

Abstract
Background
Spinal cord stimulation (SCS) is used to relieve ischemic pain and improve peripheral blood flow in selected patients with peripheral arterial diseases. Our previous studies show that antidromic activation of transient receptor potential vanilloid-1 (TRPV1) containing sensory fibers importantly contributes to SCS-induced vasodilation.
Objectives
To determine whether peripheral terminals of TRPV1 containing sensory fibers produced vasodilation that depends upon the release of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) during SCS.
Methods
A unipolar ball electrode was placed on the left dorsal column at lumbar spinal cord segments 2–3 in sodium pentobarbital anesthetized, paralyzed and ventilated rats. Cutaneous blood flow from left and right hindpaws were recorded with laser Doppler flow perfusion monitors. SCS was applied through a ball electrode at 30%, 60%, 90% and 300% of motor threshold. Resiniferatoxin (RTX; 2 μg/ml, 100 μl), an ultra potent analog of capsaicin, was injected locally into the left hindpaw to functionally inactivate TRPV-1 containing sensory terminals. In another set of experiments, CGRP 8-37, an antagonist of the CGRP-1 receptor, was injected at 0.06, 0.12, or 0.6 mg/100 μl into the left hindpaw to block CGRP responses; N-omega-nitro-L-arginine methyl ester (L-NAME), a nonselective nitric-oxide synthase (NOS) inhibitor, was injected at 0.02 or 0.2 mg/100 μl into the left hindpaw to block nitric oxide synthesis; 4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA, a neuronal NOS inhibitor, was injected at 0.02 or 0.1 mg/100 μl into the left hindpaw to block neuronal nitric oxide synthesis.
Results
SCS at all intensities produced vasodilation in the left hindpaw, but not in the right. RTX administration attenuated SCS-induced vasodilation at all intensities in the left hindpaw (P<0.05, n=7) compared with responses before RTX. CGRP 8-37 administration attenuated SCS-induced vasodilation in the left hindpaw in a dose dependent manner (linear regression, P<0.05) compared with responses before CGRP 8-37. In addition, L-NAME at a high dose, but not 4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA, decreased SCS-induced vasodilation (P<0.05, n=5).
Conclusion
While TRPV1, CGRP and NO are known to be localized in the same nerve terminals, our data indicate that SCS-induced vasodilation depends on CGRP release, but not NO release. NO, released from endothelial cells, may be associated with vascular smooth muscle relaxation and peripheral blood flow increase in response to SCS.
Keywords: Spinal cord stimulation (SCS), Vasodilation, Transient receptor potential vanilloid-1 (TRPV1), Calcitonin gene-related peptide (CGRP), Nitric oxide (NO)
1. Introduction
Spinal cord stimulation (SCS) was used for the first time to treat patients with pain in 1967 (Shealy et al., 1967). Cook was the first to observe that SCS increased blood flow to lower limbs in patients (Cook et al, 1976). SCS is an excellent alternative therapy for treating patients with peripheral arterial disease (PAD) including occlusive and vasospastic conditions, particularly when the diseases are unsuitable for the conventional revascularization treatments. The promising benefits of SCS include improvement of blood flow in the microcirculation, relief of ischemic pain and reduction of amputation rate (Linderoth and Foreman, 1999; Erdek and Staats, 2003). Furthermore, SCS is minimally invasive and has few serious complications. Approximately 70% of patients experience benefits with SCS, although it is considered as the last resort to treat inoperable patients with PAD (Cameron, 2004). Annually more than 14,000 SCS implantations are performed worldwide (Linderoth and Foreman, 2006). Recent reports also suggest that transcutaneous oxygen measurement is used as a predictor for treatment success of SCS (Petrakis and Sciacca, 2000). Benefits of SCS on ischemia in the limbs and feet are largely dependent on the increase of blood flow during SCS. Two theories are proposed to explain SCS-induced vasodilation. One theory is that SCS decreases sympathetic outflow, subsequently reduces vascular constriction, and produces peripheral vasodilation (Linderoth et al., 1991 a, b; Linderoth et al., 1994). Another theory is that SCS produces the release of vasodilators from the sensory terminals into the vascular tissue via activation of sensory fibers (Croom et al., 1997 a). The antidromic and sympathetic theories are complementary. The balance of the dual mechanisms is associated with the sympathetic activity level, SCS intensity and individual patients, or animal strains in the experimental studies (Tanaka et al., 2003 b). Previous studies have indicated that depression of sympathetic activity (Linderoth and Foreman, 2006) may account for a part of the effect, but that antidromic activation of sensory fibers and subsequent release of vasodilators account for a major portion of the SCS effect (Croom et al., 1996, 1997a, b, 1998; Tanaka et al., 2001 , 2003 a, b, 2004). A recent study has shown that SCS-induced vasodilation is predominantly mediated via transient receptor potential vanilloid-1 (TRPV1) containing sensory fibers (Wu et al., 2006 a). TRPV1 and vasodilators including calcitonin gene related peptide (CGRP) and possibly nitric oxide (NO) are co-localized in the peripheral terminals of TRPV1 containing sensory fibers (Kopp et al., 2001; Collins et al., 2002; Eguchi et al., 2004; Wang et al., 2006). Activation of TRPV1 and subsequent depolarization of terminals by SCS is very likely to release these vasodilators and to produce vasodilation.
The aim of the present study was to determine whether peripheral terminals of TRPV1 containing sensory fibers produce vasodilation that depends on the release of CGRP and NO during SCS. We determined whether: (1) local hindpaw injection of resiniferatoxin (RTX), an ultrapotent TRPV1 agonist for desensitizing TRPV1 containing terminals (Pan et al., 2003; Zahner et al., 2003; Wu et al., 2006 a and b), influences SCS-induced vasodilation; (2) local hindpaw injection of CGRP 8-37, an antagonist of CGRP-1 receptor, affects SCS-induced vasodilation; (3) local hindpaw injection of 4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA, a neuronal NOS (nNOS) inhibitor, alters the effects of SCS on peripheral blood flow; and (4) local hindpaw injection of N-omega-nitro-L-arginine methyl ester (L-NAME), a nonselective nitric-oxide synthase (NOS) inhibitor, influences SCS-induced vasodilation. The experiments were performed in anesthetized, paralyzed and artificially ventilated rats. Our previous studies have confirmed that more than 95% of effects of SCS on vasodilation is via antidromic activation of sensory fibers in this experimental setup and protocol (Tanaka et al., 2003 b).
The results showed that local hindpaw injection of RTX abolished SCS-induced vasodilation. The hindpaw injection of CGRP 8-37 at the middle and high dose also largely decreased the vasodilation produced by SCS. In addition, L-NAME at a high dose, but not (4S)-N-(4-Amino-5[aminoethyl]aminopentyl) N′-nitroguanidine, TFA, reduced SCS-induced vasodilation. Our data indicate that SCS-induced vasodilation largely depends on CGRP release from the terminals of TRPV1 containing sensory fibers. NO, possibly released from endothelial cells, but not from nerve endings is also necessary in SCS-induced vasodilation.
2. Results
To determine the diffusion distance of local injections, pontamine skyblue was injected into the left hindpaw. Twenty min later, the animals were euthanized with an anesthetic overdose and the left and right hindpaws were collected for examination. The blue color showed the area that this local injection spread in the paw. The blue color was located in the ventral aspect of the left hindpaw 20 min after injection of skyblue solution (Fig 1A), but not in the dorsal aspect (Fig 1B). The cross sections also showed that blue color was detected in the middle of the left hindpaw (Fig 1C), but not near the left ankle (Fig 1D).
Fig 1. Local injection of 100 μl solution of 2 % skyblue dye into left paw.
