10 min. The heavy membrane fraction (mitochondria) was washed once again in isolation buffer by centrifugation at 600 × g for 5 min to remove any contaminating particles and then recovered by centrifugation at 7000 × g for 10 min. The isolated mitochondria were resuspended in incubation buffer (250 mM sucrose, 10 mM KHEPES, pH 7.2, 2 mM KH2PO4, 5 mM sodium succinate, 2.5 mM EGTA).
2.7. SAM and SAH Assays by High Performance Liquid Chromatography (HPLC)
Deproteinized cellular extracts, liver cytosolic or mitochondrial fractions (4% metaphosphoric acid), were prepared and SAM and SAH were determined by an HPLC method using a 5-mm Hypersil C-18 column (250 × 4.6 mm) [12, 28]. The mobile phase consisted of 40 mM ammonium phosphate, 8 mM heptane sulfonic acid (ion-pairing reagent, pH 5.0), and 6% acetonitrile, and was delivered at a flow rate of 1.0 ml/min. SAM and SAH were detected using a Waters 740 UV detector at 254 nm. An internal standard, S-adenosylethionine (SAE), was added to all samples and standard solutions to a concentration of 100 nmol/ml. Protein concentrations were measured by a protein assay kit from Bio-Rad (Bio-Rad Laboratories, Hercules, CA) in accordance with the manufacturer's instructions.
2.8. Assay of Mitochondrial SAM Transport
Uptake of SAM into mitochondria was estimated by a rapid filtration technique [22]. Incubations were performed at 37°C by rapidly mixing 100μl of mitochondrial suspension (10mg protein/ml) with 900 μl of incubation buffer containing 1μCi [3H-CH3]SAM and other compounds as indicated. Uptake was terminated at the desired time by rapid filtration on Millipore membrane filters (0.22 μm pore size) followed by two washes with 1ml ice-cold stop solution ( 100mM KCl, 100mM mannitol, 10mM potassium phosphate, pH 7.4). The filters were dried, and uptake of SAM into mitochondria was determined by scintillation counting.
2.9. TNF-α Enzyme-Linked Immunosorbent Assay (ELISA)
For the measurement of TNF-α in liver tissues, liver tissues (∼0.1g) were homogenized in RIPA lysis buffer (1:10), followed by centrifugation at 15,000 g for 30 minutes. TNF-α in homogenized tissues was quantified using ELISA kits in accordance with the manufacturer's instructions as described previously [12] The detection limit for TNF-α is 13.5 pg/mL.
2.10. Enzymatic Assay of Caspase-3 Activity in Liver Tissue
Fresh liver tissues were homogenized with a Teflon homogenizer in the extraction buffer [25 mmol/L HEPES buffer, pH 7.4, containing 5 mmol/L ethylenediaminetetraacetic acid (EDTA), 2 mmol/L dithiothreitol, and 0.1% CHAPS]. The homogenate was centrifuged at 20,000 × g for 30 minutes. The supernatant was diluted with the assay buffer (50 mmol/L HEPES, 10 mmol/L dithiothreitol, 1.0 mmol/L EDTA, 100 mmol/L NaCl, 0.1% CHAPS, and 10% glycerol, pH 7.4) and incubated at 37°C with 200 μmol/L caspase-3 substrate I (Ac-DEVD-pNA). p-Nitroaniline was used as the standard. Cleavage of the substrate was monitored at 405 nm and the specific activity was expressed in pmol of the product (nitroaniline) per minute per mg protein.
2.11. ELISA Assay for DNA Fragmentation
DNA fragmentation was quantified using a commercially available ELISA kit from Roche (Indianapolis, IN) in accordance with the manufacturer's instructions. Briefly, HepG2 cells, WRL68 cells, or rat primary hepatocytes were plated in 24-well plates and pretreated with either 0.25mM SAH or 40μM 3-deaza-adenosine (DZA) for 2 hours before TNF (500 U/mL; human recombinant TNF-α for HepG2 and WRL68 cells and rat recombinant TNF-α for rat primary hepatocytes; R&D Systems, Minneapolis, MN) was added to the media. At 20 hours after TNF addition, cell membranes were lysed for 30 minutes at 20°C in a solution containing 10mM ethylenediaminetetraacetic acid and 0.1% Tween-20. After centrifugation for 10 minutes at 200 × g to remove cellular debris and nuclear components, 20μL cytoplasmic lysate together with 80μL immunoreagent containing 5% anti-histone-biotin and 5% anti-DNA-peroxidase were added to the 96-well plate precoated with streptavidin. After a two-hour incubation, wells were rinsed and 100μL of substrate solutions were added to each well. DNA fragmentation was measured at 405 nm against substrate solution as a blank.
2.12. Statistical Analysis
All data are expressed as mean ± SD. Statistical analysis was performed using one-way ANOVA and further analyzed by Newman-Keul's test for statistical difference. Differences between treatments were considered to be statistically significant at P<0.05.
3. Results
3.1. In vivo Studies
Male C57BL/6 mice fed the Lieber-De Carli liquid diet containing alcohol for 4 weeks were used to document TNF production, histological alterations, as well as SAM and SAH changes in both cytosol and mitochondria. Chronic alcohol feeding induced elevated hepatic TNF levels (Fig. 1A); obvious hepatocyte apoptosis by TUNEL staining (Fig. 1B); and histologic finding for alcoholic fatty liver as described previously (data not shown) (12). To determine the effect of chronic alcohol consumption on the SAM and SAH levels in cytosol and mitochondria, mitochondria were isolated from liver tissues. SAM and SAH concentrations were measured by our well-established HPLC system to obtain a SAM:SAH ratio. As shown in Fig. 1C & E, alcohol feeding for 4 weeks significantly decreased cytosolic SAM levels and decreased the SAM:SAH ratio (consistent with our previous reports). More importantly, chronic alcohol consumption significantly decreased the mitochondrial SAM levels, and numerically but not significantly increased SAH concentrations, leading to significantly decreased SAM:SAH ratio in mitochondria (Fig. 1D & E).
Fig. 1.
