

Abstract
Computational analysis of eukaryotic promoters is one of the most difficult problems in computational genomics and is essential for understanding gene expression profiles and reverse-engineering gene regulation network circuits. Here I give a basic introduction of the problem and recent update on both experimental and computational approaches. More details may be found in the extended references. This review is based on a summer lecture given at Max Planck Institute at Berlin in 2005.
Background
The promoter of a gene is defined as the cis-regulatory DNA region at a specific location (the transcription start site, or TSS) that can drive the transcription of its target gene in response to environmental signals. Computationally, it is often conveniently divided into three regions: the core-promoter (~80–100 bp surrounding the TSS), the proximal-promoter (~250–1000 bp upstream of the core-promoter) and the distal-promoter (further upstream, normally excluding enhancer or other regulatory regions whose influences are position/orientation independent). The core-promoter is minimally required for the assembly of the preinitiation complex (PIC) and can drive a reporter gene at a basal level from the TSS. The proximal-promoter often contains major cis-regulatory elements for driving activated reporter gene expression with some tissue-specificity. However, the distal-promoter together with distal enhancers/silencers and insulators are often necessary for accurately reproducing the endogenous gene expression patterns in vivo, especially for early developmental genes. Distal cis-regulatory elements also occur in the introns and the downstream regions, and therefore computational studies of these regions have been difficult and often limited to only the conserved sub-regions and/or regions in which functional cis-regulatory elements form clusters. Most of our work has been focused on 1 kb proximal-promoters (defined as -700 to +300 with respect to the TSS). We have shown that DNA motifs in this region can predict tissue-specific gene expression [1]. Computational promoter analyses usually face two related problems: the localization of the core-promoter (TSS prediction) and the identification of cis-regulatory elements (motif discovery). Basic computational methods have been reviewed previously [2], here I emphasize some recent developments.
Results
New experimental developments
One recent surprise, revealed after more detailed biochemistry studies of promoter activation, is that people have underestimated the diversity and complexity of core-promoter architecture and regulation. I refer readers to the recent comprehensive review on "the general transcription machinery and general cofactors" [3].
Although several core-promoter elements have been identified (Figure 1), with each element being short and degenerate and not every element occurring in a given core-promoter, the combinatorial regulatory code within core-promoters remains elusive. Their predictive value has also been very limited, despite some weak statistical correlations among certain subsets of the elements which were uncovered recently [4,5]. Further biochemical characterization of core-promoter binding factors under various functional conditions is necessary before a reliable computational classification of core-promoters becomes possible. An example of the type of question that must be answered is how CK2 phosphorylation of TAF1 may switch TFIID binding specificity from a DCE to DPE function [6] (Figure 1).
Figure 1.
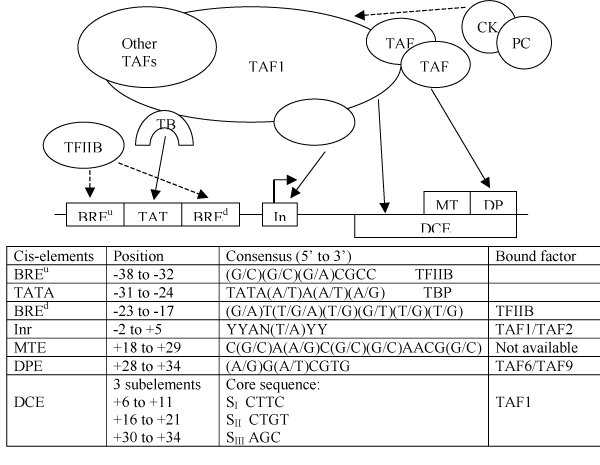
Regulation of core-promoter elements by TFIID and TFIIB (adapted from Fig. 2 of Thomas & Chiang 2006 [3]).
The most significant advance comes from the new sequencing and microarray technologies that, for the first time, can provide ample and accurate 5'UTR sequence and core-promoter/TFBS location data. In particular, large-scale 5'RACE technology at Tokyo University and 5'CAGE tag technology at Riken have provided DBTSS (Database of Transcriptional Start Sites, mainly human) [7] and Fantom (Functional Annotation of Mouse) [8,9] with an order of magnitude more promoter sequences derived from full-length 5'UTRs/cDNAs than were present in the traditional part of EPD (Eukaryotic Promoter Database) [10]. These sequences serve as the best training data for all current computational studies in promoter recognition. Many of the surprising new statistical features of the core-promoter have come from the recent analyses of such data (see [11] for a nice updated summary). One particularly interesting point made in this reference is that "Contrary to expectations, only a small fraction of RNAP II promoters appear to contain a TATA box. In contrast, a large proportion of RNAP II promoters in metazoan genomes appear to contain an INR element. Finally, about 25% of human promoters appear to lack known core promoter elements. This may point to the existence of additional core promoter sequence elements that remain to be identified and functionally characterized.". More mammalian promoter statistics are discussed in [12] which presents a comprehensive study of Fantom3 data.