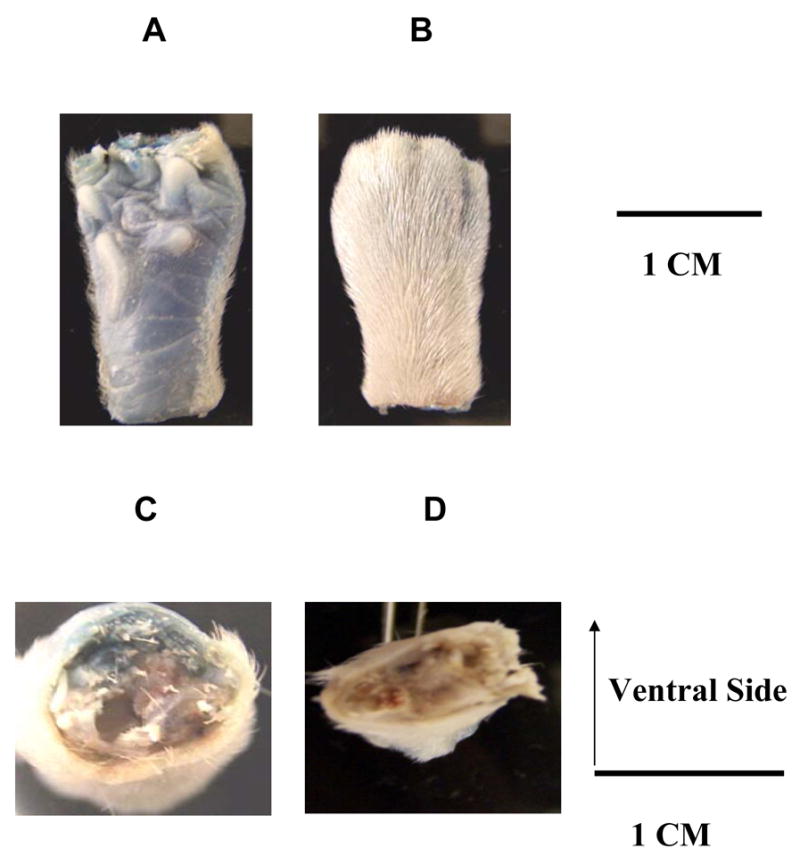
(A) Ventral side of the middle of the left paw. (B) Dorsal side of the middle of the left paw. (C) Cross section of the middle of the left paw. (D) Cross section near the left ankle.
The average motor threshold (MT) of the first group of rats was 489 ± 42 μA (n=7). As observed in our previous reports (Tanaka et al., 2001, 2003 a, b, 2004; Wu et al., 2006 a), SCS at the different intensities (30%, 60%, 90% and 300% of MT) increased the left (ipsilateral), but not the right (contralateral) blood flow (Fig 2, top panel). SCS did not elicit vasodilation in the left hindpaw ten min after local injection of RTX into the left hindpaw. However SCS at 90% of MT in the right side of spinal cord (SC) still produced a large vasodilation in the right hindpaw (Fig 2, top panel). A summary of SCS-induced vasodilation before and after RTX (Fig 2, middle panel) showed that the increase of blood flow due to SCS at different intensities was nearly abolished after RTX administration. The responses of arterial blood pressure (ABP) to SCS were variable. SCS consistently increased blood flow and decreased vascular resistance unrelated to ABP changes. The changes of vascular resistance during SCS were calculated and summarized (Fig 2, bottom panel) to show that SCS significantly reduced ipsilateral vascular resistance, coinciding with the increased ipsilateral blood flow. There were no significant changes of contralateral vascular resistance before and after RTX.
Fig 2. Changes of blood flow and vascular resistance produced by SCS before and after paw injections of RTX.
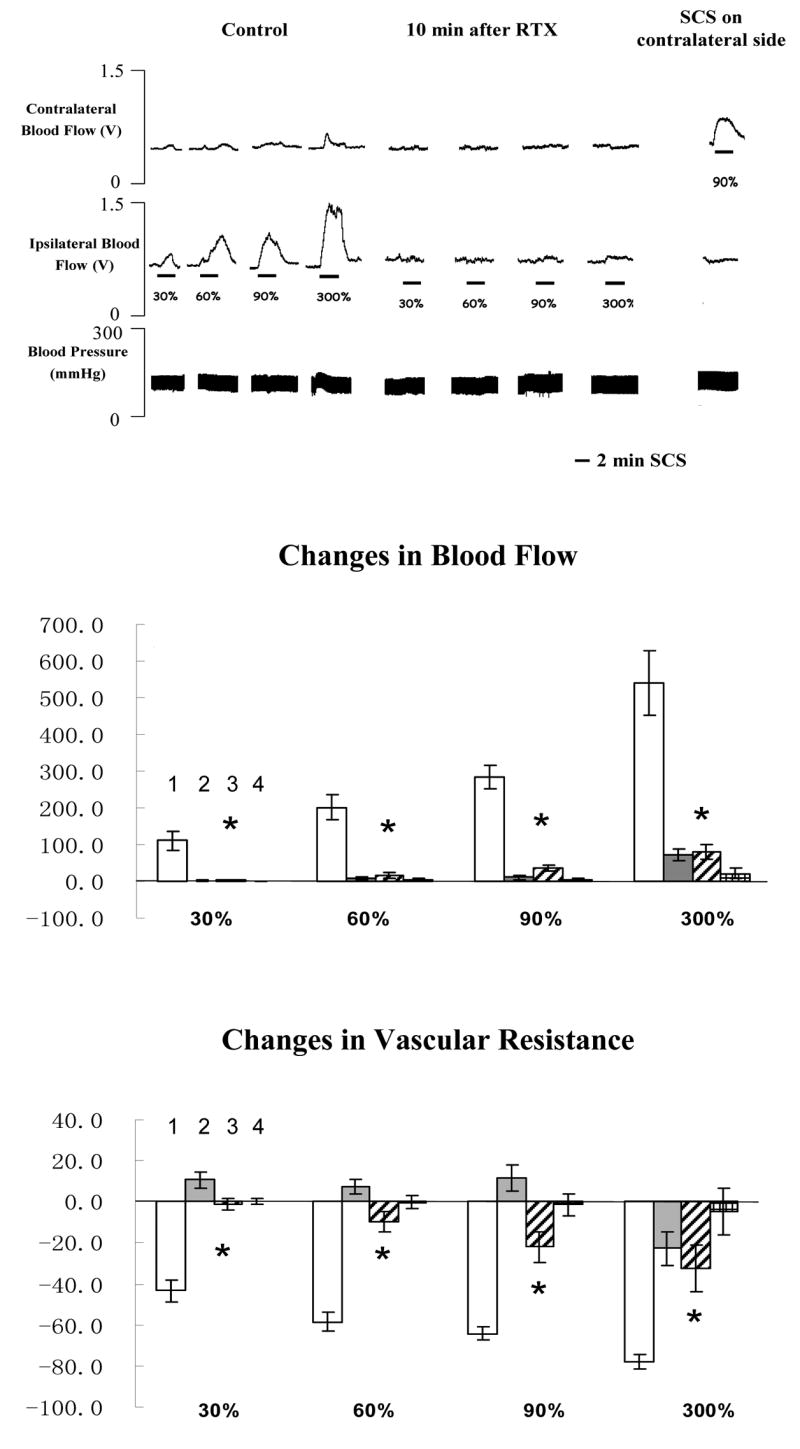
The top panel shows a representative example of ipsilateral and contralateral changes in blood flow and arterial blood pressure during SCS at different intensities, before and after RTX (100 μl saline containing 0.2 μg RTX, 0.1 μl 95% ethanol and 0.1 μl tween 80). The middle panel shows the percent changes of blood flow in the ipsilateral and contralateral hindpaws in response to 2-min SCS. (Column 1: changes in ipsilateral blood flow before RTX; Column 2: changes in contralateral blood flow before RTX; Column 3: changes in ipsilateral blood flow after RTX; Column 4: changes in contralateral blood flow after RTX). Each data point represents mean ± S.E.M (n=7). * P<0.05, compared to changes in ipsilateral blood flow during SCS before RTX. The bottom panel shows the percent changes of vascular resistance in the ipsilateral and contralateral hindpaws in response to 2-min SCS. (Column 1: changes in ipsilateral vascular resistance before RTX; Column 2: changes in contralateral vascular resistance before RTX; Column 3: changes in ipsilateral vascular resistance after RTX; Column 4: changes in contralateral vascular resistance after RTX). * P<0.05, compared to ipsilateral vascular resistance during SCS before RTX.