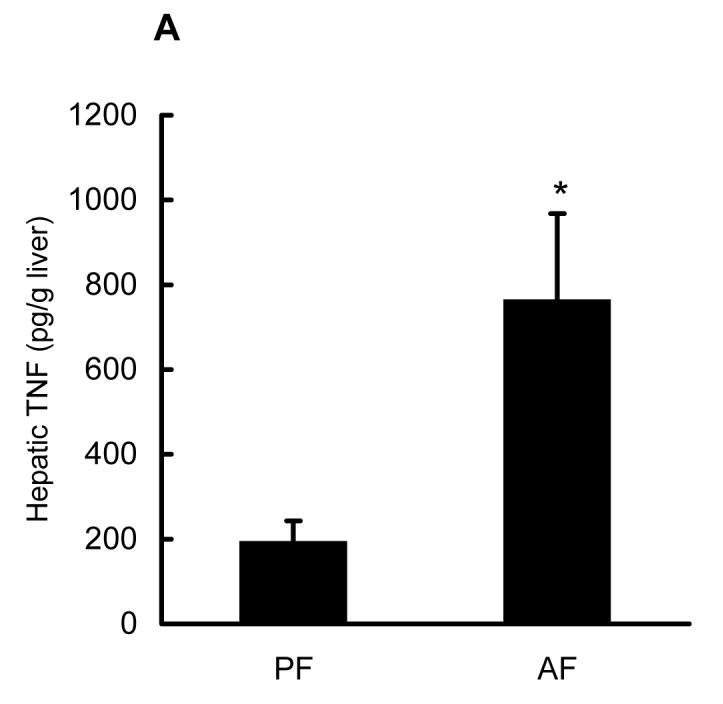


Effects of chronic alcohol consumption on hepatic TNF production, hepatocyte apoptosis, as well as SAM and SAH levels in both cytosol and mitochondria. Male C57BL/6 mice were pair-fed liquid diets with or without ethanol for four weeks. (A) Chronic alcohol feeding increased hepatic TNF production. Data represent means ± SD of n=8 animals per group. * p<0.01 compared with PF. (B) Formalin-fixed and paraffin-embedded liver sections were used in TUNEL assays to detect fragmented nuclear DNA. Liver from PF animals had very few detectable TUNEL positive hepatocytes. In contrast, livers from AF animals had more scattered hepatocytes with TUNEL positive labeling of nuclei (arrows). CV: central vein. (original magnification X 130). (C) Chronic alcohol consumption resulted in decreased cytosolic SAM, and elevated SAH levels. Data represent means ± SD of n=6 animals per group. * p<0.01 compared with PF. (D) Chronic alcohol consumption significantly decreased SAM levels with no significant effect on SAH levels in mitochondria. Data represent means ± SD of n=8 animals per group. * p<0.01 compared with PF. (E) Chronic alcohol consumption decreased SAM/SAH ratio in both cytosol and mitochondria. Data represent means ± SD of n=8 animals per group. * p<0.01 compared with PF. PF: pair feeding; AF: alcohol feeding.
DZA is a potent inhibitor of S-adenosylhomocysteine hydrolase (SAHH), the enzyme which catalyzes the reaction for the hydrolysis of SAH to homocysteine and adenosine. Because SAHH is the only enzyme that metabolizes SAH in the liver, its inhibition in vivo is expected to result in accumulation of SAH in liver. In order to examine the in vivo effect of accumulation of hepatic SAH on the sensitization to LPS/TNF induced hepatotoxicity, we injected male C57BL/6 mice with two doses of DZA followed by LPS administration. Plasma ALT levels and caspase-3 activities were measured 24 hrs later. As shown in Fig. 2, DZA injections alone had no effect on liver function. Likewise LPS injection caused only minimal liver injury. However, the combination of DZA and LPS injections led to markedly increased hepatotoxicity as demonstrated by significantly elevated ALT levels (Fig. 2A) and caspase-3 activities (Fig. 2B). The effects of DZA injections on the hepatic SAM and SAH levels were determined by measuring time course changes of hepatic SAM and SAH levels. As shown in Fig. 3A, significant SAH accumulation in liver was detected at 6 hrs after the last dose of DZA injection. Although moderately increased SAM levels were also observed 6 hrs following DZA injections (data not shown), the SAM:SAH ratio was significantly decreased at this time point due to more prominent increases in SAH levels (Fig. 3B). Both SAH and SAM:SAH ratio returned to the normal levels at 24 hrs, which may be accounted for the metabolism (removal) of DZA by the system.
Fig. 2.

Injections of 3-deaza-adenosine (DZA) sensitized to lipopolysaccharide (LPS) induced liver injury. Male C57BL/6 mice received two injections of DZA (50mg/kg BW for each) i.p. within a 3-hour interval. Two hours after the last DZA injection, a group of animals (6 mice) were i.p. injected with LPS in a dose of 2mg/kg BW. Plasma alanine aminotransferase (ALT) and liver caspase-3 activities were measured 24 hours later. (A) Plasma ALT activity changes. Data represent mean ± SD (n=6). (B) Caspase-3 activities in liver tissue. Data represent mean ± SD (n=6). Bars with different characters differ significantly, p<0.05.
Fig. 3.
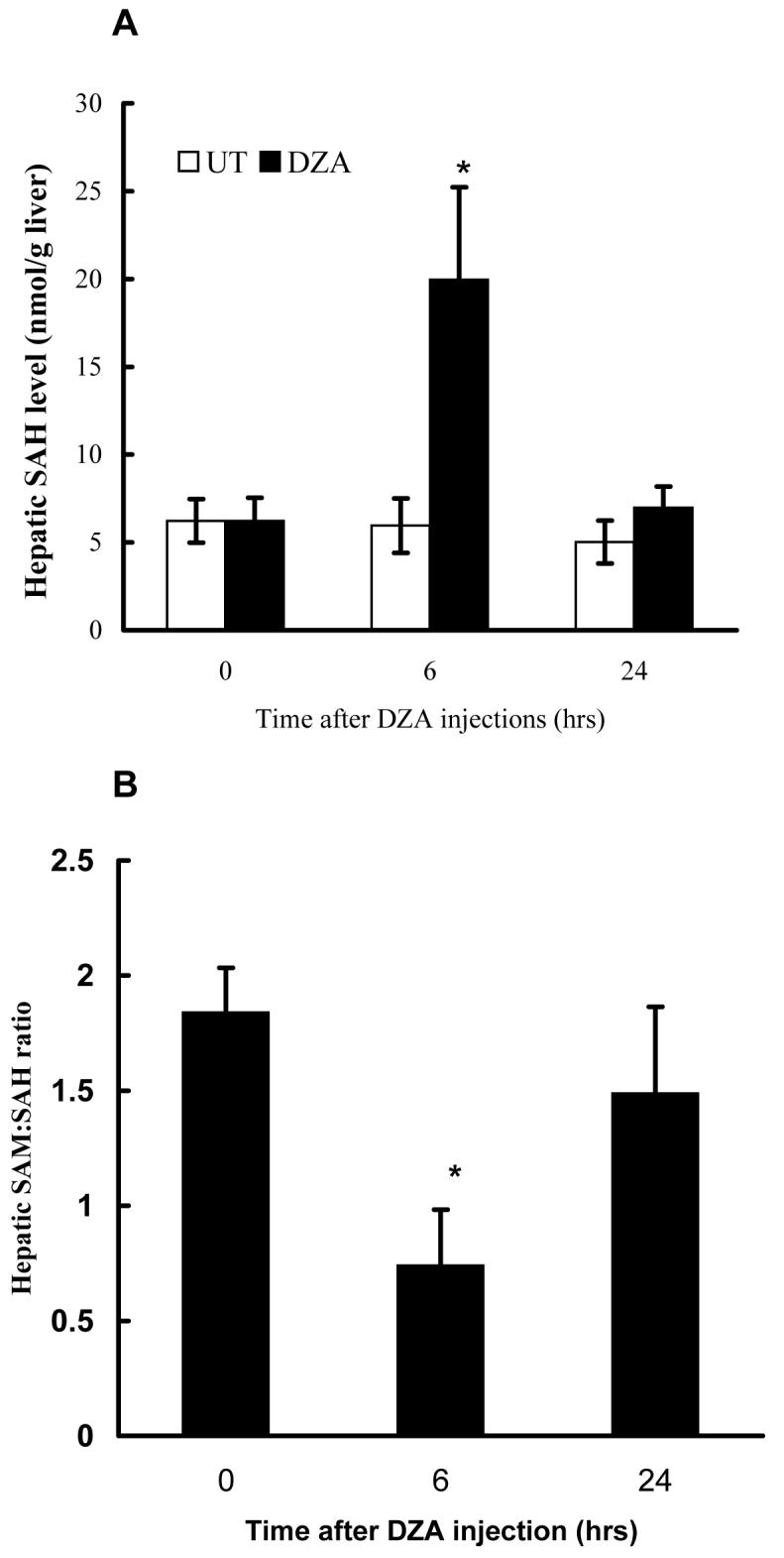
Effect of 3-deaza-adenosine (DZA) injections on hepatic S-adenosylhomocysteine (SAH) levels and SAM:SAH ratio. Male C57BL/6 mice received two injections of either vehicle (t = 0) or DZA (50mg/kg BW for each) i.p. within a 3-hour interval. Liver samples were harvested at different time points after the second injection and homogenized with 4% metaphosphoric acid. Hepatic Sadenosylmethionine (SAM) and SAH were assayed by high-performance liquid chromatography. (A) Time course change of hepatic SAH levels. Data represent mean ± SD (n=6). (B) Time course change of hepatic SAM/SAH ratio. Data represent mean ± SD (n=6). * p<0.01 compared with control group.
3.2. Intracellular Accumulation of SAH Sensitized Hepatocytes to TNF-Induced Cell Death in Vitro
