

Abstract
c-Jun N-terminal kinases (JNKs), also referred to as stress-activated kinases (SAPKs), were initially characterized by their activation in response to cell stress such as UV irradiation. JNK/SAPKs have since been characterized to be involved in proliferation, apoptosis, motility, metabolism and DNA repair. Dysregulated JNK signaling is now believed to contribute to many diseases involving neurodegeneration, chronic inflammation, birth defects, cancer and ischemia/reperfusion injury. In this review, we present our current understanding of JNK regulation and their involvement in homeostasis and dysregulation in human disease.
Keywords: c-Jun N-terminal Kinase (JNK), Stress-activated Protein Kinase (SAPK), ischemia, apoptosis, metabolic regulation, obesity, neurodegeneration, chronic inflammation
Introduction
JNK/SAPKs phosphorylate c-Jun at the NH2-terminal Ser63 and 73 residues in response to UV irradiation and other stress stimuli [1–5]. The importance of c-Jun as a member of the AP-1 transcription factors lead to an intense analysis of JNK regulation of AP-1 function. AP-1 transcription factors are heterodimers composed of Jun, Fos, Maf and ATF subunits [5]. c-Jun, ATF2 and ATF3 are substrates for phosphorylation by JNKs, which enhances AP-1 transcriptional control of specific gene expression [6].
There are three JNK genes (JNK1, JNK2, JNK3). JNK1 and JNK2 are ubiquitously expressed while JNK3 is restricted to brain, heart and testes [1, 4, 7, 8]. Differential splicing and exon usage results in multiple isoforms of the JNK1, 2 and 3 genes [9, 10]. Each JNK is expressed as a short form (46 kDa) and long form (54 kDa) [7]. The alternative forms of each JNK1, 2 and 3 appear to differ in their ability to bind and phosphorylate different substrate proteins [9, 10]. The different JNK isoforms can also be differentially activated [9, 10]. Targeted gene disruption of each JNK has also defined differential functions for JNK1, JNK2 and JNK3 in many different cell types involving gene expression, apoptosis, metabolism and other critical physiological responses [8, 11–13].
JNK/SAPK Regulatory Network
As with all MAPKs, the JNKs are part of a three kinase module. The TXY motif in the activation loop of each JNK is dually phosphorylated by specific MAPK kinases (MKKs). MKK4 and MKK7 phosphorylate the threonine and tyrosine within the activation loop TXY motif (T183 and Y185 in JNK1) resulting in JNK activation. MKKs are in turn phosphorylated at specific serine or threonine residues within their activation loop (S257 and T265 for MKK4; S271 and T275 for MKK7).
There are at least 20 MKKKs of which at least 14 activate the MKK4/MKK7-JNK/MAPK pathway. Several of the MKKKs capable of phosphorylating and activating either MKK4 and MKK7 in the JNK pathway are also capable of activating other MKKs including MKK1 and 2 for ERK1/2 activation, MKK3 and 6 for p38 activation, and MKK5 for ERK5 activation (Fig. 1). The twenty MKKKs outnumber the 7 MKKs and 11 MAPKs. The MKKKs are differentially regulated by protein-protein interactions and covalent modifications. Many MKKKs are activated by GTPases such as Ras (the Raf MKKKs) or Rho family GTPases (MLK3 by Cdc42, MEKK1 by RhoA) [14–16]. Ubiquitination may also regulate the activity of MKKKs; including MEKK1 auto-ubiquitination that inhibits its activity [17] or ubiquitination associated with assembly of signaling complexes such as the IL-1 receptor signaling complex for TAK1 [18]. Many of the MKKKs also are phosphorylated by MKKKKs such as PAK, GCK and HPK that may be involved in controlling MKKK activity, interaction with other proteins and localization [19–21]. It is the diversity of MKKK regulation resulting from differential protein-protein interaction and covalent modifications that allow the integration of MAPK activation in the cellular response to a diverse range of stimuli. These stimuli include cytokines, growth factors, antigens, cell adhesion and cell-cell interactions, toxins, pharmacological drugs, and a range of stress stimuli including heat, cold, toxicity and shear. Of the 20 defined MKKKs it is notable that at least 14 regulate the JNK pathway, demonstrating the importance of JNK pathways in the cellular response to external stimuli.
Figure 1.