The average MT of the rats in the second group was 477 ± 45 μA (n=21). Representative example of changes of blood flow and ABP during SCS before and after CGRP 8-37 at 0.06, 0.12 or 0.6 mg/100 μl are shown in the top panel of Fig 3A, 3B and 3C, respectively. Summaries of the changes of blood flow and vascular resistance (middle and bottom panels in Fig 3A, 3B and 3C) showed that CGRP 8-37 at middle and high doses reduced SCS-induced increase in blood flow and decrease in vascular resistance. In addition, the effects of CGRP 8-37 on SCS-induced increases in blood flow were dose dependent, using the analysis of linear regression (SCS at 30% of MT: R=0.5318, P<0.05; SCS at 60% of MT: R=0.5134, P<0.05; SCS at 90% of MT: R=0.6072, P<0.01). SCS at 90% of MT on the right side of spinal cord still produced vasodilation of the right hindpaw at 40 min after the administration of CGRP 8-37 (244.60 ± 37.2 %, n=4).
Fig 3. Changes of blood flow and vascular resistance produced by SCS before and after CGRP 8-37. (A) CGRP 8-37 at 0.06 mg/100 μl saline.
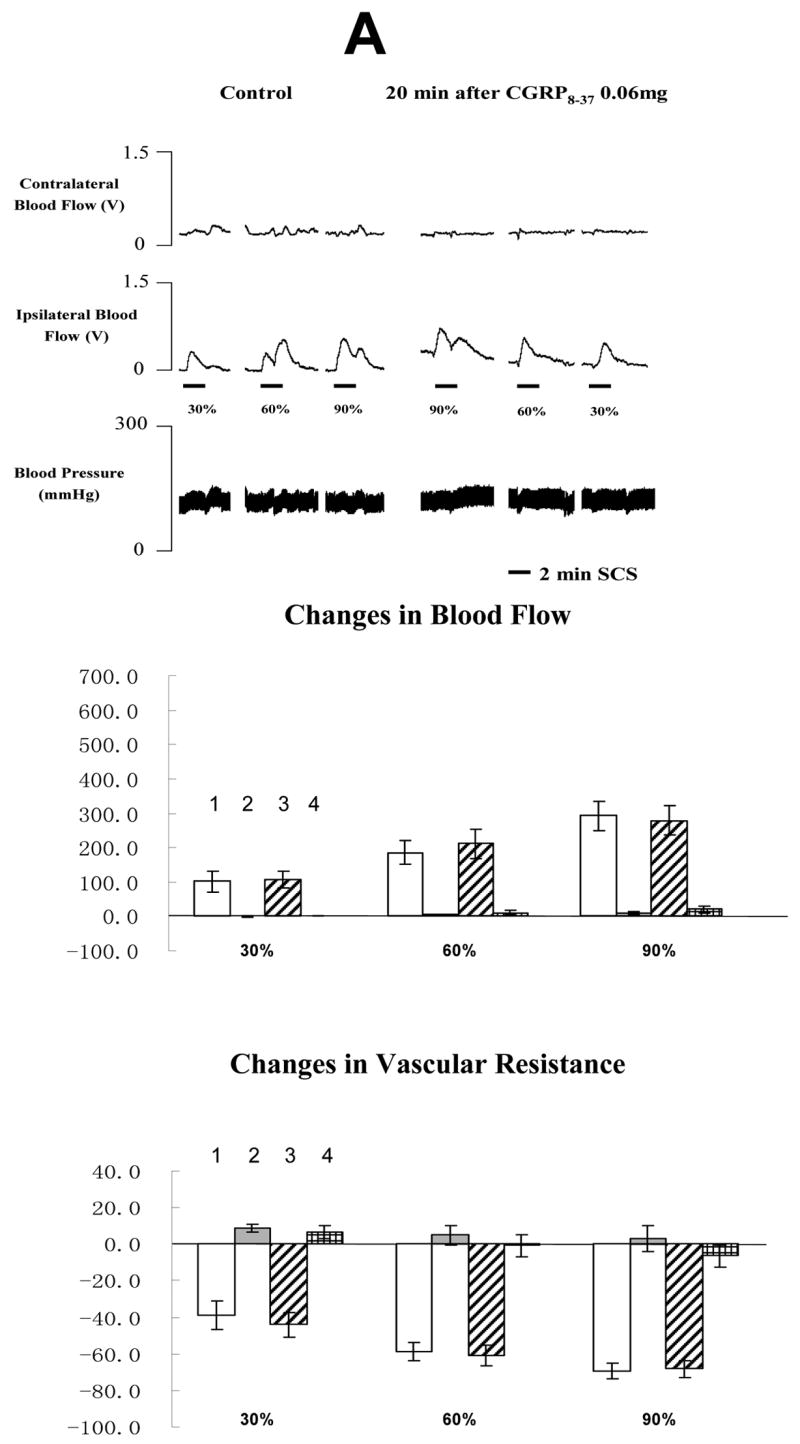
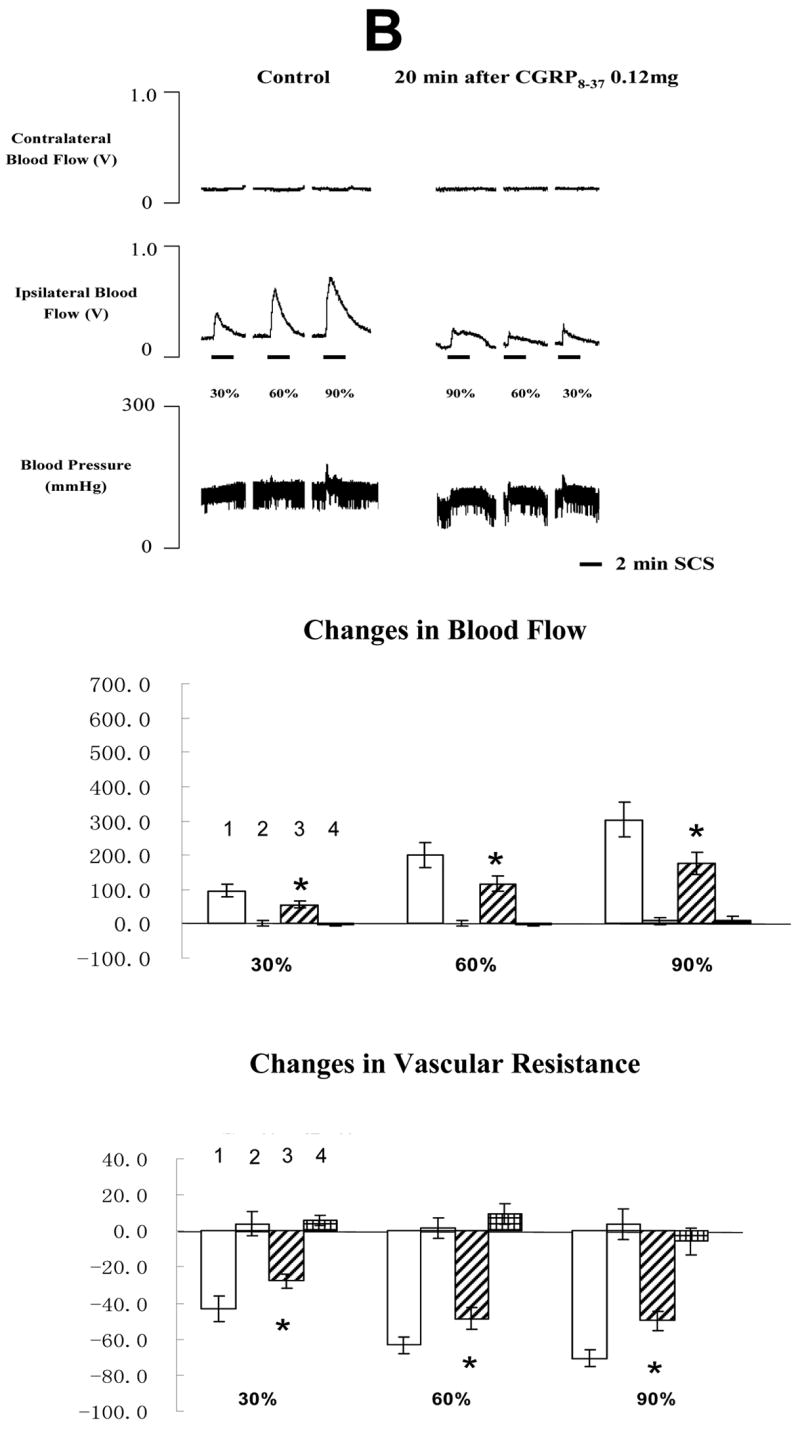
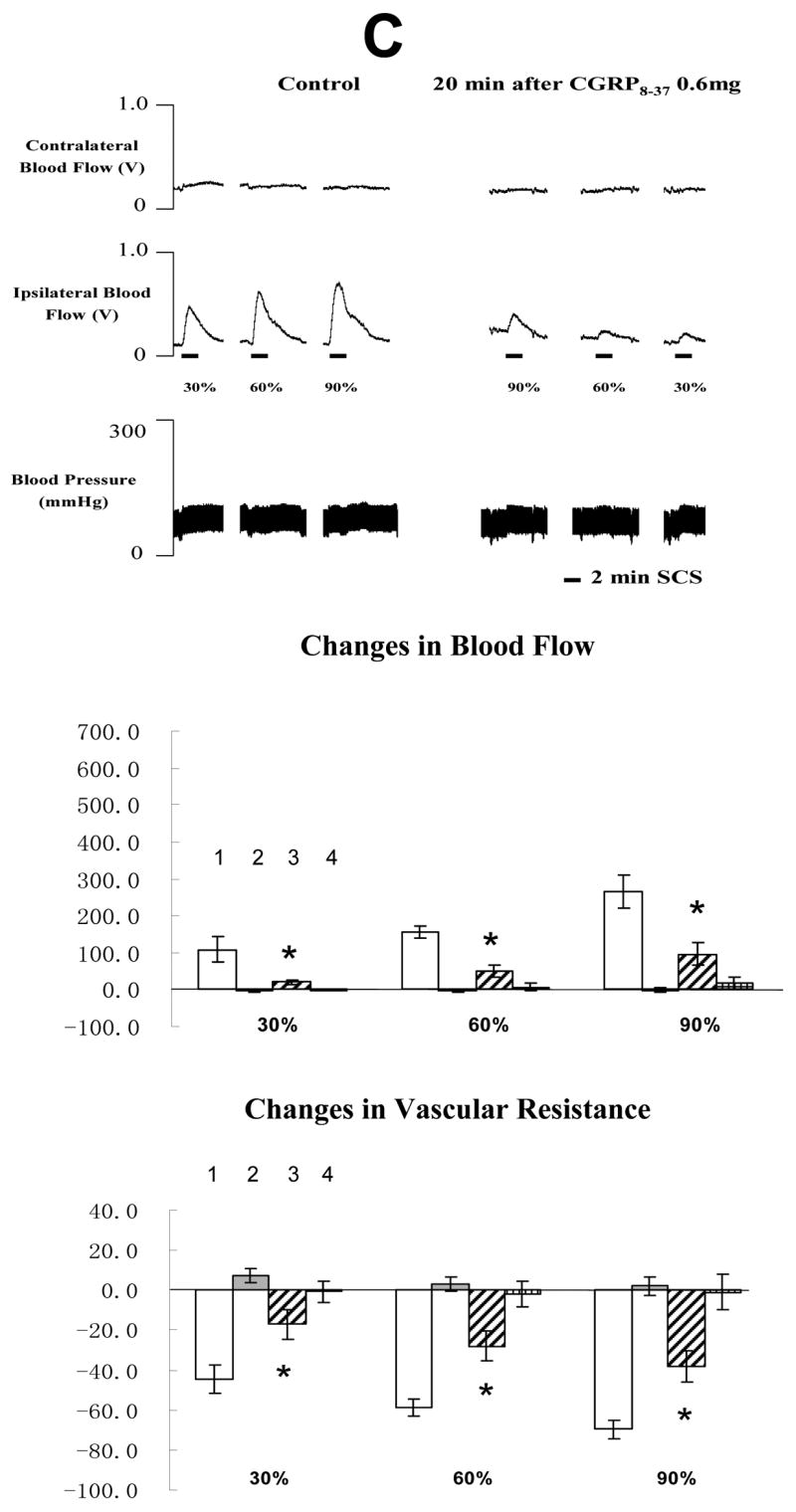
The top panel shows a representative example of ipsilateral and contralateral changes in blood flow and arterial blood pressure during SCS at different intensities, before and after CGRP 8-37 at 0.06 mg/100 μl saline. The middle panel shows the percent changes of blood flow in the ipsilateral and contralateral hindpaws in response to 2-min SCS. The bottom panel shows the percent changes of vascular resistance in the ipsilateral and