In vitro studies using the cell culture system were conducted to examine the effects of intracellular accumulation of SAH on the sensitization of hepatocytes to TNF induced cytotoxicity. Primary rat hepatocytes, HepG2 cells, and WRL68 cells were pretreated with either SAH (0.25mM) or DZA (40μM) for 2 hrs followed by TNF stimulation. Cell death was measured by DNA fragmentation ELISA assay 20 hrs later and expressed as fold over untreated cells. As shown in Fig. 4A, without TNF exposure, SAH and DZA did not cause significant cell death; however, pretreatment with either agent induced significantly enhanced TNF cytotoxicity in all three cell types, indicating that intracellular accumulation of endogenous SAH sensitized hepatocytes to TNF induced cytotoxicity. Dose-dependent effects of DZA on TNF-induced hepatotoxicity were examined by the pretreatment of HepG2 cells with varying concentrations of DZA for 2 hrs followed by the stimulation with 500 U/ml TNF. Cell death was measured by DNA fragmentation ELISA 20 hrs later. As shown in Fig. 4B, pretreatment with DZA induced a dose-dependent increase in TNF induced cell death. To determine the involvement of caspase activation in this TNF induced cell death, HepG2 cells were treated with 40μM DZA for 90 minutes followed by the addition of either z-IETD-FMK, a relatively specific inhibitor of caspase-8, or z-DQMD-FMK, a relatively specific inhibitor of caspase-3, each at a dose of 100μM. TNF at 500U/ml was added 30 minutes later. Our results showed that both inhibitors attenuated TNF induced cell death (Fig. 4C), indicating that apoptosis is involved in DZA sensitization to TNF induced cell death.
Fig. 4.


DZA pretreatment sensitized hepatocytes to tumor necrosis factor-α (TNF)-induced cytotoxicity in vitro. (A) DZA pretreatment induced significantly increased cell death. Hepatocytes were cultured in media alone (UT, untreated), TNF alone, S-adenosylhomocysteine (SAH)-enhancing agents alone (SAH at 0.25 mM or 3-deazaadenosine (DZA) at 40μM), or the combination of TNF and one of the agents. Hepatocytes were pretreated with SAH or DZA for 2 hrs followed by stimulation of TNF (500U/ml). Cell death was measured by quantity DNA fragmentation ELISA assay 20 hrs later and expressed as fold of UT. Data represent a mean of at least three experiments ± SD. This modest amount of TNF by itself caused minimal toxicity. Moreover, neither SAH nor DZA alone caused major toxicity. However, in combination with TNF, they caused severe cell death. Bars with different characters differ significantly, p<0.05. (B) Dose response of cell death to TNF-induced cytotoxicity in HepG2 cells pretreated with DZA. HepG2 cells were pretreated with different doses of DZA for 2 hrs followed by stimulation of TNF at 500U/ml for 20 hrs. Cell death was measured by quantity DNA fragmentation ELISA assay and expressed as fold of UT. Data represent a mean of at least three experiments ± SD. (C) Inhibitors for caspase-3 or caspase-8 attenuated TNF induced cell death in HepG2 cells sensitized by DZA. HepG2 cells were pretreated with DZA (40μM) for 90 minutes followed by addition of either z-IETD-FMK, a relatively specific inhibitor of caspase-8, or z-DQMD-FMK, a relatively specific inhibitor of caspase-3, each at a dose of 100μM. Thirty minutes later, TNF at 500U/ml was added. Cell death was measured 20 hrs later by quantity DNA fragmentation ELISA assay and expressed as fold of UT. Data represent a mean of at least three experiments ± SD. Bars with different characters differ significantly, p<0.05.
3.3. DZA Induced Cytosolic SAH Accumulation and Subsequent Mitochondrial SAM Depletion in vitro
Time course effects of DZA on SAM, SAH and the SAM:SAH ratio in both cytosol and mitochondria were examined using HepG2 cells. As shown in Fig. 5, DZA increased SAH accumulation in cytosol over a 24-hour period (Fig. 5A). Although cytosolic SAM levels were also elevated by DZA treatment (Fig. 5B), the increase of SAH levels in the cytosol was much more prominent, resulting in a significantly lower SAM:SAH ratio during the entire 24 hrs study period (Fig. 5C). In parallel with SAH accumulation and the SAM:SAH decrease in cytosol, mitochondrial SAM levels were significantly decreased by DZA treatment (Fig. 5D).
Fig. 5.