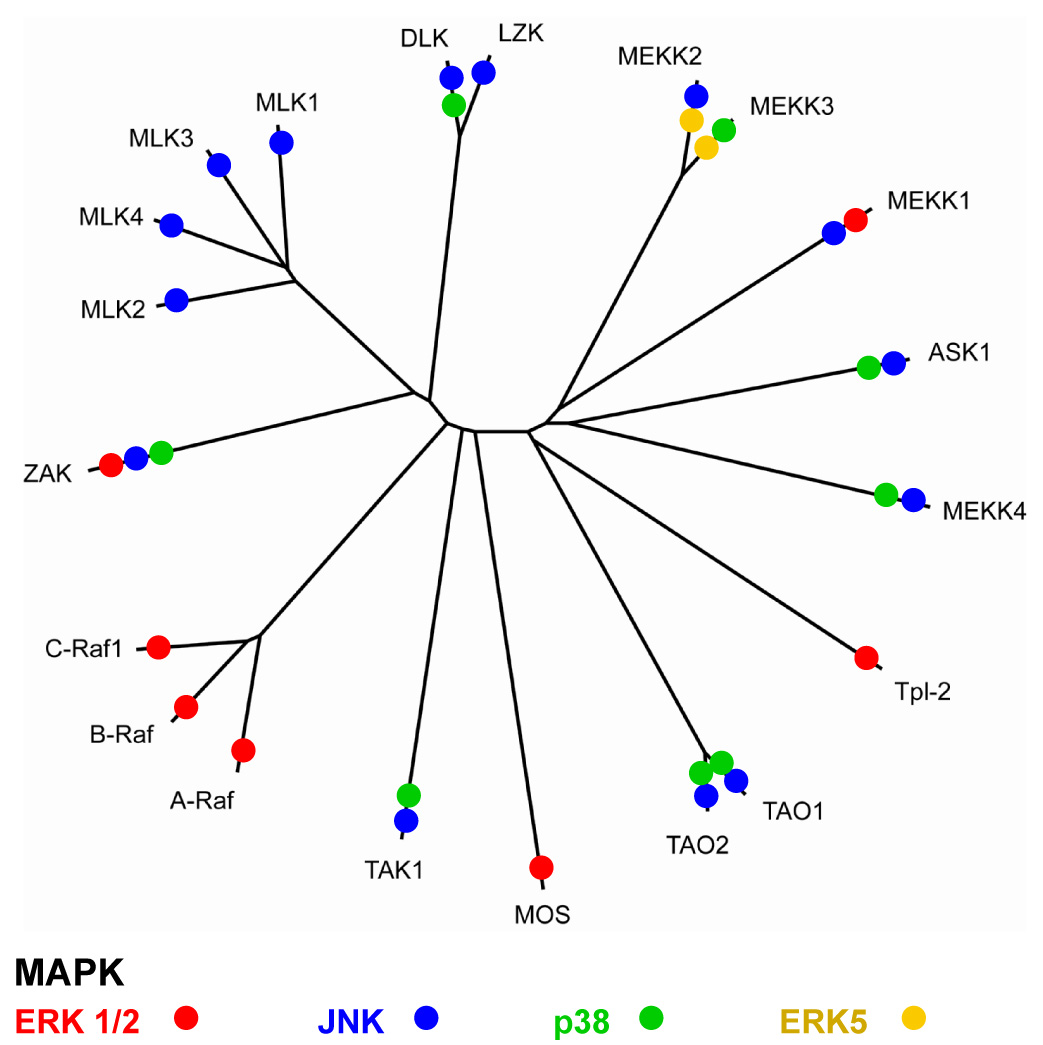
Dendrogram showing the relationships between the kinase domains of MKKKs and their ability to activate the JNK/SAPK pathway. MKKK regulation of different MAPK pathways is accumulated from literature survey with an emphasis on MKKK knockout data when available.
Organization of the JNK signaling module
JNK signaling modules are organized by two different mechanisms: i. recognition motif between MKKK and MKK and MKK and MAPKs; and ii. Scaffold proteins that assemble the MKKK-MKK-MAPK module in larger protein complexes (Fig. 2&Fig. 3).
Figure 2.
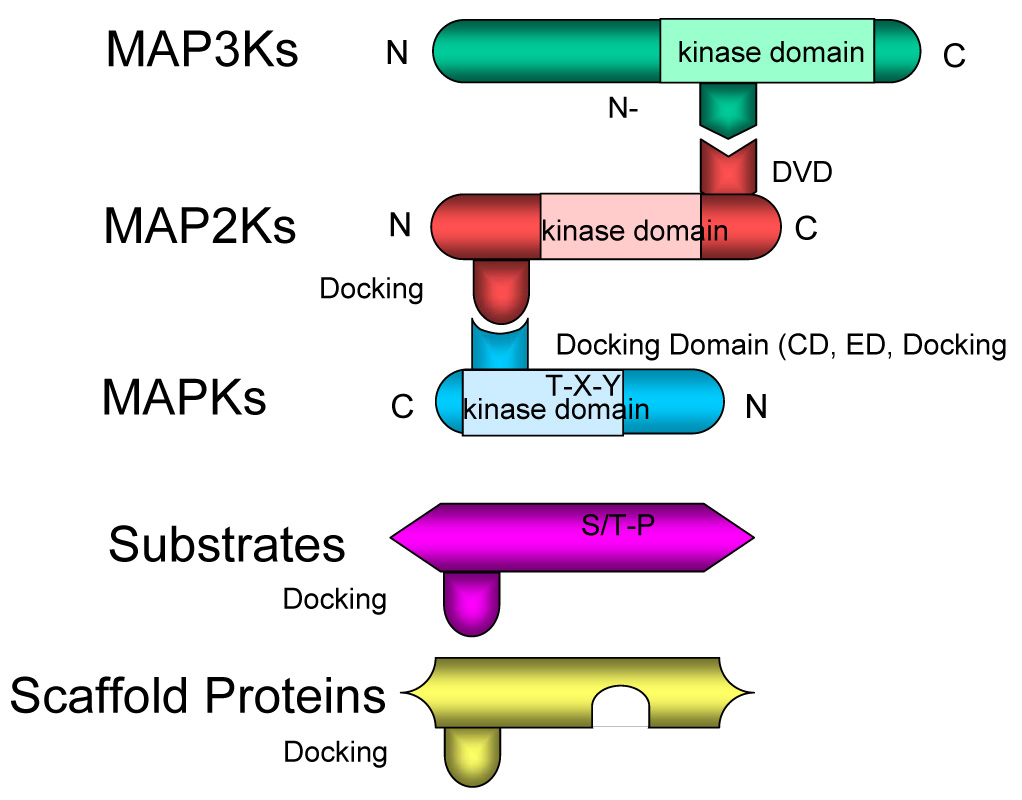

A) Organization of MKKK-MKK-MAPK interactions controlled by docking sites, docking domains (CD, ED and docking groove), DVD site and N lobe. See text for discussion. B) CD (common docking) domain (bottom) characterized by acidic residues in C-terminus of MAPK. ED site (middle) is far from CD domain in the primary sequence, the ED site and CD domain are in the same groove on the opposite side of catalytic center [25]. Crystal structures of p38 and MKK3b or MEF2A define a role of another docking groove (top) in binding to φXφ motif in docking sites [28]. Shown in red are the experimentally proved critical residues for docking site interaction.
Figure 3.