contralateral hindpaws in response to 2-min SCS. (B) CGRP8-37 at 0.12 mg/100 μl saline. The figure legends are the same as those in Fig 3 A. * P<0.05, compared to ipsilateral blood flow or ipsilateral vascular resistance during SCS before the administration of CGRP 8-37 at 0.12 mg/100 μl saline. (C) CGRP8-37 at 0.6 mg/100 μl saline. The figure legends are the same as those in Fig 3 A and B. * P<0.05, compared to ipsilateral blood flow or ipsilateral vascular resistance during SCS before the administration of CGRP 8-37 at 0.12 mg/100 μl saline. The representations of columns are similar to Fig 2, except for the different names of chemical agents
The average MT of the rats in the third group was 456 ± 51 μA (n=10). The typical changes of blood flow and ABP responses to SCS before and after neuronal NOS blocker 4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA) are showed in the top panel of Fig 4A and 4B. Basically the administration of neuronal NOS blocker did not alter SCS-induced vasodilation. Summaries of blood flow and vascular resistance showed no statistical difference when comparing values before and after injection of neuronal NOS blocker at the low or high doses (in the middle and bottom panels of Fig 4A and 4B). 0.1 mg/100 μl is the highest dose of 4S)-N-(4-Amino-5[aminoethyl]aminopentyl) -N′-nitroguanidine, TFA that can be dissolved into saline at room temperature.
Fig 4. Changes of blood flow and vascular resistance produced by SCS before and after neuronal NOS blocker (4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA). (A) Neuronal NOS blocker at 0.02 mg/100μl saline.