In addition to sequence data, ChIP-chip technologies (e.g. see review [13]) provide genome-wide in vivo mapping of protein-DNA binding regions which provide the best experimental data for all current computational studies in cis-regulatory motif discovery. Most of the important data for promoter prediction has come from the ChIP-chip localization of PIC at active core-promoters in the whole genome at sub-100 bp resolution [14]. When more such data are produced for different tissues/cells and development stages, it will transform the field of computational promoter prediction and genome regulation networks (further discussed below).
Advances in motif discovery
The traditional approach for finding cis-elements is to collect a set of (target gene) promoter sequences believed to be enriched by some common TFBS motifs. They may either be collected from the literature or from systematic experiments (such as SELEX, etc.). There are many de novo TFBS motif finding algorithms available. For a recent review on computational TFBS finding methods, see e.g., [15]. For a recent benchmark of some popular motif finders, see [16]. In addition to the classical alignment-based motif finding algorithms, such as CONSENSUS [17], EM [18]/MEME [19] and the Gibbs sampler [20] which have been reviewed previously [21], most modern approaches have tried to extend either to the discovery of motif combinations (called cis-regulatory modules or CRMs), the use of evolutionary conservation information (with either phylogenetic footprinting or shadowing approaches), or a combination of both approaches. One can also increase specificity by incorporating structural information, for example, if the protein binds as a homodimer, one could restrict the search to only the palindromic motifs.
More powerful and flexible motif finders can take the advantage of a separate sequence set called a background set, serving as a negative control. The goal is to search only for motifs that are most discriminating, i.e. only those enriched in the foreground set relative to the background set. Examples of such motif finders, called discriminant motif finders, include: ANN-Spec [22], DMOTIFS [23], DWE [24] and DME [25]. DME is particularly novel and powerful; it can enumerate all possible (discretized) weight matrices above user-defined minimum information content. A newer version (called DME-B [26]) of DME can optimize the classification ability of the identified motifs based on whether or not the sequence contains at least one occurrence of the motif. This technology has been used to systematically catalog of mammalian tissue-specific TFBS motifs [27,28].
The most powerful generalization of this idea would be to turn motif finding into a feature selection problem in regression analysis by asking what is the set of features X (some functions of the motifs or CRMs) that can best explain the microarray data Y (e.g. expression scores). This is very similar to the general problem in genetics: Y represents the phenotype (mRNA expression) and X represents the genotype (promoter DNA elements). One would like to learn a model (function f) so that f(X) can best predict Y. When "best" is measured by the average squared error based on the distribution Pr(X, Y), the solution is the conditional expectation (also known as the regression function, see, e.g. [29]): f(X) = E (Y| X = x). REDUCE was the first successful motif selection algorithm based on linear regression [30]. It has now been generalized to include cross-interaction terms [31], to use nucleotide weight matrices discovered by MDscan (Motif Regressor [32]), to apply logistic regression [33] and to a nonlinear model based on regression trees called MARSMotif [34,35]. The matrix version of REDUCE (called MatrixREDUCE [36]) and of MARSMotif (called MARSMotif-M [37]) are becoming important motif discovery tools for mammalian promoter analyses. Almost all the tools developed for analyzing expression microarray data can also be easily applied to the analysis of localization data, such as ChIP-chip data. Although ChIP-chip is a global measurement for in vivo binding of proteins to chromatin DNA and hence is potentially capable of revealing direct target genes (most targets identified in expression arrays are not direct targets); due to the current resolution and to non-specific or non-functional cross-links, not all putative targets are functional or possess functional cis-elements. ChIP-chip data have also been used to further refine motifs found by expression data (e.g. using a boosting approach [38]).
Better promoter prediction
A number of statistical and machine learning approaches that can discriminate between the known promoter and some non-promoter sequences have been applied to TSS prediction. In a recent large scale comparison [39], eight prediction algorithms were compared. Among the most successful algorithms were Eponine [40] (which trains Relevant Vector Machines to recognize a TATA-box motif in a G+C rich domain and uses Monte Carlo sampling), McPromoter [41] (based on Neural Networks, interpolated Markov models and physical properties of promoter regions), FirstEF [42] (based on quadratic discriminant analysis of promoters, first exons and the first donor site) and DragonGSF [43,39] (based on artificial neural networks). However, DragonGSF is not publicly available and uses additional binding site information based on the TRANSFAC database [44], exploiting specific information that is typically not available for unknown promoters.