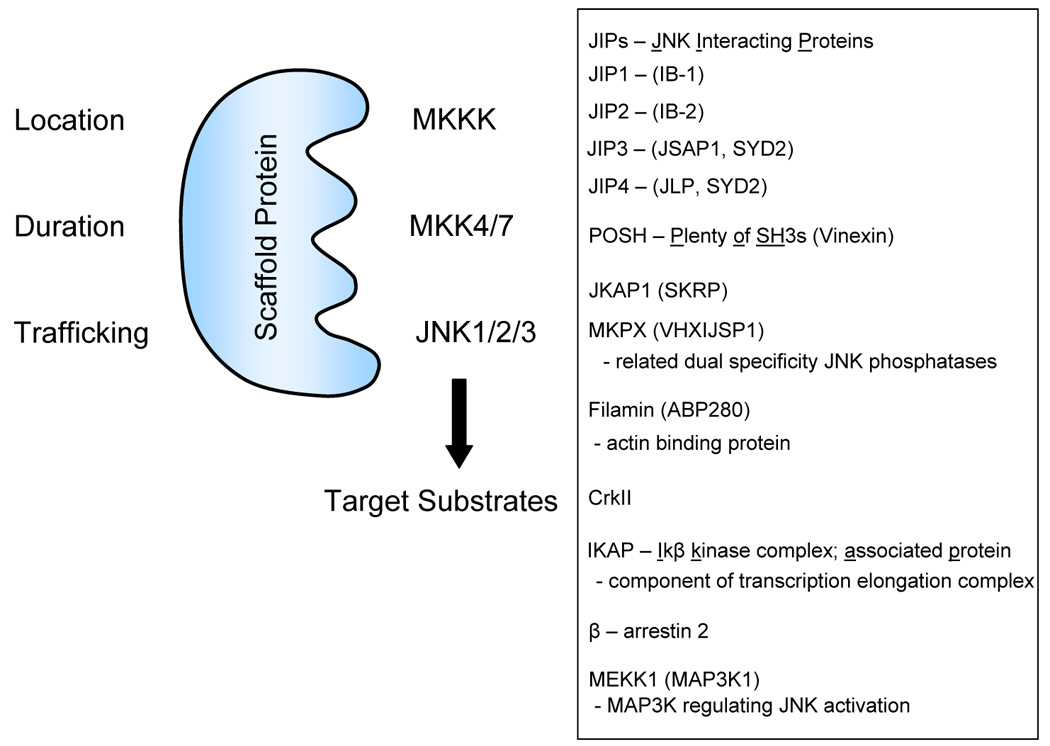
Function of JNK/SAPK scaffolds in organizing the MKKK-MKK4/MKK7- JNK signaling module. Known JNK scaffold proteins are listed in the box. See text for discussion.
Docking interactions are achieved via conserved sequences in the MAPKs (JNK, ERK1/2, p38, ERK5) that are selective for the specific upstream MKK (MKK4 and 7 for JNKs) and substrates such as c-Jun, ATF2 and others (see Table 1 for a list of representative JNK substrates). Fig 2 depicts the interaction between the different motifs in MKKKs, MKK4/7 and JNKs. On all MAPKs, including JNKs, there is a cluster of negatively charged amino acids C-terminal to the kinase domain sequence (Fig. 2B). This site is referred to as the common docking (CD) domain and is used in the docking of MKKs, MAPK phosphatases and specific substrates and scaffold proteins [22–27]. A second conserved motif referred to as the ED (Glu-Asp) domain, also contributes to the docking interactions of MAPKs [22–26]. Docking site sequences in MKKs, substrates, MAPK phosphatases and scaffolds share a conserved motif of R/K-X4-ØA-X-ØB (where ØA and ØB are the hydrophobic residues Leu, Ile or Val) [28]. Whereas the basic residues in this docking domain (DD) bind the acidic residues of the MAPK CD domain, the hydrophobic residues reside in a docking groove that engages the ØA-X-ØB hydrophobic motif of the DD motif. These docking site interactions provide the specificity for MAPK interactions and a role in the activation of the kinase [28].
Table 1. SAPK/JNK Substrates.
Table 1 represents a list of known JNK substrates. The list is not comprehensive but representative.
| Transcription |
| c-Jun |
| JunD |
| ATF2 |
| ATF3 |
| Elk 1 |
| Elk-3 |
| .p53 |
| RXRα |
| RARα |
| Androgen receptor |
| NFAT4 |
| HSF-1 |
| c-Myc |
| Nuclear Pore Complex |
| Nup214 |
| Apoptosis |
| Smac |
| Bim |
| Bmf |
| Bcl-2 |
| Bcl-XL |
| Mcl-1 |
| Microtubules |
| Tau |
| MAP1 |
| MAP2A |
| DCX (doublecortin) |
| Amyloid β Precursor Protein |
| Signaling |
| IRS-1 |
| Paxillin |
| 14-3-3 |
In addition to the docking interactions of MAPKs with MKKs, substrates and regulators there are specific motifs that control the interactions of MKKKs and MKKs (Figs. 2A&B) [29]. A docking site termed DVD (domain for versatile docking) is found in several MKKs including MKK4/7 for JNK regulation. The DVD site is near the extreme C-terminus of the MKK and was shown to be involved in binding MKKKs including MEKK1, MEKK4 (MTK1), ASK1, Tao2 and Tak1, all of which are able to activate the JNK pathway. The N lobe within the kinase domain of the MKKK was shown to bind to the MKK DVD site.
An additional aspect of JNK control is the organization of signaling complexes by scaffolding proteins (Fig. 3). Scaffolding proteins generally have no catalytic function themselves, but encode docking sites for binding members of the MAPK module-MKKKs, MKKs and MAPKs [27]. In general, scaffolds bind additional proteins through interaction motifs such as SH2, SH3, PTB, and other domains that target MAPK signaling complexes to different locations in the cell. It is often the scaffold protein and specific MKKK that provides the selectivity and spatio-temporal dynamics of MAPK activation by different stimuli. Fig 3 lists known scaffold proteins for the JNKs. Among the different scaffolds, JIPs (JNK Interacting Proteins) bind to specific kinesins and the MLK group of MKKKs and appear to be particularly important in cytoskeletal tracking of the JNK module in cells such as neurons [27, 30–32]. JNK scaffolds such as arrestins are recruited to phosphorylated G protein-coupled receptors [33], while POSH has multiple SH3 domains and is involved in JNK signaling in apoptosis in mammalian cells as well as Drosophila [34–36]. Crk II scaffolds JNK signaling at focal adhesions [37]. Finally, the MKKK, MEKK1, has been shown to bind both MKK4 and JNK1/2, indicating it has a scaffold-like function itself, much like PBS2 in the yeast response pathway for hyperosmotic stress [38, 39].