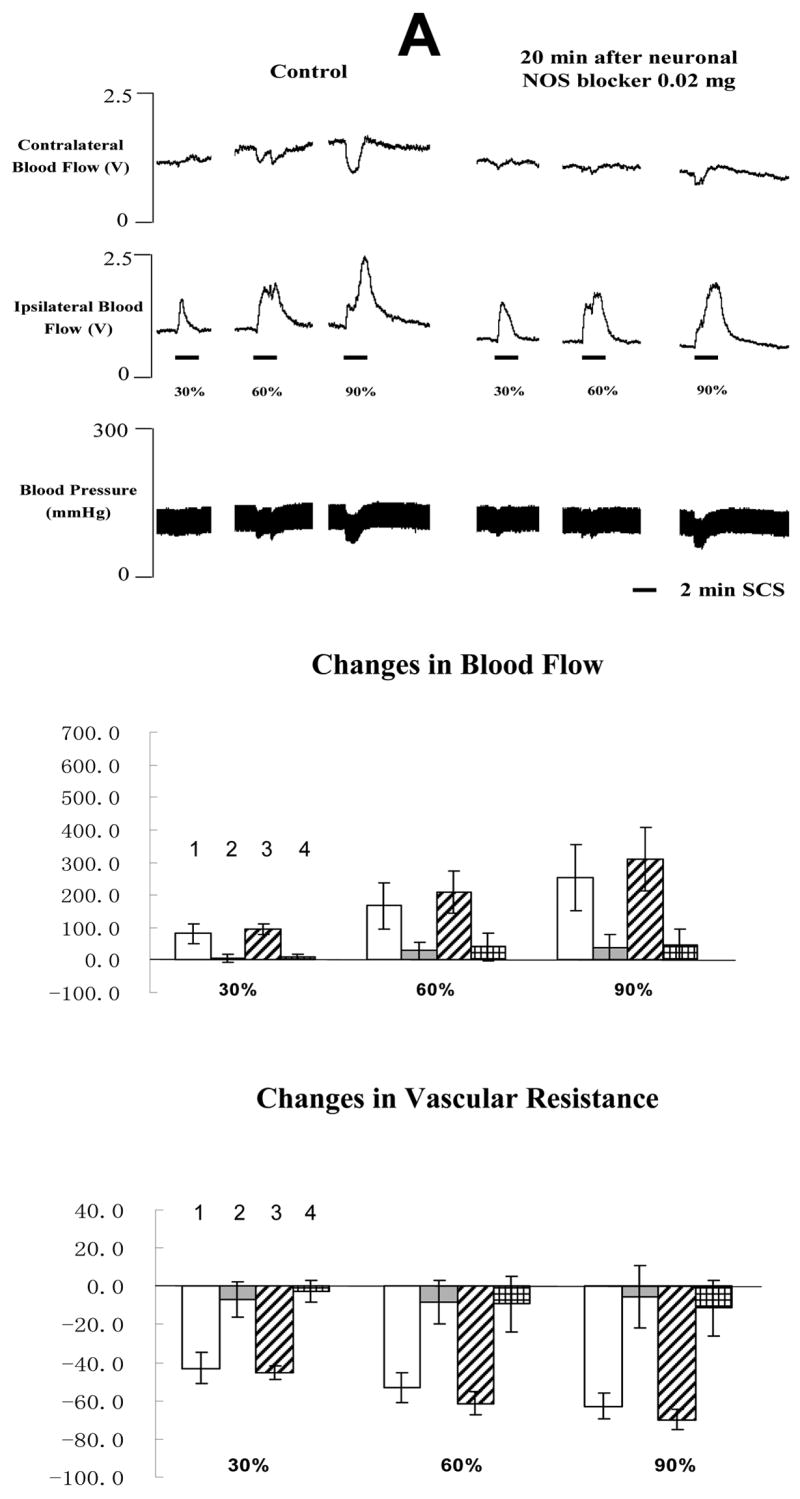
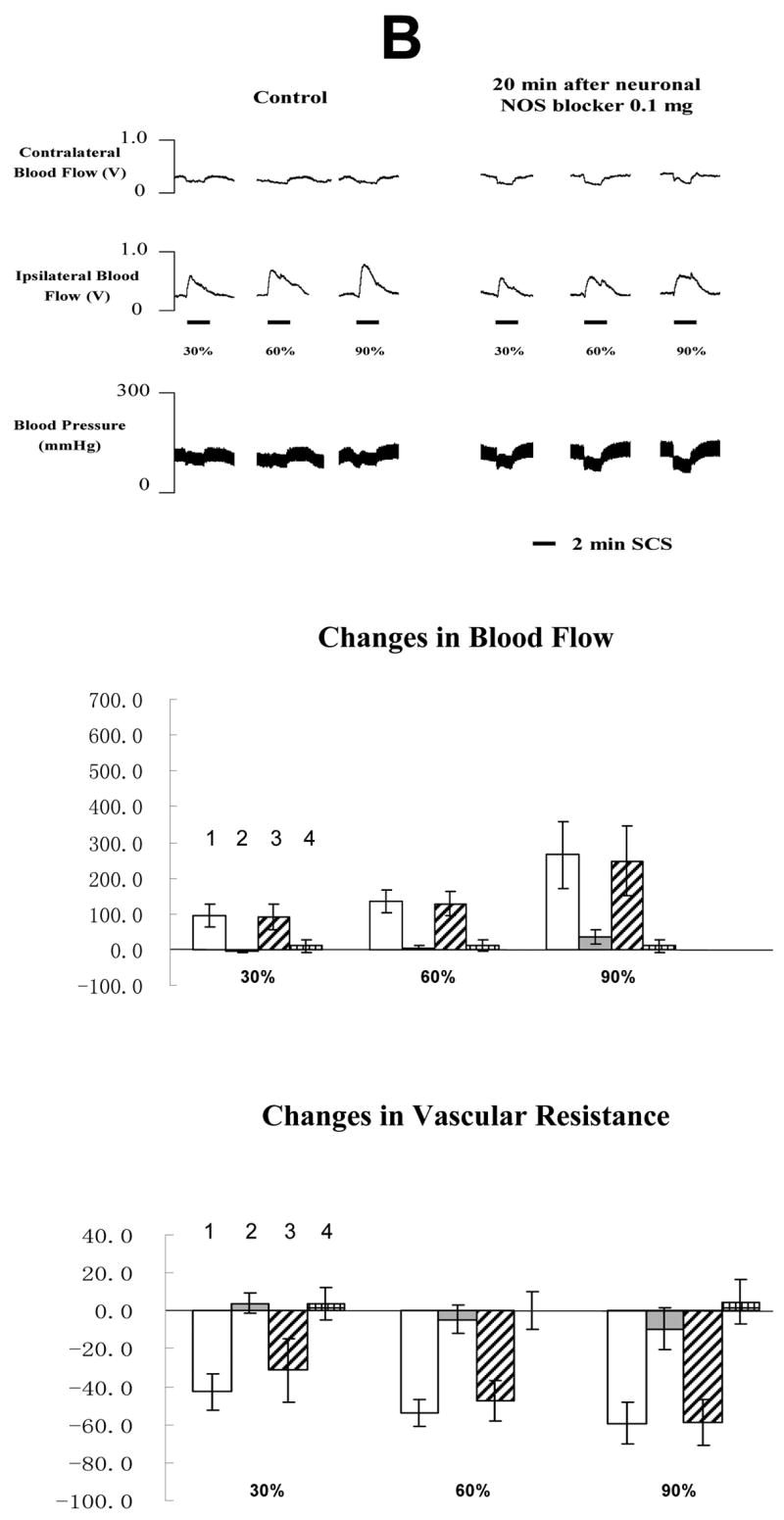
The top panel shows a representative example of ipsilateral and contralateral changes in blood flow and arterial blood pressure during SCS at different intensities, before and after neuronal NOS blocker at 0.02 mg/100 μl saline. The middle panel shows the percent changes of blood flow in the ipsilateral and contralateral hindpaws in response to 2-min SCS. Each data point represents mean ± S.E.M (n=5). The bottom panel shows the percent changes of vascular resistance in the ipsilateral and contralateral hindpaws in response to 2-min SCS. Each data point represents mean ± S.E.M (n=5). (B) Neuronal NOS blocker at 0.1 mg/100μl saline. The figure legends are the same as those in Fig 4 A. The representations of columns are similar to Fig 2, except for the different names of chemical agents
The average MT of the rats in the fourth group was 498 ± 53 μA (n=10). An example of the effects of SCS on blood flow and ABP before and after L-NAME administration is shown in the top panel of Fig 5A and 5B. L-NAME at 0.02 mg/100 μl did not influence the effects of SCS on blood flow and vascular resistance (in the middle and bottom panels of Fig 5A). However, L-NAME 0.2 mg/100 μl dramatically reduced SCS-induced increases in blood flow and decreases in vascular resistance (in the middle and bottom panels of Fig 5B). Saline did not affect SCS-induced vasodilation (before vs after: 224.6 ± 34.9 % vs 209.4 ± 25.6 %, n=3). Another vehicle (saline containing 0.1 μl 95% ethanol and 0.1 μl tween 80) also did not alter SCS-induced vasodilation (before vs after: 233.1 ± 37.2 % vs 218.8 ± 29.7 %, n=3).
Fig 5. Changes of blood flow and vascular resistance produced by SCS before and after L-NAME. (A) L-NAME at 0.02 mg/100μl saline.