Two new de novo promoter prediction algorithms have emerged that further improve in accuracy. One is ARTS [45], which is based on Support Vector Machines with multiple sophisticated sequence kernels. It claims to find about 35% true positives at a false positive rate of 1/1000, where the above mentioned methods find only about half as many true positives (18%). ARTS uses only downstream genic sequences as the negative set (non-promoters), and therefore it may get more false-positives from upstream non-genic regions. Furthermore, ARTS does not distinquish if a promoter is CpG-island related or not and it is not clear how ARTS may peform on non-CpG-island related promoters. Another novel TSS prediction algorithm is CoreBoost [46] which is based on simple LogitBoosting with stumps. It has a false positive rate of 1/5000 at the same sensitivity level (Zhao, personal communication). CoreBoost uses both immediate upstream and downstream fragments as negative sets and trains separate classifiers for each before combining the two. The training sample is 300 bp fragments (-250, +50), hence it is more localized than ARTS which has training sample of 2 kb fragments (-1 kb, +1 kb). The ideal application of TSS prediction algorithms is to combine them with gene prediction algorithms [21] and/or with the ChIP-chip PIC mapping data [14].
Future direction: epigenetics and chromatin states
Although much progress has been made in promoter prediction and cis-regulatory motif discovery, false-positives are still the main problem when scanning through the whole genome. Fundamentally this is because the information about chromatin structure is still missing in all our models! Protein-DNA binding specificity is partly determined by the energetics and partly determined by "entropy", which depends on how much of the genome is accessible to the DNA binding protein [47] Without knowing which regions of chromatin are open or closed (and to what degree), researchers have to assume the whole genome is accessible for binding, which is obviously wrong and will lead to more false positives (and false negatives because of the extra noise). This is clearly shown by recent genome-wide ChIP-chip data as well as DNase I Hypersensitivity mapping data. There is a necessity for higher order prediction algorithms that are capable of predicting chromatin states based upon, perhaps, genome-wide epigenetic measurements, CpG-islands and repeat characteristics in addition to genomic sequences. It is fortunate that such kinds of data are rapidly being generated [48-54] and the corresponding analysis tools [55-57] are also coming along. The days of more realistic dynamic modeling of chromatin structure and its relation to expression and regulation are finally coming.
Acknowledgments
Acknowledgements
I would like to thank Dr. Dustin Schones for his careful proof-reading of the manuscript. Work in my lab is partially supported by grants from NSF, NIH and Dart NeuroGenomics Alliance.
This article has been published as part of BMC Bioinformatics Volume 8 Supplement 6, 2007: Otto Warburg International Summer School and Workshop on Networks and Regulation. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2105/8?issue=S6
References
- Smith AD, Sumazin P, Xuan Z, Zhang MQ. DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc Natl Acad Sci USA. 2006;103:6275–6280. doi: 10.1073/pnas.0508169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MQ. In: Computational Methods for Promoter Recognition. Jiang T, Xu Y, Zhang MQ, editor. MIT Press, Cambridge, Massachusetts; pp. 249–268. [Google Scholar]
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–78. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- Jin VX, Singer GA, Agosto-Perez FJ, Liyanarachchi S, Davuluri RV. Genome-wide analysis of core promoter elements from conserved human and mouse orthologous pairs. BMC Bioinformatics. 2006;7:114. doi: 10.1186/1471-2105-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershenzon NI, Trifonov EN, Ioshikhes IP. The features of Drosophila core promoters revealed by statistical analysis. BMC Genomics. 2006;7:161. doi: 10.1186/1471-2164-7-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Sims RJ, 3rd, Lane WS, Reinberg D. Functional characterization of core promoter elements: DPE-specific transcription requires the protein kinase CK2 and the PC4 coactivator. Mol Cell. 2005;18:471–481. doi: 10.1016/j.molcel.