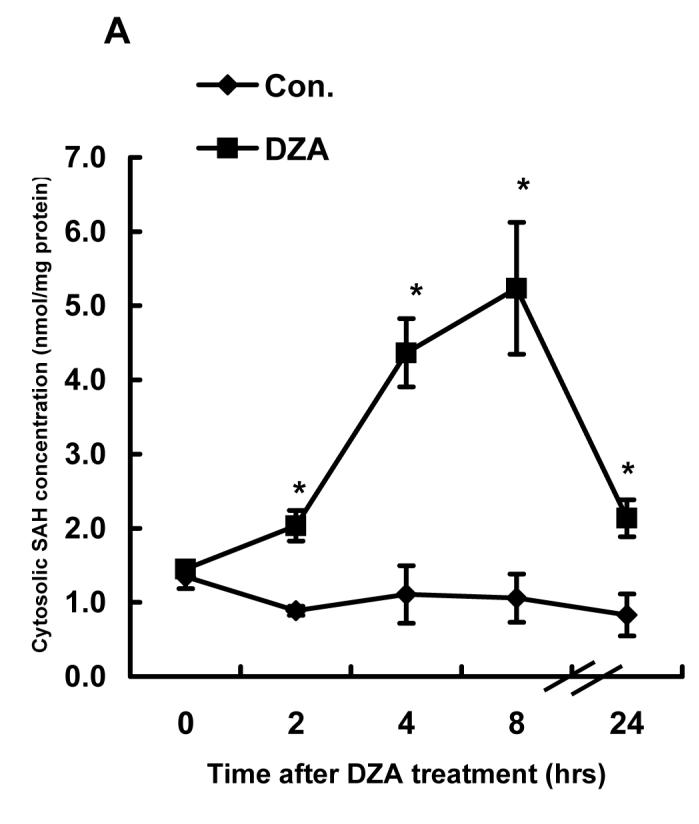
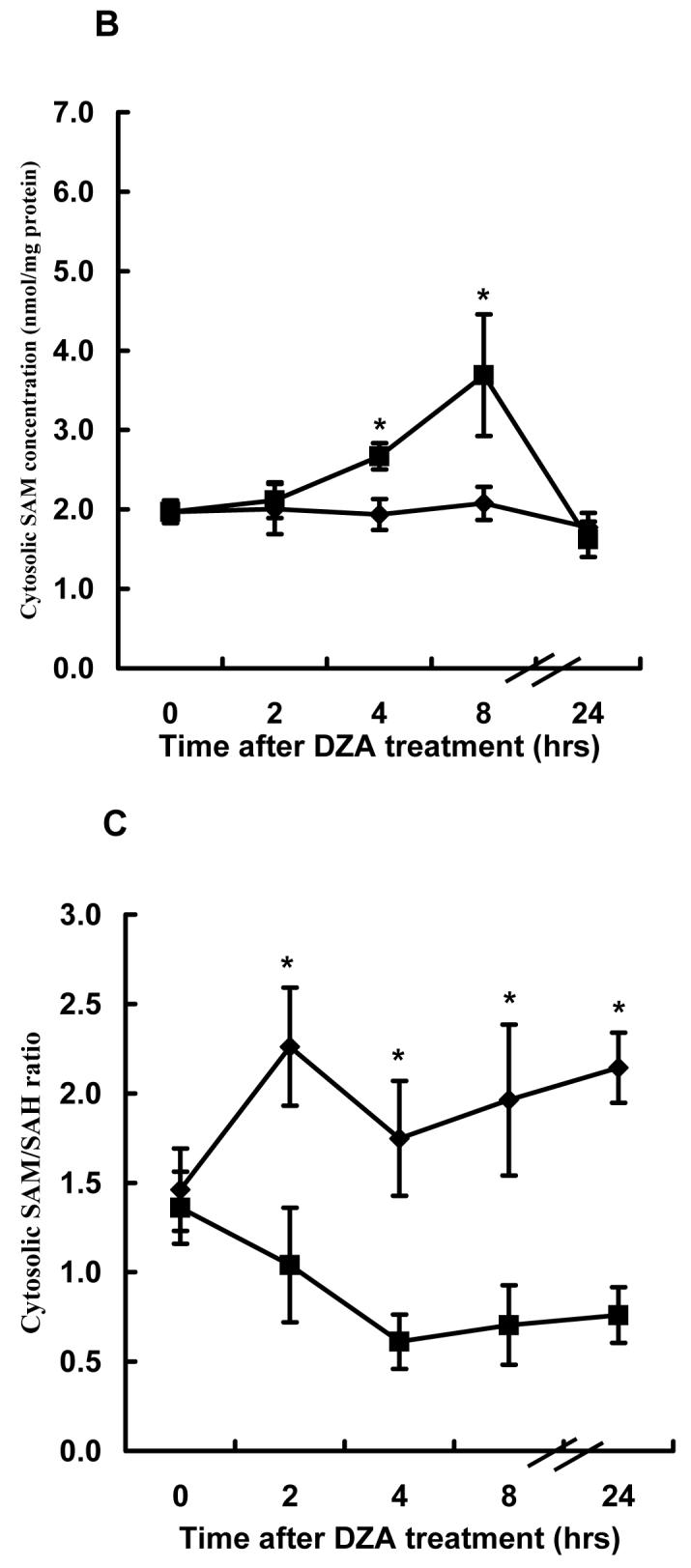
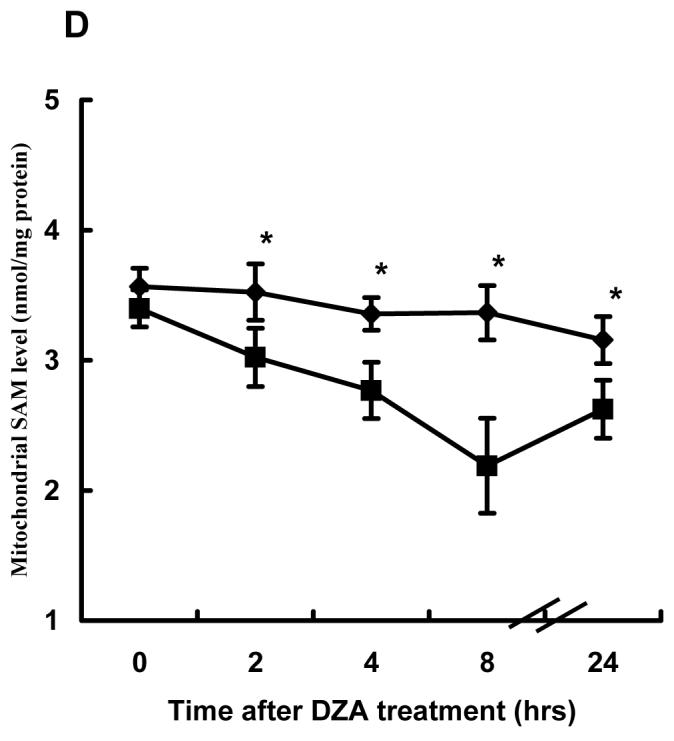
Effect of 3-deaza-adenosine (DZA) on S-adenosylmethionine (SAM) and Sadenosylhomocysteine (SAH) levels in both cytosol and mitochondria. HepG2 cells were treated with 40μM DZA for varying periods. Mitochondria and cytosol were separated by differential centrifugation as described in Materials and Methods. SAM and SAH in both cytosol and mitochondria were measured by high-performance liquid chromatography. (A) Time course changes of cytosolic SAH concentration. Data represent a mean of at least three experiments ± SD. (B) Time course changes of cytosolic SAM concentration. Data represent a mean of at least three experiments ± SD. (C) Time course changes of cytosolic SAM:SAH ratio. Data represent a mean of at least three experiments ± SD. (D) Time course changes of mitochondrial SAM concentration. Data represent a mean of at least three experiments ± SD. * p<0.01 compared with corresponding control values.