JNK/SAPK in disease
Roles for JNK/SAPK signaling in physiology and disease stems mostly from the knowledge gained from biochemical studies and mouse models where the different JNKs or upstream regulators have been deleted by targeted homologous recombination-gene knockouts [40]. These studies have provided the rationale for targeting JNKs and their upstream regulators including specific MKKKs for therapeutic intervention with small molecule or peptide inhibitors [41–43]. The use of JNK/SAPK inhibitors has further strongly supported an important role for the JNK/SAPK pathway in several different diseases. In particular, JNK inhibitors for the prevention of cell death induced by ischemia and other stress-induced apoptotic responses has shown significant therapeutic potential [41–44]. Below the evidence for JNK/SAPK inhibitors for therapeutic benefit in human disease is presented.
JNK signaling in excitotoxicity of hippocampal neurons
One of the first demonstrations for the role of JNKs in apoptosis was made with JNK3 knockout mouse [8]. JNK3 is highly expressed in the fore- and hind- brain regions in the mouse. JNK3 is also expressed in the heart, but unlike JNK1 and JNK2, it is not required for embryonic development. Normally, treatment of mice with excitotoxic agents like kainic acid causes a marked apoptotic death of hippocampal neurons. JNK 3−/− mice are resistant to glutamate-induced excitotoxicity, clearly demonstrating that JNK3 activation is a pro-apoptotic pathway in hippocampal neurons. Consistent with a role for JNK3 in excitotoxic neuronal cell death, mice expressing a mutant c-Jun having the JNK phosphorylation sites S63 and S73 mutated to alanines are resistant to glutamate-induced excitotoxicity. In contrast, JNK1−/− and JNK2−/− mice are similar to wild-type animals in their susceptibility to excitotoxic neuronal cell death [5, 44].
The role of JNK3 in excitotoxicity suggested that JNK/SAPK pathways would be involved in other neuronal death responses and neurodegenerative diseases. A clear role of JNK/SAPKs in ischemia-induced cell death has been demonstrated in mice. Borsello, et al [45, 46] used two different models of cerebral artery occlusion: a transient occlusion in adult animals and a permanent occlusion in 2 week old pups. Inhibition of JNK activity in both models protected against neuronal cell death. A similar study in gerbils using a transient ischemia model showed that intracerebral administration of SB203580, a rather non-selective JNK inhibitor, reduced ischemic cell death [47].
JNK in liver ischemia/reperfusion models:
The role of JNK/SAPK pathways in ischemia-induced cell injury goes beyond that in the CNS. Ischemia/reperfusion (I/R) injury is a major clinical problem in several organs including brain, heart, kidney and liver. Hepatic injury due to I/R occur during several conditions including transplantation, liver tumor resection and circulatory shock. A recent study by Uehara, et al [48] demonstrated that JNK/SAPK activity was a major mediator of hepatic I/R injury. In this study, three ATP-competitive, reversible and highly selective JNK inhibitors (CC0209766, CC0223105, and CC-401 from Signal Pharmaceuticals, Inc.) decreased both necrosis and apoptosis of hepatocytes and sinusoidal endothelial cells. The findings are consistent with JNK inhibitors blocking hepatocyte apoptosis in culture [48].
It has also been shown that the JNK inhibitor AS601245 decreases cardiomyocyte apoptosis and infarct size in a rat cardiac I/R model in rats [49]. JNK activation in this model occurs primarily in the reperfusion phase in response to the generation of reactive oxygen species. Similarly, the JNK inhibitor SP600125 was shown to improve I/R injury in the transplantation of rat lungs [50]. In each of these models, JNK is probably mediating the release of pro-apoptotic factors from mitochondria, may directly phosphorylate pro-apoptotic Bcl family members such as Bak and Bid, and induce the expression of pro-inflammatory cytokines such as TNFα, IL-1 and IL-6.
JNK in neurodegenerative diseases
JNK3 may also prove to be a therapeutic target for neurodegenerative diseases including Parkinson’s and Alzheimer’s disease [44, 46, 51–53]. The neurotoxin 1-methyl-4-pheny-1, 2, 4, 6- tetrahydropyridine (MPTP) induces a neuropathology with loss of dopaminergic neurons similar to that observed with Parkinson’s disease in humans [54]. Mice treated MPTP have activated c-Jun, as shown by phospho-c-Jun immunostaining. A similar activation of c-Jun is seen in dopaminergic neurons from Parkinson’s disease patients. JNK3−/− and JNK2−/−, but not JNK1−/− mice are protected from MPTP dopaminergic cell death. The double knockout JNK2−/−/JNK3−/− mice have a greater dopaminergic cell protection than either knockout alone. An important downstream target from JNK2 and JNK3-mediated dopaminergic cell death was the expression of cyclooxygenase (COX) 2.
Alzheimer’s disease is characterized by diffuse plaques primarily composed of β-amyloid peptide (Aβ) and neurofibrillary tangles composed of hyper-phosphorylated Tau protein [55–63]. JNK3−/− are resistant to Aβ-induced apoptosis [55]. In this model, JNK3 activates AP-1 dependent expression of Fas-ligand [55]. There is a correlation with JNK2 and JNK3 activity with neurofibriallary pathology including JNK phosphorylation of Tau and the formation of Tau fibrils induced by JNK3 [64–70]. There is some controversy as to the exact role of c-Jun phosphorylation by JNKs versus other substrates including Nap214, a nuclear pore complex protein, in neurodegenerative models [71].