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Yamashita R, Sugano S, Nakai K. DBTSS, DataBase of Transcriptional Start Sites: Progress Report 2004. Nucleic Acids Res. 2004;32:D78–81. doi: 10.1093/nar/gkh076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Kasukawa T, Oyama R, Gough J, Frith M, Engstrom PG, Lenhard B, Aturaliya RN, Batalov S, Beisel KW, Bult CJ, Fletcher CF, Forrest AR, Furuno M, Hill D, Itoh M, Kanamori-Katayama M, Katayama S, Katoh M, Kawashima T, Quackenbush J, Ravasi T, Ring BZ, Shibata K, Sugiura K, Takenaka Y, Teasdale RD, Wells CA, Zhu Y, Kai C, Kawai J, Hume DA, Carninci P, Hayashizaki Y. Transcript annotation in FANTOM3: mouse gene catalog based on physical cDNAs. PLoS Genet. 2006;2:e62. doi: 10.1371/journal.pgen.0020062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, Forrest AR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y. Genomewide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Schmid CD, Perier R, Praz V, Bucher P. EPD in its twentieth year: towards complete promoter coverage of selected model organisms. Nucleic Acids Res. 2006;34:D82–5. doi: 10.1093/nar/gkj146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross P, Oelgeschlager T. Core promoter-selective RNA polymerase II transcription. Biochem Soc Symp. 2006;73:225–36. doi: 10.1042/bss0730225. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Tan SL, Christoffels A, Schonbach C, Lipovich L, Yang L, Hofmann O, Kruger A, Hide W, Kai C, Kawai J, Hume DA, Carninci P, Hayashizaki Y. Mice and men: their promoter properties. PLoS Genet. 2006;2:e54. doi: 10.1371/journal.pgen.0020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Ren B. Genome-Wide Analysis of Protein-DNA Interactions. Annu Rev Genomics Hum Genet. 2006;7:81–102. doi: 10.1146/annurev.genom.7.080505.115634. [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–80. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5:276–87. doi: 10.1038/nrg1315. [DOI] [PubMed] [Google Scholar]
- Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Regnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005;23:137–44. doi: 10.1038/nbt1053. [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Hartzell GW, 3rd, Stormo GD. Identification of consensus patterns in unaligned DNA sequences known to be functionally related. Comput Appl Biosci. 1990;6:81–92. doi: 10.1093/bioinformatics/6.2.81. [DOI] [PubMed] [Google Scholar]
- Lawrence CE, Reilly AA. An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins. 1990;7:41–51. doi: 10.1002/prot.340070105. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. The value of prior knowledge in discovering motifs with MEME. Proceedings of the International Conference on Intelligent Systems for Molecular Biology. 1995;3:21–9. [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–14. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. Computational Prediction of Eukaryotic Protein-Coding Genes. Nat Rev Genet. 2002;3:698–709. doi: 10.1038/nrg890. [DOI] [PubMed] [Google Scholar]
- Workman CT, Stormo GD. ANN-Spec: A method for discovering transcription factor binding sites with improved specificity. Pacific Symposium on Biocomputing. 2002. pp. 467–78. [DOI] [PubMed]
- Sinha S. Discriminative motifs. J Comput Biol. 2003;10:599–615. doi: 10.1089/10665270360688219. [DOI] [PubMed] [Google Scholar]
- Sumazin P, Chen G, Hata N, Smith AD, Zhang T, Zhang MQ. DWE: Discriminating word enumerator. Bioinformatics. 2005;21:31–8. doi: 10.1093/bioinformatics/bth471. [DOI] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc Natl Acad Sci USA. 2005;102:1560–5. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Das D, Zhang MQ. Mining ChIP-chip data for transcription factor and cofactor binding sites. Bioinformatics. 2005;21:i403–12. doi: 10.1093/bioinformatics/bti1043. [DOI] [PubMed] [Google Scholar]
- Martinez MJ, Smith AD, Li B, Zhang MQ, Harrod KS. Computational prediction of novel components of lung transcriptional networks. Bioinformatics. 2007;23:21–29. doi: 10.1093/bioinformatics/btl531. [DOI] [PubMed] [Google Scholar]
- Smith AD, Sumazin P, Zhang MQ. Tissue-specific regulatory elements in mammalian promoters. Mol Syst Biol. 2007;3:73. doi: 10.1038/msb4100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. Springer, New York; 2001. [Google Scholar]
- Bussemaker HJ, Li H, Siggia ED. Regulatory element detection using correlation with expression. Nat Genet. 2001;27:167–71. doi: 10.1038/84792. [DOI] [PubMed] [Google Scholar]
- Keles S, van der Laan M, Eisen MB. Identification of regulatory elements using a feature selection method. Bioinformatics. 2002;18:1167–75. doi: 10.1093/bioinformatics/18.9.1167. [DOI] [PubMed] [Google Scholar]
- Conlon EM, XS Liu, JD Lieb, JS Liu. Integrating regulatory motif discovery and genome-wide expression analysis. Proc Natl Acad Sci USA. 2003;100:3339–44. doi: 10.1073/pnas.0630591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keles S, van der Laan MJ, Vulpe C. Regulatory motif finding by logic regression. Bioinformatics. 2004;20:2799–811. doi: 10.1093/bioinformatics/bth333. [DOI] [PubMed] [Google Scholar]