3.4. Inhibition of SAM Transport into Mitochondria Sensitized HepG2 Cells to TNF Induced Cytotoxicity
Although no specific inhibitors for the mitochondrial SAM transporter have been identified and purified so far, PLP (pyridoxal 5-phosphate) and sinefungin, a SAM analogue, have been reported to significantly inhibit the mitochondrial SAM transporter. We first measured the effects of PLP and sinefungin on mitochondrial SAM transporter activity in HepG2 cells by incubating isolated mitochondria with [3H-CH3]SAM in the presence/absence of 200μM PLP or sinefungin for 60 minutes. The mitochondria were separated from residual buffer solution after rapid filtration through 22μm filters as outlined in Materials and Methods, and radioactivity was then measured by scintillation counter. The presence of both PLP and sinefungin in the incubation buffer resulted in a significant decrease in mitochondrial SAM transporter activities (Fig. 6A), with PLP causing approximately 70% reduction and sinefungin about 22%. To determine if the inhibitors of mitochondrial SAM transporter might sensitize HepG2 cells to TNF induced cell death, we treated HepG2 cells with 200μM PLP or sinefungin for 2 hrs followed by TNF (500U/mL) stimulation. Cell viability was measured by DNA fragmentation ELISA assay. As shown in Fig. 6B, neither PLP nor sinefungin induced obvious cell death by themselves; however, pretreatment with these agents significantly increased TNF induced cell death, suggesting that the inhibition of the mitochondrial SAM transporter and subsequent mitochondrial SAM depletion could sensitize to TNF induced hepatotoxicity.
Fig. 6.
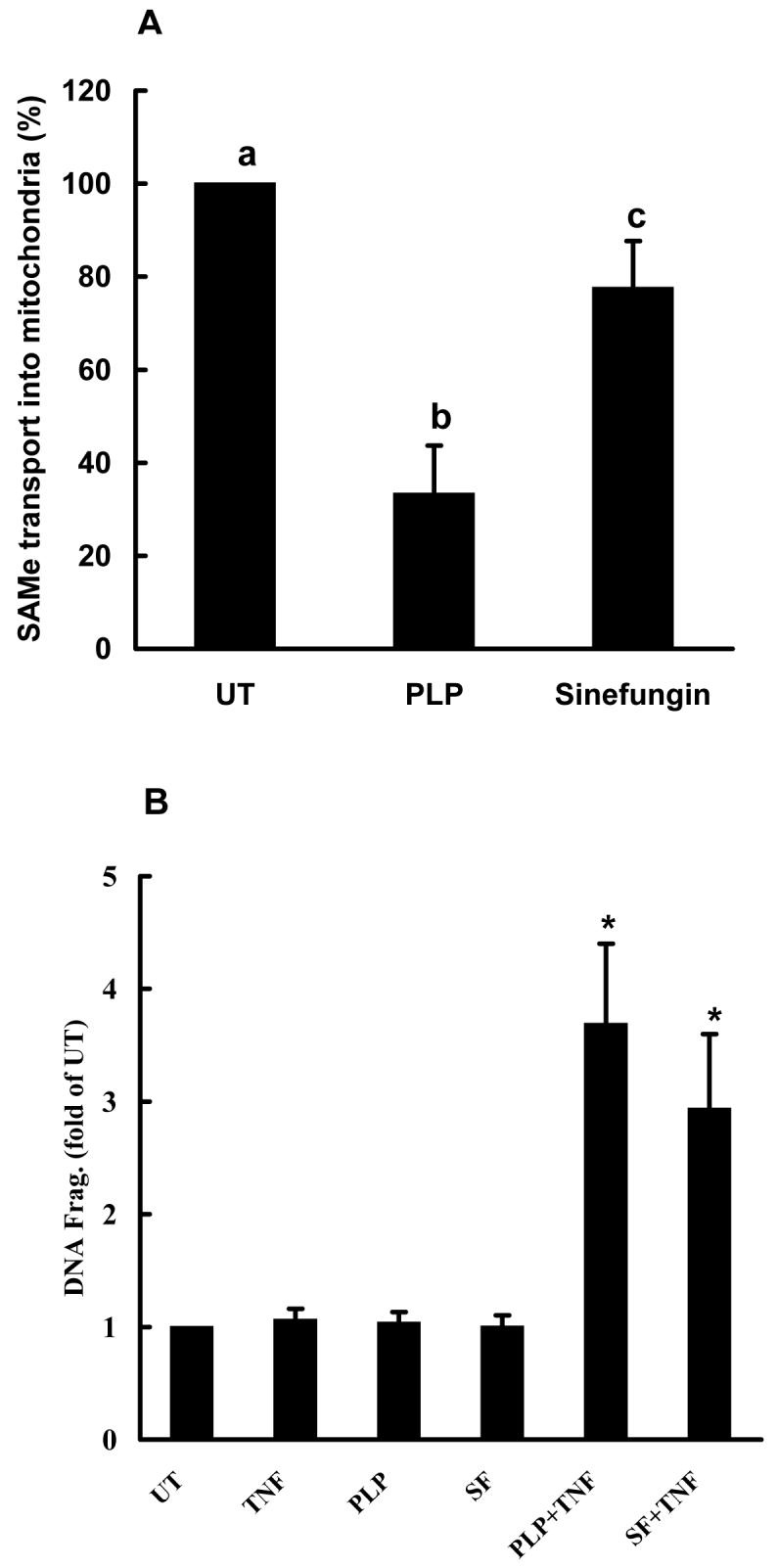
Inhibitors of mitochondrial S-adenosylmethionine (SAM) transporter sensitized HepG2 cells to tumor necrosis factor-α (TNF) cytotoxicity. (A) Both pyridoxal 5-phosphate (PLP) and sinefungin inhibited SAM transport into mitochondria. Mitochondria (10mg/ml) were isolated from cultured HepG2 cells as described in Materials and Methods and incubated with 900 μl of incubation buffer containing 1μCi [3H-CH3]SAM with/without the presence of 200μM PLP or sinefungin at 37°C. Uptake was terminated at the 1 hr by rapid filtration on 25-mm Millipore membrane filters (0.22 μm pore size) and uptake of SAM into mitochondria was determined by scintillation counting. Data represent a mean of at least three experiments ± SD. (B) PLP and sinefungin sensitized HepG2 cells to TNF cytotoxicity. HepG2 cells were stimulated with TNF (500U/ml) in the presence or absence of either 200μM PLP or sinefungin for 20 hrs. Cell death was measured by quantity DNA fragmentation ELISA assay and expressed as fold of UT. Data represent a mean of at least three experiments ± SD. Bars with different characters differ significantly, p<0.05.