Hearing loss/deafness
JNK/SAPKs appear to play an important role in acquired deafness [72]. Exposure of hair cells in the cochlea to acoustic trauma, aminoglycoside antibiotics and cancer chemotherapy can lead to both necrosis and apoptosis, ultimately leading to deafness. JNK inhibition using the D-JNK-1 inhibitor peptide provided otoprotective benefit in organ cultures of neonatal mouse cochlea and in adult guinea pigs exposed to acoustic trauma [73, 74], suggesting JNK/SAPK inhibitors may be important therapeutic targets for the prevention of cochlear hair cell loss and deafness.
Neural tube birth defects
Concurrent loss of JNK1 and JNK2 in the JNK1−/−/JNK2−/− double knockout results in defective neural tube development manifested primarily as exencephaly [11, 13]. The neurotube defect is not seen in either JNK−/− or JNK2−/− animals indicating a compensatory function in neural tube development for the two JNK/SAPKs. Exencephaly in the JNK1/2 double knockout embryos was due to enhanced apoptosis in the forebrain and decreased apoptosis in the hindbrain [11].
Further examination of the JNK1−/− and JNK2−/− mice demonstrated that the anterior commissure axons are absent in JNK1 but not JNK2 knockout animals [12]. This is due to a diminished microtubule-associated protein (MAP) 1 and 2 phosphorylation, where MAP1 and MAP2 are preferential JNK1 substrates relative to JNK2 [12]. Phosphorylated MAP1 and MAP2 promote microtubule polymerization. Thus, loss of their phosphorylation causes shortening of neuronal microtubules.
JNK/SAPK signaling pathway in cancer
The most insightful evidence for a role of JNK/SAPK signaling modules in cancer has come from the identification of MKK4 as a putative tumor suppressor [75–79]. MKK4 is a MKK capable of phosphorylating both JNKs and p38 MAPKs. This distinguishes MKK4 from MKK7 which has only been shown to phosphorylate JNKs and not p38 MAPKs. Genetic inactivation of the MKK4 gene on chromosome 17p has been reported for several different tumor types including a subset of breast, biliary and pancreatic carcinomas [75, 76]. In addition, in ovarian and prostatic carcinomas epigenetic loss of MKK4 expression has been shown to be correlated with metastasis [77–79]. More recently, studies with SKOV3ip.1 cells expressing kinase-inactive MKK4 showed suppression of experimental metastasis in nude mice [80]. In this study, overexpression of MKK6 (p38 pathway) but not MKK7 (JNK pathway) suppressed the SKOV3 cell growth in nude mice. The results were interpreted that the p38 pathway and not the JNK pathway, was the critical MAPK for the tumor suppressor activity of MKK4. Additional models using MKK4 and MKK6 deficient cells will be required to substantiate this interpretation.
In a mouse model, JNK1 deficiency has been shown to significantly decrease susceptibility to diethylnitrosamine-induced hepatocarcinogenesis [81]. JNK1−/− but not JNK2−/− mice were shown to have decreased expression of cyclin D and VEGF, diminished hepatocyte proliferation and reduced tumor neovascularization. The findings suggest that JNK1 inhibition may be useful for therapeutic intervention and chemoprevention of hepatocellular carcinoma.
Several studies suggest JNK can promote tumor cell growth and therefore, is a useful anti-tumor target [82–85]. Furthermore, JNK inhibitors block the induction of DNA repair genes in cells treated with DNA damaging drugs [86]. However, JNK signaling has also been shown to be critical for inducing apoptosis in response to chemotherapeutic drugs [87]. Thus, JNKs have opposing roles in promoting proliferation and transformation and inducing apopotosis [88]. It seems that the effectiveness of JNK inhibitors in treating human cancers may be very context dependent for the specific tumor and cell origin [88].
JNK/SAPK signaling in chronic inflammatory diseases
JNK/SAPKs appear to have a significant role in chronic inflammatory diseases involving the expression of specific proteases and cytokines [89, 90]. For example, JNK signaling appears to be involved in the expression of metalloproteases, which contributes to joint destruction in rheumatoid arthritis [91]. JNKs also appear to contribute to the stimulation of TNFα expression, a major pro-inflammatory cytokine in rheumatoid arthritis. JNK inhibitors such as SP600125 and JNK1 deficiency (JNK1−/− mice) suppressed metalloproteinase expression and protected mice from joint damage in rheumatoid arthritis animal models [91]. JNK2 deficiency (JNK2−/−) also suppressed matrix degradation in joints, but did not significantly decrease inflammation [92].
Atherosclerosis is a second chronic inflammatory disease that appears to involve JNK/SAPK signaling [93, 94]. Using the ApoE−/− mouse model for atherosclerosis, it was shown that deficiency of JNK2 (ApoE−/−/JNK2−/−) animals developed less atherosclerosis than control ApoE−/− animals. JNK1 deficiency did not reduce the level of atherosclerosis. JNK inhibitors were also able to reduce the level of atherosclerosis. The mechanism appears to be JNK2-dependent phosphorylation of the modified lipid-binding and internalizing scavenger A receptor (SR-A) that promotes uptake of lipids in macrophages contributing to foam cell formation. The findings suggest specific JNK2 inhibitors may decrease atherogenesis.