- Friedman J. Multivariate adaptive regression splines. Ann Stat. 1991;19:1–141. [Google Scholar]
- Das D, Banerjee N, Zhang MQ. Interacting models of cooperative gene regulation. Proc Natl Acad Sci USA. 2004;101:16234–9. doi: 10.1073/pnas.0407365101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foat BC, Morozov AV, Bussemaker HJ. Statistical mechanical modeling of genome-wide transcription factor occupancy data by MatrixREDUCE. Bioinformatics. 2006;22:e141–9. doi: 10.1093/bioinformatics/btl223. [DOI] [PubMed] [Google Scholar]
- Das D, Nahle Z, Zhang MQ. Adaptively inferring human transcriptional subnetworks. Mol Syst Biol. 2006;2 doi: 10.1038/msb4100067. 2006.0029. Epub Jun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong P, Liu XS, Zhou Q, Lu X, Liu JS, Wong WH. A boosting approach for motif modeling using ChIP-chip data. Bioinformatics. 2005;21:2636–43. doi: 10.1093/bioinformatics/bti402. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Tan SL, Suzuki Y, Sugano S. Promoter prediction analysis on the whole human genome. Nat Biotechnol. 2004;22:1467–73. doi: 10.1038/nbt1032. [DOI] [PubMed] [Google Scholar]
- Down TA, Hubbard TJ. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 2002;12:458–61. doi: 10.1101/gr.216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Liao GC, Niemann H, Rubin GM. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-12-research0087. RESEARCH0087. Epub 2002 Dec 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Grosse I, Zhang MQ. Computational identification of promoters and first exons in the human genome. Nat Genet. 2001;29:412–417. doi: 10.1038/ng780. Erratum: Nat Genet 2002, 32(3):459. [DOI] [PubMed] [Google Scholar]
- Bajic VB, Seah SH. Dragon Gene Start Finder identifies approximate locations of the 5' ends of genes. Nucleic Acids Res. 2003;31:3560–3. doi: 10.1093/nar/gkg570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, Voss N, Stegmaier P, Lewicki-Potapov B, Saxel H, Kel AE, Wingender E. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–10. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg S, Zien A, Ratsch G. ARTS: accurate recognition of transcription starts in human. Bioinformatics. 2006;22:e472–80. doi: 10.1093/bioinformatics/btl250. [DOI] [PubMed] [Google Scholar]
- Zhao X, Xuan Z, Zhang MQ. Boosting with stumps for predicting transcription start sites. Genome Biol. 2007;8:R17. doi: 10.1186/gb-2007-8-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MJ, Lieb JD. A chromatin-mediated mechanism for specification of conditional transcription factor targets. Nat Genet. 2006;38:1446–51. doi: 10.1038/ng1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebert DJ, Bernstein BE. Genomic views of chromatin. Curr Opin Genet Dev. 2005;15:476–81. doi: 10.1016/j.gde.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Yuan GC, Liu YJ, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ. Genome-scale identification of nucleosome positions in S. cerevisiae. Science. 2005;309:626–30. doi: 10.1126/science.1112178. [DOI] [PubMed] [Google Scholar]
- Rollins RA, Haghighi F, Edwards JR, Das R, Zhang MQ, Ju J, Bestor TH. Large-scale structure of genomic methylation patterns. Genome Res. 2006;16:157–63. doi: 10.1101/gr.4362006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze SR, Wallrath LL. Gene Regulation by Chromatin Structure: Paradigms Established in Drosophila melanogaster. Annu Rev Entomol. 2007;52:171–92. doi: 10.1146/annurev.ento.51.110104.151007. [DOI] [PubMed] [Google Scholar]
- Cavalli G. Chromatin and epigenetics in development: blending cellular memory with cell fate plasticity. Development. 2006;133:2089–94. doi: 10.1242/dev.02402. [DOI] [PubMed] [Google Scholar]
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, Weaver M, Shafer A, Lee K, Neri F, Humbert R, Singer MA, Richmond TA, Dorschner MO, McArthur M, Hawrylycz M, Green RD, Navas PA, Noble WS, Stamatoyannopoulos JA. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–8. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J. CpG island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA structure. PLoS Genet. 2006;2:e26. doi: 10.1371/journal.pgen.0020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Dimitrova N, Xuan Z, Rollins RA, Haghighi F, Edwards JR, Ju J, Bestor TH, Zhang MQ. Computational prediction of methylation status in human genomic sequences. Proc Natl Acad Sci USA. 2006;103:10713–6. doi: 10.1073/pnas.0602949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang JP, Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–8. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]