4. Discussion
In ALD, both increased TNF production by monocytes/Kupffer cells and ‘sensitization’ to TNF-induced hepatocyte cytotoxicity occur [1, 5]. Moreover, abnormal methionine metabolism in hepatocytes represents a fundamental metabolic abnormality [2, 3]. Decreased hepatic SAM and increased homocysteine levels in ALD have been extensively reported. In addition to its effects on SAM and homocysteine levels, chronic alcohol consumption results in a significant increase in hepatic SAH levels [3, 12, 13]. Importantly, we showed that accumulation of intracellular SAH sensitized hepatocytes to TNF induced cytotoxicity in vitro, which we postulate is a clinically relevant metabolic alteration in the pathogenesis of ALD [12]. The goal of this research was to further investigate interaction between SAM/SAH and the mitochondria in TNF-induced hepatotoxicity. We hypothesized that inhibition of mitochondrial SAM transport and subsequent mitochondrial SAM depletion by intracellular SAH accumulation plays a critical role in SAH-induced sensitization to TNF hepatotoxicity in ALD. Consistent with our hypothesis, we demonstrate here that chronic alcohol feeding not only increased cytosolic SAH concentration, but more importantly, significantly decreased the mitochondrial SAM pool. Moreover, we show that hepatic accumulation of SAH via DZA inhibition of SAHH sensitized to LPS/TNF induced hepatotoxicity in vivo, further supporting our previous in vitro observations. Our current in vitro results using both primary rat hepatocytes and human hepatocyte cell lines demonstrate that intracellular accumulation of SAH decreased the mitochondrial SAM concentration and sensitized to TNF induced hepatotoxicity. Together with our observation that inhibitors of the mitochondrial SAM transporter decreased SAM transport into mitochondria and sensitized hepatyctes to TNF cytotoxicity, our study suggests that inhibition of the mitochondrial SAM transporter and subsequent depletion of the mitochondrial SAM pool by increased intracellular SAH are important in SAH induced sensitization to TNF hepatotoxicity.
SAM is produced from methionine and ATP by methionine adenosyltransferase (MAT). Alcohol-induced inhibition of gene expression and enzyme activity in liver-specific MAT has been reported both in patients and experimental animal models [2, 29], and involves both oxidative stress and endotoxemia[30, 31]. SAM is the source of the methyl groups in almost all biological methylation reactions. Transmethylation is a process by which the methyl group of SAM is transferred to a large variety of acceptors resulting in the formation of, among others, creatine, sarcosine, and methylated phospholipids, nucleic acids and proteins. The direct by-product of transmethylation reactions is SAH, which is subsequently split into adenosine and homocysteine by SAHH, the only reversible reaction in hepatic methionine metabolism [32, 33]. Since the thermodynamics of SAHH favor the synthesis of SAH, the reaction in vivo procedes in the direction of hydrolysis only if the products, adenosine and homocysteine, are rapidly removed [2, 34]. Therefore, alcohol-induced inhibition in methionine synthase activity [35], an enzyme for the remethylation of homocysteine to methionine, may at least in part account for the increased accumulation of intracellular homocysteine and SAH. Although SAM is produced only in the cytosol, transmethylation reactions occur both in the cytosol and in mitochondria. Since mitochondria have a relatively large pool of SAM and lack the enzyme required for SAM synthesis, MAT, a SAM transporter is needed to maintain normal mitochondrial SAM levels [21, 22]. Compartmentalization of SAM in isolated rat hepatocytes was first reported more than 2 decades ago [21], however, it was only recently demonstrated that rat liver mitochondria had a transport system for SAM whose activity can be inhibited by a close structural analog of SAM, such as SAH or sinefungin [24]. In a more recent study, Palmieri's group identified the human mitochondrial SAM transporter through over-expressing a human cDNA sequence of the mitochondrial SAM transporter in bacteria followed by purification and reconstitution of its product into phospholipid vesicles [23]. Their results showed that the SAM transporter gene was expressed in various human tissues including liver and was localized to the mitochondria. The physiological role of the SAM transporter is to exchange cytosolic SAM for mitochondrial SAH, and its activity can be inhibited by increased SAH levels in the incubation buffer. In the current study, we examined the effects of accumulation of cytoplasmic SAH on mitochondrial SAM levels using an intact human hepatocyte culture system. We found that inhibition of SAHH activity by DZA caused endogenous accumulation of cytoplasmic SAH, which subsequently led to depletion of the mitochondrial SAM pool. These results provide new evidence supporting the concept that alteration of cytoplasmic SAH levels plays a critical role in modulating intra-mitochondrial SAM levels.
Mitochondrial dysfunction has been implicated in a variety of liver diseases including ALD [7, 8, 36-40]. Selective depletion of the hepatocyte mitochondrial GSH pool by alcohol and the subsequent sensitization to TNF induced cytotoxicity has been postulated to be one etiologic factor in the development of ALD [7, 8]. Our current study is the first to report that chronic alcohol consumption caused depletion of the mitochondrial SAM pool. Furthermore, inhibition of the mitochondrial SAM transporter and subsequent depletion of the mitochondrial SAM pool by increased SAH levels, as observed in ALD, sensitized hepatocytes to TNF induced cell death. Since SAM is the major methyl group donor for almost all methylation reactions inside mitochondria [17, 18], maintenance of normal intra-mitochondrial SAM concentrations plays a pivotal role in regulating mitochondrial functions.