Obesity and insulin-resistant diabetes
A therapeutic benefit may be the inhibition of JNKs in metabolic regulation [95–97]. Obesity-induced insulin resistance and diabetes are now worldwide disorders and a growing health crisis. Insulin activation of its receptor stimulates the receptors tyrosine kinase activity. Insulin Receptor Substrate 1 (IRS1) is phosphorylated on tyrosines by the insulin receptor. The tyrosine phosphorylated IRS1 functions as a complex scaffolding protein recruiting numerous signaling proteins in the control of insulin signaling and metabolic regulation. The JNKs phosphorylate IRS1 at S307 that results in reduced insulin receptor catalyzed phosphorylation of IRS1 [98]. JNKs are activated by high fat diet and obesity, in part due to elevated levels of TNFα and IL-1 [95, 97]. JNK1−/− mice and JIP1−/− mice have improved insulin sensitivity that correlates with reduced S307 phosphorylation [95, 99]. JNK2−/− mice are similar to wild type mice in their response to a high fat diet. JIP1, is a scaffolding protein for JNK signaling, and interestingly, a JIP1 gene mutation has been found in patients with a form of type II diabetes [100]. Consistent with these findings, a cell permeable JNK inhibitory peptide was shown to maintain insulin sensitivity in the obese mouse model [42]. These findings indicate JNK1 may be an important target in the treatment of obesity and type II diabetes.
Conclusions
JNK/SAPKs are clearly involved in ischemia-induced cell death and reperfusion injury in several different tissues and the control of insulin sensitivity in metabolic regulation. There are many other suggestions in the literature that link JNK/SAPK signaling to additional human diseases such as type I diabetes, osteosarcoma, ataxia and immune system dysfunction. JNKs probably play a role in chronic inflammation, airway hyperesponsiveness and protease-directed tissue remodeling. It is likely that selective JNK1, JNK2 and JNK3 inhibitors will be needed for specificity and lack of toxicity. It may also be useful to develop specific MKKK inhibitors to selectively block JNK activation in response to different upstream inputs [19].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 2.Smeal T, Binetruy B, Mercola D, Grover-Bardwick A, Heidecker G, Rapp UR, Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Molecular and Cellular Biology. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 5.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nature genetics. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 6.Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 7.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 8.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 9.Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes & Development. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- 10.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. The EMBO Journal. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 11.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 12.Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Developmental Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 13.Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mechanisms of Development. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- 14.Gallagher ED, Gutowski S, Sternweis PC, Cobb MH. RhoA binds to the amino terminus of MEKK1 and regulates its kinase activity. The Journal of Biological Chemistry. 2004;279:1872–1877. doi: 10.1074/jbc.M309525200. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Cobb MH. Activation of MEKK1 by Rho GTPases. Methods in Enzymology. 2006;406:468–478. doi: 10.1016/S0076-6879(06)06035-6. [DOI] [PubMed] [Google Scholar]
- 16.Gallo KA, Johnson GL. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Cell Molecular Nature Reviews. 2002;3:663–672. doi: 10.1038/nrm906. [DOI] [PubMed] [Google Scholar]
- 17.Witowsky JA, Johnson GL. Ubiquitylation of MEKK1 inhibits its phosphorylation of MKK1 and MKK4 and activation of the ERK1/2 and JNK pathways. The Journal of Biological Chemistry. 2003;278:1403–1406. doi: 10.1074/jbc.C200616200. [DOI] [PubMed] [Google Scholar]
- 18.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 19.Johnson GL, Dohlman HG, Graves LM. MAPK kinase kinases (MKKKs) as a target class for small-molecule inhibition to modulate signaling networks and gene expression. Current Opinion in Chemical Biology. 2005;9:325–331. doi: 10.1016/j.cbpa.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiological Reviews. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 21.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends in Biochemical Sciences. 2006;31:268–275. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 22.Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. The Journal of Biological Chemistry. 2006;281:13169–13179. doi: 10.1074/jbc.M601010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes & Development. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- 24.Mooney LM, Whitmarsh AJ. Docking interactions in the c-Jun N-terminal kinase pathway. The Journal of Biological Chemistry. 2004;279:11843–11852. doi: 10.1074/jbc.M311841200. [DOI] [PubMed] [Google Scholar]
- 25.Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nature Cell Biology. 2000;2:110–116. doi: 10.1038/35000065. [DOI] [PubMed] [Google Scholar]
- 26.Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends in Biochemical Sciences. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- 27.Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annual Review of Cell and Developmental Biology. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 28.Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Molecular Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- 29.Takekawa M, Tatebayashi K, Saito H. Conserved docking site is essential for activation of mammalian MAP kinase kinases by specific MAP kinase kinase kinases. Molecular cell. 2005;18:295–306. doi: 10.1016/j.molcel.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Kelkar N, Standen CL, Davis RJ. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Molecular and Cellular Biology. 2005;25:2733–2743. doi: 10.1128/MCB.25.7.2733-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Molecular and Cellular Biology. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pellet JB, Haefliger JA, Staple JK, Widmann C, Welker E, Hirling H, Bonny C, Nicod P, Catsicas S, Waeber G, Riederer BM. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. The European Journal of Neuroscience. 2000;12:621–632. doi: 10.1046/j.1460-9568.2000.00945.x. [DOI] [PubMed] [Google Scholar]