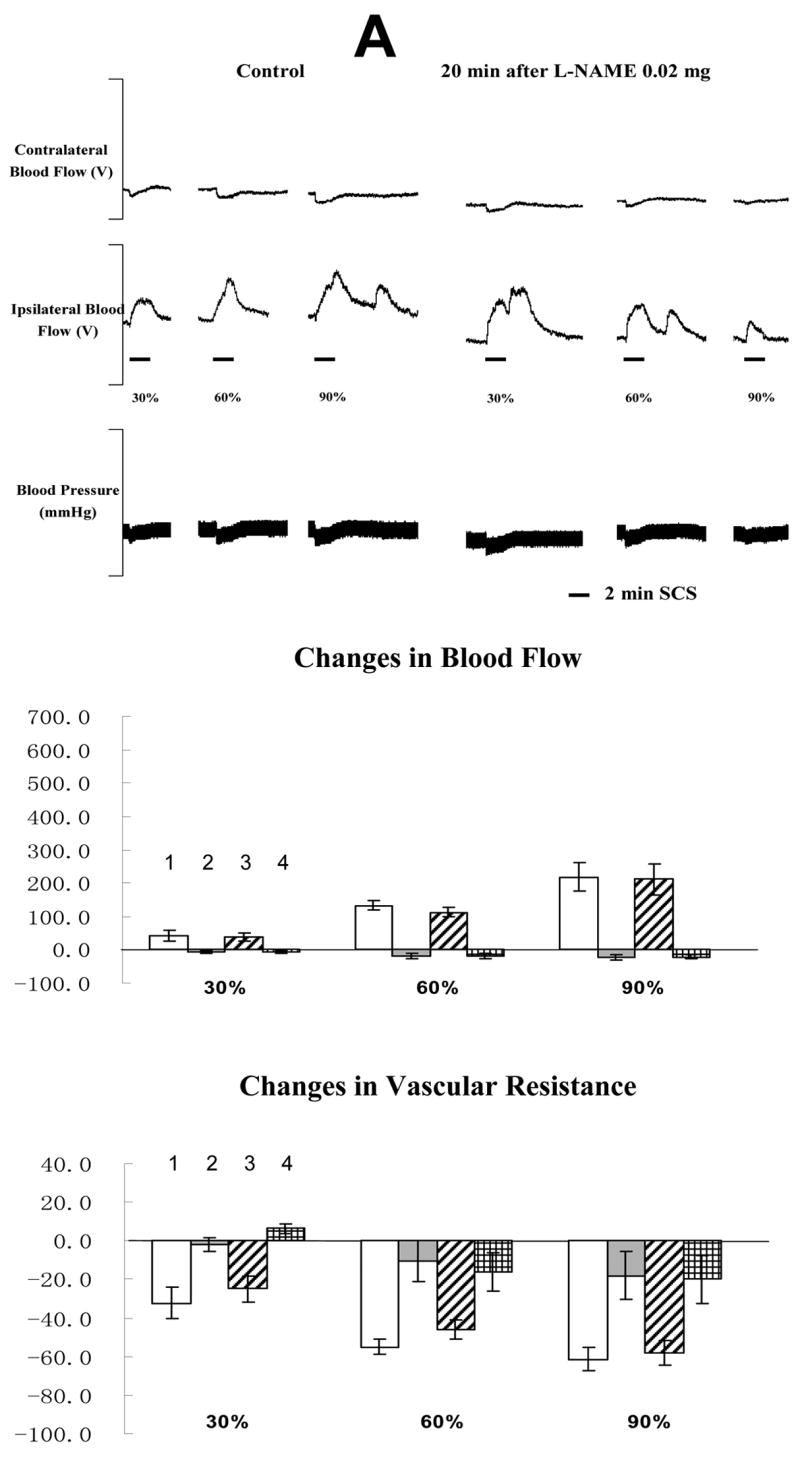
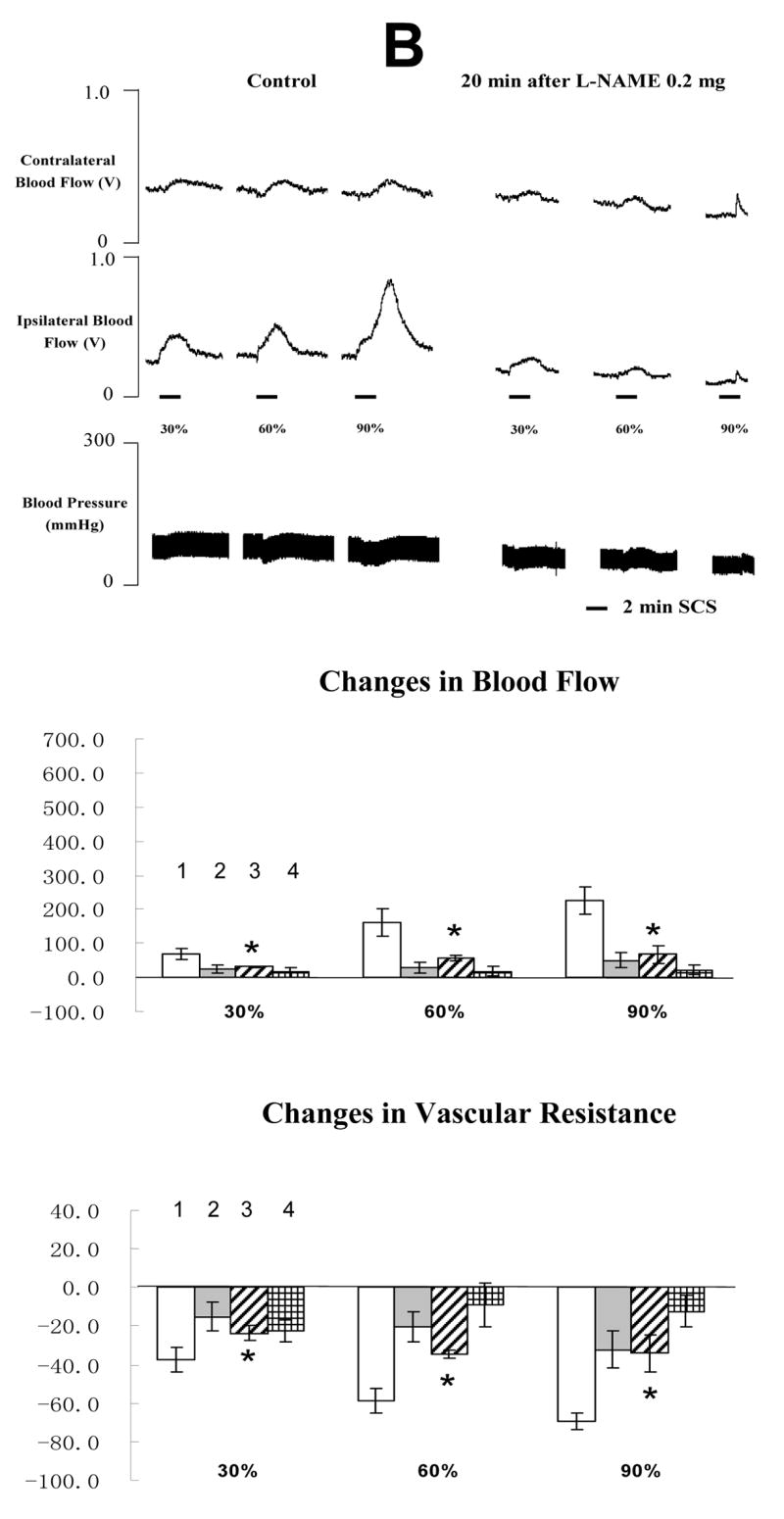
The top panel shows a representative example of ipsilateral and contralateral changes in blood flow and arterial blood pressure during SCS at different intensities, before and after neuronal NOS blocker at 0.02 mg/100 μl saline. The middle panel shows the percent changes of blood flow in the ipsilateral and contralateral hindpaws in response to 2-min SCS. Each data point represents mean ± S.E.M (n=5). The bottom panel shows the percent changes of vascular resistance in the ipsilateral and contralateral hindpaws in response to 2-min SCS. (B) L-NAME at 0.2 mg/100μl saline. The figure legends are the same as those in Fig 5 A. * P<0.05, compared to ipsilateral blood flow or ipsilateral vascular resistance during SCS before L-NAME at 0.2 mg/100 μl saline. The representations of columns are similar to Fig 2, except for the different names of chemical agents
3. Discussion
The data from the present study supports the mechanism that SCS antidromically activates TRPV1 containing sensory fibers and produces the release of CGRP, but not NO from TRPV1 containing peripheral terminals. Subsequently CGRP either directly relaxes smooth muscle cells by binding CGRP-1 receptor of smooth muscle cells, or binds CGRP-1 receptor of endothelial cells to synthesize and release NO into smooth muscle cells, also causing relaxation of smooth muscle cells which leads to vasodilation.