There are several functions for SAM after its transport into mitochondria. One of its main functions is to provide methyl groups for the synthesis of phosphatidylcholine from phosphatidylethanolamine [26]. Phosphatidylcholine is the major phospholipid component in the mitochondrial membrane, and its composition plays a critical role in maintaining membrane fluidity and integrity [41, 42]. The influence of alcohol on the mitochondrial membrane has been reported and postulated to be one of mechanisms explaining alcohol-induced selective mitochondrial GSH depletion and sensitization to TNF hepatotoxicity [7, 43].Studies using mitochondria from rat hepatocytes have shown that inhibition of the mitochondrial SAM transporter inhibited incorporation of the methyl group from SAM into phospholipid [24]. Therefore, it is possible that the depletion of the mitochondrial SAM pool by SAH causes the loss of integrity of the mitochondrial membrane leading to the sensitization to TNF induced hepatotoxicity. In addition to the synthesis of phospholipids, SAM is essential for the methylation of various types of nucleic acids present in the mitochondria. In mammals, methylation of DNA, tRNA and rRNA has been documented, and some have been shown to be essential for mitochondrial function [44-47]. SAM is also necessary for post-translational modifications of some mitochondrial proteins. Studies on knockout mice deficient in hepatic SAM synthetase (MAT1A-/-), and in cultured rat hepatocytes using a medium without methionine or in the presence of cycloleucine, a known inhibitor of MAT1 activity, have shown a post-translational downregulation of prohibitin (PHB1) and of subunits I and II of cytochrome c oxidase without any decrease in their mRNA transcripts. Furthermore, it has been shown that the decrease in PHB1 levels correlates with a loss of mitochondrial function [48]. PHB1 functions as a chaperone-like protein for the correct folding and assembly of subunits of the mitochondrial respiratory-chain complexes, and it must be methylated inside the mitochondria before it functions [49]. Therefore, mitochondrial SAM depletion could affect electron transfer along the mitochondrial respiratory-chain and result in oxidative stress inside mitochondria, representing another possible mechanism involved in sensitization to TNF hepatotoxicity by intracellular SAH accumulation.
In conclusion, our research demonstrates that chronic alcohol consumption caused not only hepatic SAH accumulation, but also depletion of the mitochondrial SAM pool in the liver. Inhibition of SAHH caused intra-hepatic accumulation of SAH and sensitized to LPS/TNF induced liver injury in vivo. In vitro studies demonstrated that intracellular accumulation of SAH sensitized hepatocytes to TNF cytotoxicity and that the hepatocyte death was associated with mitochondrial SAM depletion. These data provide important new information concerning potential interactions between abnormal hepatic methionine metabolism, mitochondrial dysfunction and TNF-induced hepatotoxicity in ALD. These data further support the concept that removal of intracellular SAH may be a potential therapy for ALD and deserves further investigation.
Acknowledgments
This research was supported by the National Institutes of Health (Z. Song, Z. Zhou, I. Deaciuc, and C. J. McClain), and the Department of Veterans Affairs (C. J. McClain).
Abbreviations
- SAH
S-adenosylhomocysteine
- SAM
S-adenosylmethionine
- MAT
methionine adenosyltranserase
- DZA
3'-deazaadenosine
- PLP
pyridoxal 5-phosphate
- SAHH
S-adenosylhomocysteine hydrolyse
- TUNEL
Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling
- GSH
glutathione
- ALD
alcoholic liver disease
- TNF
tumor necrosis factor α
- HPLC
high performance liquid chromatography
- LPS
lipopolysaccharide
- ELISA
enzyme-linked immunosorbent assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic hepatitis. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 2.Lu SC, Huang ZZ, Yang H, Mato JM, Avila MA, Tsukamoto H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in alcoholic rat liver. Am J Physiol Gastrointest Liver Physiol. 2000;279:G178–185. doi: 10.1152/ajpgi.2000.279.1.G178. [DOI] [PubMed] [Google Scholar]
- 3.Barak AJ, Beckenhauer HC, Kharbanda KK, Tuma DJ. Chronic ethanol consumption increases homocysteine accumulation in hepatocytes. Alcohol. 2001;25:77–81. doi: 10.1016/s0741-8329(01)00168-9. [DOI] [PubMed] [Google Scholar]
- 4.Diehl AM. Cytokine regulation of liver injury and repair. Immunol Rev. 2000;174:160–171. doi: 10.1034/j.1600-0528.2002.017411.x. [DOI] [PubMed] [Google Scholar]
- 5.Pastorino JG, Hock JB. Ethanol potentiates tumor necrosis factor alpha cytotoxicity in hepatoma cells and primary rat hepatocytes by promoting induction of the mitochondrial permeability transition. Hepatology. 2000;31:1141–1152. doi: 10.1053/he.2000.7013. [DOI] [PubMed] [Google Scholar]
- 6.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 7.Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 8.Matsumaru K, Ji C, Kaplowitz N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology. 2003;37:1425–1434. doi: 10.1053/jhep.2003.50230. [DOI] [PubMed] [Google Scholar]
- 9.Kim HH, Kim K. Enhancement of TNF-alpha-mediated cell death in vascular smooth muscle cells through cytochrome c-independent pathway by the proteasome inhibitor. FEBS Lett. 2003;535:190–194. doi: 10.1016/s0014-5793(02)03894-2. [DOI] [PubMed] [Google Scholar]
- 10.Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost. 2000;26:219–225. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 11.Lu SC, Tsukamoto H, Mato JM. Role of abnormal methionine metabolism in alcoholic liver injury. Alcohol. 2002;27:155–162. doi: 10.1016/s0741-8329(02)00226-4. [DOI] [PubMed] [Google Scholar]
- 12.Song Z, Zhou Z, Uriarte S, Wang L, Kang YJ, Chen T, Barve S, McClain CJ. Sadenosylhomocysteine sensitizes to TNF-alpha hepatotoxicity in mice and liver cells: a possible etiological factor in alcoholic liver disease. Hepatology. 2004;40:989–997. doi: 10.1002/hep.20412. [DOI] [PubMed] [Google Scholar]
- 13.Halsted CH, Villanueva JA, Devlin AM, Niemela O, Parkkila S, Garrow TA, Wallock LM, Shigenaga MK, Melnyk S, James SJ. Folate deficiency disturbs hepatic methionine metabolism and promotes liver injury in the ethanol-fed micropig. Proc Natl Acad Sci USA. 2002;99:10072–10077. doi: 10.1073/pnas.112336399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lieber CS, Robins SJ, Leo MA. Hepatic phosphatidylethanolamine methyltransferase activity is decreased by ethanol and increased by phosphatidylcholine. Alcohol Clin Exp Res. 1994;18:592–595. doi: 10.1111/j.1530-0277.1994.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 15.Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res. 2004;28:173–181. doi: 10.1097/01.ALC.0000108654.77178.03. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein JD. Methionine metabolism in liver diseases. Am J Clin Nutr. 2003;77:1094–1095. doi: 10.1093/ajcn/77.5.1094. [DOI] [PubMed] [Google Scholar]