- 33.Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends in endocrinology and metabolism: TEM. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 34.Tapon N, Nagata K, Lamarche N, Hall A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. The EMBO Journal. 1998;17:1395–1404. doi: 10.1093/emboj/17.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kukekov NV, Xu Z, Greene LA. Direct interaction of the molecular scaffolds POSH and JIP is required for apoptotic activation of JNKs. The Journal of Biological Chemistry. 2006;281:15517–15524. doi: 10.1074/jbc.M601056200. [DOI] [PubMed] [Google Scholar]
- 36.Xu Z, Kukekov NV, Greene LA. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. The EMBO Journal. 2003;22:252–261. doi: 10.1093/emboj/cdg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girardin SE, Yaniv M. A direct interaction between JNK1 and CrkII is critical for Rac1-induced JNK activation. The EMBO Journal. 2001;20:3437–3446. doi: 10.1093/emboj/20.13.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260:315–319. doi: 10.1126/science.8385802. [DOI] [PubMed] [Google Scholar]
- 39.Xu S, Robbins DJ, Christerson LB, English JM, Vanderbilt CA, Cobb MH. Cloning of rat MEK kinase 1 cDNA reveals an endogenous membrane-associated 195-kDa protein with a large regulatory domain; Proceedings of the National Academy of Sciences of the United States of America; 1996. pp. 5291–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuida K, Boucher DM. Functions of MAP kinases: insights from gene-targeting studies. Journal of Biochemistry. 2004;135:653–656. doi: 10.1093/jb/mvh078. [DOI] [PubMed] [Google Scholar]
- 41.Bogoyevitch MA. Therapeutic promise of JNK ATP-noncompetitive inhibitors. Trends in Molecular Medicine. 2005;11:232–239. doi: 10.1016/j.molmed.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 42.Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochimica et Biophysica Acta. 2004;1697:89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Waetzig V, Herdegen T. Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage. Trends in Pharmacological Sciences. 2005;26:455–461. doi: 10.1016/j.tips.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Resnick L, Fennell M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discovery Today. 2004;9:932–939. doi: 10.1016/S1359-6446(04)03251-9. [DOI] [PubMed] [Google Scholar]
- 45.Borsello T, Bonny C. Use of cell-permeable peptides to prevent neuronal degeneration. Trends in Molecular Medicine. 2004;10:239–244. doi: 10.1016/j.molmed.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 46.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature Medicine. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 47.Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, Nishida E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. Journal of Neuroscience. 2000;20:4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uehara T, Bennett B, Sakata ST, Satoh Y, Bilter GK, Westwick JK, Brenner DA. JNK mediates hepatic ischemia reperfusion injury. Journal of Hepatology. 2005;42:850–859. doi: 10.1016/j.jhep.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 49.Ferrandi C, Ballerio R, Gaillard P, Giachetti C, Carboni S, Vitte PA, Gotteland JP, Cirillo R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. British Journal of Pharmacology. 2004;142:953–960. doi: 10.1038/sj.bjp.0705873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishii M, Suzuki Y, Takeshita K, Miyao N, Kudo H, Hiraoka R, Nishio K, Sato N, Naoki K, Aoki T, Yamaguchi K. Inhibition of c-Jun NH2-terminal kinase activity improves ischemia/reperfusion injury in rat lungs. Journal of Immunolnology. 2004;172:2569–2577. doi: 10.4049/jimmunol.172.4.2569. [DOI] [PubMed] [Google Scholar]
- 51.Kuan CY, Burke RE. Targeting the JNK signaling pathway for stroke and Parkinson's diseases therapy. Current Drug Targets. 2005;4:63–67. doi: 10.2174/1568007053005145. [DOI] [PubMed] [Google Scholar]
- 52.Silva RM, Kuan CY, Rakic P, Burke RE. Mixed lineage kinase-c-jun N-terminal kinase signaling pathway: a new therapeutic target in Parkinson's disease. Movement Disorders. 2005;20:653–664. doi: 10.1002/mds.20390. [DOI] [PubMed] [Google Scholar]
- 53.Bonny C, Borsello T, Zine A. Targeting the JNK pathway as a therapeutic protective strategy for nervous system diseases. Reviews in the Neurosciences. 2005;16:57–67. doi: 10.1515/revneuro.2005.16.1.57. [DOI] [PubMed] [Google Scholar]
- 54.Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease; Proceedings of the National Academy of Sciences of the United States of America; 2004. pp. 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. Journal of Neuroscience. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith WW, Gorospe M, Kusiak JW. Signaling mechanisms underlying Abeta toxicity: potential therapeutic targets for Alzheimer's disease. CNS & Neurological Disorders Drug Targets. 2006;5:355–361. doi: 10.2174/187152706784111515. [DOI] [PubMed] [Google Scholar]
- 57.Muresan Z, Muresan V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-beta precursor protein. Journal of Neuroscience. 2005;25:3741–3751. doi: 10.1523/JNEUROSCI.0152-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kimberly WT, Zheng JB, Town T, Flavell RA, Selkoe DJ. Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. Journal of Neuroscience. 2005;25:5533–5543. doi: 10.1523/JNEUROSCI.4883-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D'Adamio L. Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's beta-amyloid precursor protein (APP) The Journal of Biological Chemistry. 2002;277:3767–3775. doi: 10.1074/jbc.M108357200. [DOI] [PubMed] [Google Scholar]
- 60.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiological Reviews. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 61.Pearson AG, Byrne UT, MacGibbon GA, Faull RL, Dragunow M. Activated c-Jun is present in neurofibrillary tangles in Alzheimer's disease brains. Neuroscience Letters. 2006;398:246–250. doi: 10.1016/j.neulet.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 62.Yao M, Nguyen TV, Pike CJ. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. Journal of Neuroscience. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, Smith MA. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. Journal of Neurochemistry. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 64.Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Current Topics in Medicinal Chemistry. 2006;6:579–595. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- 65.Ferrer I. Stress kinases involved in tau phosphorylation in Alzheimer's disease, tauopathies and APP transgenic mice. Neurotoxicity Research. 2004;6:469–475. doi: 10.1007/BF03033283. [DOI] [PubMed] [Google Scholar]