Antidromic mechanism of SCS-induced vasodilation was initially suggested by Bayliss based on his observation that SCS at high intensity produced vasodilation mediated via the thin fibers (Bayliss, 1901). Recent studies showed that dorsal rhizotomy of L3-5 abolished vasodilation produced by SCS, suggesting that SCS antidromically activates afferent fibers in the dorsal roots to produce vasodilation (Croom et al., 1997 a). The types of sensory fibers involved in SCS effects on vasodilation were also investigated by using intravenous injection or local nerve application of RTX (Wu et al., 2006 a). The results showed that RTX eliminated the vasodilation produced by SCS at all intensities. Another finding was that antidromic compound action potentials (CAPs) of C-fibers in dorsal roots were only evoked by SCS at ≥90% of MT (Tanaka et al., 2003 a). Overall these studies indicated that the sensory fibers activated by SCS are TRPV1 containing fibers, including unmyelinated C-fibers and myelinated Aδ fibers. Also SCS-induced vasodilation at ≤60% of MT may be mediated via only myelinated fibers, whereas vasodilation at ≥90% of MT may also involve unmyelinated C-fibers.
Since TRPV1 is expressed in the peripheral terminals of TRPV1 containing sensory fibers (Wang, 2005; Vaishnava and Wang, 2003), these studies were performed to determine the roles of peripheral terminals containing TRPV1 in SCS-induced vasodilation. RTX is a very useful chemical agent to desensitize TRPV1. A low dose of RTX is able to desensitize TRPV1 and block the release of the molecules from TRPV1 containing nerve fibers within 10 mins. Additionally, the effect of RTX on TRPV1 is very stable during the entire experiment (Qin et al., 2006; Wu et al., 2006 a, b). To limit systemic absorption of RTX, SCS was repeated a short time (10 min) after RTX was injected into the left hindpaw. Local injections of RTX nearly abolished the vasodilation produced by SCS, particularly at the intensities below MT (30–90%). Furthermore, SCS still was unable to increase blood flow 90 min after RTX (data not shown). Also, SCS on the right side of spinal cord produced vasodilation of the right hindpaw 40–90 min after RTX, indicating RTX mainly has a local effect when a relatively low dose was injected into the left hindpaw. Overall the results strongly suggest that TRPV1 activation of peripheral terminals during SCS is necessary to produce vasodilation.
It is well known that CGRP and other molecules are co-localized in TRPV1 containing sensory terminals (Eguchi et al, 2004). The activation of TRPV1 by SCS and subsequent depolarization of terminals are likely to produce the release of the vasodilators into the vascular tissues including endothelial cells and smooth muscle cells. Among the vasodilators, CGRP is the most powerful microvascular vasodilator that binds to CGRP-1 receptors in endothelial cells and smooth muscle cells (Brain et al., 1985). The vasodilatory effect of CGRP is approximately 10 fold greater than prostaglandins and 100–1000 times greater than other typical vasodilators including acetylcholine, adenosine and Substance P (SP) (Brain and Grant, 2004). Previous studies have reported that intravenous injection of CGRP 8-37, an antagonist of CGRP-1 receptor, eliminated vasodilation produced by SCS (Croom et al., 1997 a; Tanaka et al., 2004). CGRP is also widely expressed in the cardiovascular system and spinal cord, in addition to peripheral terminals of TRPV1 containing sensory fibers (Brain and Grant, 2004). Blockade of CGRP functions by intravenous injection of CGRP 8-37 was likely to eliminate SCS-induced vasodilation via multiple effects on the spinal cords, cardiovascular functions and peripheral terminals. In the present study, CGRP 8-37 was locally injected into the left paw to determine the roles of CGRP released from nerve endings in SCS-induced vasodilation. CGRP 8-37, at the higher doses, significantly decreased the vasodilation produced by SCS. Overall the data indicated that the release of CGRP from nerve endings and subsequently binding to CGRP-1 receptor is a major pathway for SCS-induced peripheral vasodilation.
Another potential vasodilator involved in SCS-induced vasodilation is NO. Previous studies indicated that intravenous injection of L-NAME, a non-selective NO synthase (NOS) blocker, markedly attenuated vasodilation caused by SCS at doses of 2 or 10 mg/kg (Croom et al., 1997 b). There are three sources for NO synthesis: neuronal, endothelial and inducible. It is unlikely that inducible NOS is involved in the SCS-induced peripheral vasodilation since this study investigated the close association between SCS and almost simultaneous induction of cutaneous vasodilation (Kleinert et al., 2004). More time would be required to induce NO. Therefore, NO is possibly synthesized and released from either nerve terminals or endothelial cells during SCS. In this study, even a high dose of neuronal NOS blocker (0.1 mg/100 μl) did not decrease SCS-induced vasodilation, suggesting that NO was not released from nerve endings during SCS. Furthermore, a high dose of L-NAME clearly attenuated vasodilation produced by SCS. A comparison of the data from the two groups shows that NO were not released from sensory nerve terminals, but from endothelial cells during SCS. The possibility is that the release of CGRP during SCS may bind to CGRP-1 receptors of endothelial cells and activate the intracellular pathways to synthesize NO and be released into vascular smooth muscle cells. Subsequently NO activates intracellular pathways of smooth muscle cells to decrease the calcium concentration and open potassium channels resulting in relaxation of smooth muscle cells (Brain and Grant, 2004).
SCS also showed variable effects on ABP, including increase, decrease and no change. When vascular resistance was calculated using the ratio of blood flow and ABP, the increase of blood flow coincided with the decrease of vascular resistance during SCS. Therefore we concluded that SCS-induced increase in blood flow was the result of vasodilation and not changes in ABP.
In this study, we used a local injection of 100 μl solution of different chemical agents into the left paw. The goal of this injection was to administer the chemical agents directly into the peripheral tissue. Therefore we were able to investigate the vasodilation pathways of SCS in the peripheral tissue. This injection did not influence the whole system since SCS on the right side of spinal cord still produced vasodilation in the right hindpaw after the local injection of chemical agents into the left hindpaw, i.e., RTX. Also pontamine skyblue stained a localized area of the left hindpaw.
SCS-induced vasodilation depended upon TRVP1-containing sensory fibers. Further, blockade of CGRP effects at the paw diminished the response as did a non specific, but not neuronal NOS blocker. These results are consistent with the idea that CGRP release from TRVP1-containing fibers could have mediated the response through increased NO production by endothelial cells. Immunohistochemical evaluations will be necessary to confirm this pathway and to determine whether these data also apply to the unanesthetized humans subject must also be explored
4. Experimental procedures
The protocol of this study was approved by the University of Oklahoma Health sciences Center Institutional Animal Care and Use Committee. Experimental design of this study is similar to previous studies from our group (Tanaka et al., 2001, 2003 a, b, 2004; Wu et al., 2006 a). Male Sprague-Dawley rats (300–400g; Charles Rivers, MA) were anesthesized with an intraperitoneal injection of sodium pentobarbital at a dose of 60 mg/kg. Body temperature was maintained between 36 and 38 ºC with a heating pad. A cannula (PE-50) was inserted into the left common jugular vein for a constant infusion of supplemental pentobarbital (19–25 mg/Kg/h). Pancuronium (boluses 0.2 mg/h/) was also injected via this cannula for muscle paralysis. A second cannula (PE-50) was inserted into the left common carotid artery to monitor arterial blood pressure. The rats were tracheotomized and artificially ventilated with room air (CWE ventilator model SAR-830). A laminectomy was performed to expose the dorsal surface of lumbar spinal segments 2–3 (L2–L3). A spring-loaded unipolar ball electrode was placed on the left side of the subdural surface at L2–L3.
The motor threshold (MT) stimulus intensity was determined in each animal at 50 Hz, 0.2 ms duration (parameters similar to those in clinical use) by slowly increasing the SCS current from zero until a clear retraction of the left hindlimb was observed. MT was used as a method to calculate the different intensities of SCS. Then pancuronium was injected for muscle paralysis. Before paralysis, animals were monitored for appropriate anesthesia by using withdrawal and corneal reflexes and stability of arterial blood pressure. After paralysis stability of arterial blood pressure level was used to monitor anesthesia. Experimental SCS was performed for 2 min at 30%, 60%, 90% or 300% of MT in random order of stimulus intensities with at least 5 min between consecutive stimuli.
Cutaneous blood flow in the hindlimb footpads of the rats was measured with laser Doppler flow perfusion monitors (Model 403A; Vasomedic, St. Paul, MN). ABP was also recorded during the entire experiment. Vascular resistance was calculated as the ratio of blood flow and ABP. Responses to SCS were determined as percent change from the baseline of blood flow and vascular resistance.