- 17.Dubin DT, Green CM, Prince DL. In: Biochemis-try of S-Adenosylmethionine and Related Compounds. Usdin E, Borchardt RT, Creveling CR, editors. McMil-lan Press; London: 1982. pp. 289–296. [Google Scholar]
- 18.Gantt R, Schneider WC. In: Transmethylation. Usdin E, Borchardt RT, Creveling CR, editors. Elsevier/North-Holland; New York: 1979. pp. 465–471. [Google Scholar]
- 19.Crott JW, Choi SW, Branda RF, Mason JB. Accumulation of mitochondrial DNA deletions is age, tissue and folate-dependent in rats. Mutat Res. 2005;570:63–70. doi: 10.1016/j.mrfmmm.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Shadel GS. Coupling the mitochondrial transcription machinery to human disease. Trends Genet. 2004;20:513–519. doi: 10.1016/j.tig.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Farooqui JZ, Lee HW, Kim S, Paik WK. Studies on compartmentation of S-adenosyl-Lmethionine in Saccharomyces cerevisiae and isolated rat hepatocytes. Biochim Biophys Acta. 1983;757:342–351. [PubMed] [Google Scholar]
- 22.Sheid B, Bilik E. S-adenosylmethionine synthetase activity in some normal rat tissues and transplantable hepatomas. Cancer Res. 1968;28:2512–2515. [PubMed] [Google Scholar]
- 23.Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F. Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J. 2004;379:183–190. doi: 10.1042/BJ20031664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horne DW, Holloway RS, Wagner C. Transport of S-adenosylmethionine in isolated rat liver mitochondria. Arch Biochem Biophys. 1997;343:201–206. doi: 10.1006/abbi.1997.0167. [DOI] [PubMed] [Google Scholar]
- 25.Colell A, Garcia-Ruiz C, Morales A, Ballesta A, Ookhtens M, Rodes J, Kaplowitz N, Fernandez-Checa JC. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology. 1997;26:699–708. doi: 10.1002/hep.510260323. [DOI] [PubMed] [Google Scholar]
- 26.Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, Kharbanda KK, Ballinger S, Darley-Usmar V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol. 2006;291:G857–867. doi: 10.1152/ajpgi.00044.2006. [DOI] [PubMed] [Google Scholar]
- 27.Lieber CS, DeCarli LM, Sorrell MF. Experimental methods of ethanol administration. Hepatology. 1989;10:501–510. doi: 10.1002/hep.1840100417. [DOI] [PubMed] [Google Scholar]
- 28.Song Z, Zhou Z, Chen T, Hill D, Kang J, Barve S, McClain C. S-adenosylmethionine (SAMe) protects against acute alcohol induced hepatotoxicity in mice. J Nutr Biochem. 2003;14:591–597. doi: 10.1016/s0955-2863(03)00116-5. [DOI] [PubMed] [Google Scholar]
- 29.Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballeria J, Rodes J, Mato JM. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 30.Corrales F, Ochoa P, Rivas C, Martin-Lomas M, Mato JM, Pajares MA. Inhibition of glutathione synthesis in the liver leads to S-adenosyl-L-methionine synthetase reduction. Hepatology. 1991;14:528–533. [PubMed] [Google Scholar]
- 31.Avila MA, Mingorance J, Martinez-Chantar ML, Casado M, Martin-Sanz P, Bosca L, Mato JM. Regulation of rat liver S-adenosylmethionine synthetase during septic shock: role of nitric oxide. Hepatology. 1997;25:391–396. doi: 10.1002/hep.510250222. [DOI] [PubMed] [Google Scholar]
- 32.Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32:391–395. doi: 10.1016/s1357-2725(99)00139-9. [DOI] [PubMed] [Google Scholar]
- 33.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 34.Avila MA, Garcia-Trevijano ER, Martinez-Chantar ML, Latasa MU, Perez-Mato I, Martinez-Cruz LA, del Pino MM, Corrales FJ, Mato JM. S-Adenosylmethionine revisited: its essential role in the regulation of liver function. Alcohol. 2002;27:163–167. doi: 10.1016/s0741-8329(02)00228-8. [DOI] [PubMed] [Google Scholar]
- 35.Halsted CH, Villanueva J, Chandler CJ, Stabler SP, Allen RH, Muskhelishvili L, James SJ, Poirier L. Ethanol feeding of micropigs alters methionine metabolism and increases hepatocellular apoptosis and proliferation. Hepatology. 1996;23:497–505. doi: 10.1002/hep.510230314. [DOI] [PubMed] [Google Scholar]
- 36.Garcia-Ruiz C, Morales A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Checa JC, Ookhtens M, Kaplowitz N. Effects of chronic ethanol feeding on rat hepatocytic glutathione. Relationship of cytosolic glutathione to efflux and mitochondrial sequestration. J Clin Invest. 1989;83:1247–1252. doi: 10.1172/JCI114008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 39.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 40.Bardag-Gorce F, French BA, Dedes J, Li J, French SW. Gene expression patterns of the liver in response to alcohol: in vivo and in vitro models compared. Exp Mol Pathol. 2006;80:241–251. doi: 10.1016/j.yexmp.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol. 2004;82:113–128. doi: 10.1139/o03-073. [DOI] [PubMed] [Google Scholar]
- 42.Wright MM, Howe AG, Zaremberg V. Cell membranes and apoptosis: role of cardiolipin, phosphatidylcholine, and anticancer lipid analogues. Biochem Cell Biol. 2004;82:18–26. doi: 10.1139/o03-092. [DOI] [PubMed] [Google Scholar]
- 43.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Semin Liver Dis. 1998;18:389–401. doi: 10.1055/s-2007-1007172. [DOI] [PubMed] [Google Scholar]
- 44.Helm M, Brul'e H, Degoul F, Cepanec C, Leroux JP, Gieg'e R, Florentz C. The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA. Nucleic Acids Res. 1998;26:1636–1643. doi: 10.1093/nar/26.7.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Helm M, Gieg'e R, Florentz C. A Watson–Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry. 1999;38:13338–13346. doi: 10.1021/bi991061g. [DOI] [PubMed] [Google Scholar]
- 46.Pintard L, Bujnicki JM, Lapeyre B, Bonnerot C. MRM2 encodes a novel yeast mitochondrial 21 S rRNA methyltransferase. EMBO J. 2002;21:1139–1147. doi: 10.1093/emboj/21.5.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sirum-Connolly K, Mason TL. Functional requirement of a site-specific ribose methylation in ribosomal RNA. Science. 1993;262:1886–1889. doi: 10.1126/science.8266080. [DOI] [PubMed] [Google Scholar]
- 48.Santamaria E, Avila MA, Latasa MU, Rubio A, Martin-Duce A, Lu SC, Mato JM, Corrales FJ. Functional proteomics of nonalcoholic steatohepatitis: mitochondrial proteins as targets of Sadenosylmethionine. Proc Natl Acad Sci USA. 2003;100:3065–3070. doi: 10.1073/pnas.0536625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial PHB complex: roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]