- 66.Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Current Alzheimer Research. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 67.Lambourne SL, Sellers LA, Bush TG, Choudhury SK, Emson PC, Suh YH, Wilkinson LS. Increased tau phosphorylation on mitogen-activated protein kinase consensus sites and cognitive decline in transgenic models for Alzheimer's disease and FTDP-17: evidence for distinct molecular processes underlying tau abnormalities. Molecular and Cellular Biology. 2005;25:278–293. doi: 10.1128/MCB.25.1.278-293.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reynolds CH, Utton MA, Gibb GM, Yates A, Anderton BH. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. Journal of Neurochemistry. 1997;68:1736–1744. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- 69.Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A. Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. The Journal of Biological Chemistry. 2002;277:42060–42065. doi: 10.1074/jbc.M202241200. [DOI] [PubMed] [Google Scholar]
- 70.Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) Journal of Neurochemistry. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]
- 71.Besirli CG, Wagner EF, Johnson EM., Jr The limited role of NH2-terminal c-Jun phosphorylation in neuronal apoptosis: identification of the nuclear pore complex as a potential target of the JNK pathway. The Journal of Cell Biology. 2005;170:401–411. doi: 10.1083/jcb.200501138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eshraghi AA, Van de Water TR. Cochlear implantation trauma and noise-induced hearing loss: Apoptosis and therapeutic strategies. The Anatomical Record. 2006;288:473–481. doi: 10.1002/ar.a.20305. [DOI] [PubMed] [Google Scholar]
- 73.Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. Journal of Neuroscience. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zine A, van de Water TR. The MAPK/JNK signalling pathway offers potential therapeutic targets for the prevention of acquired deafness. Current Drug Targets. 2004;3:325–332. doi: 10.2174/1568007043337166. [DOI] [PubMed] [Google Scholar]
- 75.Su GH, Hilgers W, Shekher MC, Tang DJ, Yeo CJ, Hruban RH, Kern SE. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Research. 1998;58:2339–2342. [PubMed] [Google Scholar]
- 76.Su GH, Song JJ, Repasky EA, Schutte M, Kern SE. Mutation rate of MAP2K4/MKK4 in breast carcinoma. Human Mutation. 2002;19:81. doi: 10.1002/humu.9002. [DOI] [PubMed] [Google Scholar]
- 77.Xin W, Yun KJ, Ricci F, Zahurak M, Qiu W, Su GH, Yeo CJ, Hruban RH, Kern SE, Iacobuzio-Donahue CA. MAP2K4/MKK4 expression in pancreatic cancer: genetic validation of immunohistochemistry and relationship to disease course. Clinical Cancer Research. 2004;10:8516–8520. doi: 10.1158/1078-0432.CCR-04-0885. [DOI] [PubMed] [Google Scholar]
- 78.Yamada SD, Hickson JA, Hrobowski Y, Vander Griend DJ, Benson D, Montag A, Karrison T, Huo D, Rutgers J, Adams S, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Research. 2002;62:6717–6723. [PubMed] [Google Scholar]
- 79.Yoshida BA, Dubauskas Z, Chekmareva MA, Christiano TR, Stadler WM, Rinker-Schaeffer CW. Mitogen-activated protein kinase kinase 4/stress-activated protein/Erk kinase 1 (MKK4/SEK1), a prostate cancer metastasis suppressor gene encoded by human chromosome 17. Cancer Research. 1999;59:5483–5487. [PubMed] [Google Scholar]
- 80.Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, Yamada SD. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Research. 2006;66:2264–2270. doi: 10.1158/0008-5472.CAN-05-3676. [DOI] [PubMed] [Google Scholar]
- 81.Sakurai T, Maeda S, Chang L, Karin M. Inaugural Article: Loss of hepatic NF-{kappa}B activity enhances chemical hepatocarcinogenesis through sustained c-Jun N-terminal kinase 1 activation; Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 10544–10551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zenz R, Wagner EF. Jun signalling in the epidermis: From developmental defects to psoriasis and skin tumors. The International Journal of Biochemistry & Cell Biology. 2006;38:1043–1049. doi: 10.1016/j.biocel.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 83.Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS, Depatie C, Mercola D. C-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clinical Cancer Research. 2003;9:391–401. [PubMed] [Google Scholar]
- 84.Papachristou DJ, Batistatou A, Sykiotis GP, Varakis I, Papavassiliou AG. Activation of the JNK-AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone. 2003;32:364–371. doi: 10.1016/s8756-3282(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 85.Hess P, Pihan G, Sawyers CL, Flavell RA, Davis RJ. Survival signaling mediated by c-Jun NH(2)-terminal kinase in transformed B lymphoblasts. Nature Genetics. 2002;32:201–205. doi: 10.1038/ng946. [DOI] [PubMed] [Google Scholar]
- 86.Hayakawa J, Mittal S, Wang Y, Korkmaz KS, Adamson E, English C, Ohmichi M, McClelland M, Mercola D. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Molecular Cell. 2004;16:521–535. doi: 10.1016/j.molcel.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 87.Potapova O, Basu S, Mercola D, Holbrook NJ. Protective role for c-Jun in the cellular response to DNA damage. The Journal of Biological Chemistry. 2001;276:28546–28553. doi: 10.1074/jbc.M102075200. [DOI] [PubMed] [Google Scholar]
- 88.Heasley LE, Han SY. JNK regulation of oncogenesis. Molecules and Cells. 2006;21:167–173. [PubMed] [Google Scholar]
- 89.Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- 90.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 91.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. The Journal of Clinical Investigation. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis and Rheumatism. 2002;46:818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 93.Sumara G, Belwal M, Ricci R. "Jnking" atherosclerosis. Cell Molecular Life Sci. 2005;62:2487–2494. doi: 10.1007/s00018-005-5253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A, Hersberger M, Eriksson U, Eberli FR, Becher B, Boren J, Chen M, Cybulsky MI, Moore KJ, Freeman MW, Wagner EF, Matter CM, Luscher TF. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306:1558–1561. doi: 10.1126/science.1101909. [DOI] [PubMed] [Google Scholar]
- 95.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 96.Hotamisligil GS. Role of Endoplasmic Reticulum Stress and c-Jun NH2-Terminal Kinase Pathways in Inflammation and Origin of Obesity and Diabetes. Diabetes. 2005;54 Suppl 2:S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 97.Musi N, Goodyear LJ. Insulin resistance and improvements in signal transduction. Endocrine. 2006;29:73–80. doi: 10.1385/ENDO:29:1:73. [DOI] [PubMed] [Google Scholar]
- 98.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) The Journal of Biological Chemistry. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 99.Jaeschke A, Czech MP, Davis RJ. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes & Development. 2004;18:1976–1980. doi: 10.1101/gad.1216504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Waeber G, Delplanque J, Bonny C, Mooser V, Steinmann M, Widmann C, Maillard A, Miklossy J, Dina C, Hani EH, Vionnet N, Nicod P, Boutin P, Froguel P. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nature Genetics. 2000;24:291–295. doi: 10.1038/73523. [DOI] [PubMed] [Google Scholar]