Four groups were used to address the aims of this study. 1) Rats (n=7) were used to determine the roles of TRPV1 containing terminals in vasodilation produced by SCS at 30–300% of MT. RTX solution (100 μl saline containing 0.2 μg RTX, 0.1 μl 95% ethanol and 0.1 μl tween 80) was locally injected into the left paw. Since the clinical stimulation of SCS is below MT, the applied intensities of SCS were from 30 to 90 % of MT in all the following studies. 2) Rats (n=21) were used to determine whether the release of CGRP from nerve endings by SCS, and subsequent bindings to CGRP-1 receptors of peripheral tissue including endothelial cells and vascular smooth muscle cells, is a main pathway to produce vasodilation. CGRP 8-37, an antagonist of CGRP-1 receptor, was locally injected at 0.06, 0.12, or 0.6 mg/100μl into the left hindpaw. Seven rats were tested with each dose for a total of 21 rats in the group. 3) Rats (n=10) were used to determine whether SCS produces the release of NO from the nerve endings during SCS-induced vasodilation. 4S)-N-(4-Amino-5[aminoethyl]aminopentyl)-N′-nitroguanidine, TFA, a neuronal NOS (nNOS) inhibitor, was injected at 0.02 or 0.1 mg/100 μl into the left hindpaw. 4) Rats (n=10) were used to determine whether SCS activates the release of NO from peripheral terminals, including endothelial cells and nerve endings, to elicit vasodilation during SCS. N-omega-nitro-L-arginine methyl ester (L-NAME), a nonselective nitric-oxide synthase (NOS) inhibitor, was injected at 0.02 or 0.2 mg/100μl into the left hindpaw. The third and fourth groups were performed to determine where NO was released and its roles in SCS-induced vasodilation.
Responses to vehicles (100 μl saline with or without 0.1 μl 95% ethanol and 0.1 μl tween 80) were also studied to determine its effects on SCS-induced vasodilation as a control. Three rats were used for each vehicle.
To investigate peripheral vasodilation pathways in SCS-induced vasodilation, the solution containing different chemical agents (100 μl) was locally injected into the left hindpaw from dorsal side into ventral side where the blood flow was measured (Fig 1). To identify the spread of this local hindpaw injection, 100 μl solution containing 2% pontamine skyblue was injected into the left hindpaw. Twenty min later, the animals were killed by anesthetic overdose and the left and right hindpaws were collected for examination.
5. Statistical Analysis
Changes in peripheral blood flow were presented as the percentage of changes of the peak amplitude from baseline and reported as mean ± S.E.M. Mean blood pressure was calculated using the formula 2/3 × diastolic blood pressure (BP) + 1/3 × Systolic BP. Vascular resistance was calculated by dividing mean blood pressure by blood flow. Differences in blood flow and vascular resistance before and after local hindpaw injections of different chemical agents were examined with a one-way ANOVA. In addition, linear regression was used to examine whether the effects of CGRP 8-37 on SCS-induced vasodilation were dose dependent. The level of significance was set at P < 0.05.
Acknowledgments
We thank Dr. S. Benyajati for helpful comments as well as D. Holston for her expert technical assistance. We are grateful to Drs. X. Yang (Xi’an Jiaotong University) and J. Gao (Heilongjiang University of Chinese Medicine) for their advice in this manuscript. This study was supported by NIH grant HL075524 & NS35471 (R.D.F), 2005 University of Oklahoma Health Sciences Center Graduate Student Association Research Grant (M.W), American Heart Association (AHA) Predoctoral Fellowship 0615642Z (M.W), and Presbyterian Health Foundation grant 1401 (N.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature reference
- Bayliss WM. On the origin from the spinal cord of the vasodilator fibers of the hind-limb and on the nature of these fibers. J Physiol. 1901;26:173–209. doi: 10.1113/jphysiol.1901.sp000831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brain SD, Grant AD. Vascular actions of calcitonin gene-related peptide and adrenomedullin. Physiol Rev. 2004;84:903–934. doi: 10.1152/physrev.00037.2003. [DOI] [PubMed] [Google Scholar]
- Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313:54–56. doi: 10.1038/313054a0. [DOI] [PubMed] [Google Scholar]
- Cameron T. Safety and efficacy of spinal cord stimulation for the treatment of chronic pain: a 20-year literature review. J Neurosurg. 2004;100:254–267. doi: 10.3171/spi.2004.100.3.0254. [DOI] [PubMed] [Google Scholar]
- Collins JJ, Usip S, McCarson KE, Papka RE. Sensory nerves and neuropeptides in uterine cervical ripening. Peptides. 2002;23:167–183. doi: 10.1016/s0196-9781(01)00593-9. [DOI] [PubMed] [Google Scholar]
- Cook AW, Oygar A, Baggenstos P, Pacheco S, Kleriga E. Vascular disease of extremities: Electrical stimulation of spinal cord and posterior roots. New York State J of Med. 1976;76:366–368. [PubMed] [Google Scholar]
- Croom JE, Foreman RD, Chandler MJ, Barron KW. Cutaneous blood flow increases in the rat hindpaw during dorsal column stimulation. Brain Res. 1996;728:281–6. doi: 10.1016/0006-8993(96)00554-9. [DOI] [PubMed] [Google Scholar]
- Croom JE, Foreman RD, Chandler MJ, Barron KW. Cutaneous vasodilation during dorsal column stimulation is mediated by dorsal roots and CGRP. Am J Physiol. 1997a;272:950–957. doi: 10.1152/ajpheart.1997.272.2.H950. [DOI] [PubMed] [Google Scholar]
- Croom JE, Foreman RD, Chandler MJ, Koss MC, Barron KW. Role of nitric oxide in cutaneous blood flow increases in the rat hindpaw during dorsal column stimulation. Neurosurgery. 1997b;40:565–70. doi: 10.1097/00006123-199703000-00027. discussion 571. [DOI] [PubMed] [Google Scholar]
- Croom JE, Foreman RD, Chandler MJ, Barron KW. Reevaluation of the role of sympathetic nervous system in cutaneous vasodilation during dorsal spinal cord stimulation: Are multiple mechanisms active? Neuromodulation. 1998;1:91–101. doi: 10.1111/j.1525-1403.1998.tb00022.x. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Tezuka S, Hobara N, Akiyama S, Kurosaki Y, Kawasaki H. Vanilloid receptors mediate adrenergic nerve- and CGRP-containing nerve-dependent vasodilation induced by nicotine in rat mesenteric resistance arteries. Br J Pharmacol. 2004;142:1137–1146. doi: 10.1038/sj.bjp.0705773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdek MA, Staats PS. Spinal cord stimulation for angina pectoris and peripheral vascular disease. Anesthesiol Clin North America. 2003;21:797–804. doi: 10.1016/s0889-8537(03)00090-7. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–66. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Kopp UC, Cicha MZ, Smith LA, Hokfelt T. Nitric oxide modulates renal sensory nerve fibers by mechanisms related to substance P receptor activation. Am J Physiol Regul Integr Comp Physiol. 2001;281:R279–290. doi: 10.1152/ajpregu.2001.281.1.R279. [DOI] [PubMed] [Google Scholar]
- Linderoth B, Fedorcsak I, Meyerson BA. Peripheral vasodilatation after spinal cord stimulation: animal studies of putative effector mechanisms. Neurosurgery. 1991a;28:187–195. [PubMed] [Google Scholar]
- Linderoth B, Foreman RD. Physiology of spinal cord stimulation: review and update. Neuromodulation. 1999;2:150–164. doi: 10.1046/j.1525-1403.1999.00150.x. [DOI] [PubMed] [Google Scholar]
- Linderoth B, Foreman RD. Mechanisms of spinal cord stimulation in painful syndromes: role of animal models. Pain Med. 2006;7:S14–26. [Google Scholar]
- Linderoth B, Gunasekera L, Meyerson BA. Effects of sympathectomy on skin and musclemicroculation during dorsal column stimulation: animal studies. Neurosurgery. 1991b;29:874–879. doi: 10.1097/00006123-199112000-00012. [DOI] [PubMed] [Google Scholar]
- Linderoth B, Herregodts P, Meyerson BA. Sympathetic mediation of peripheral mediation of peripheral vasodilation induced by spinal cord stimulation: animal studies of the role of cholinergic and adrenergic receptor subtypes. Neurosurgery. 1994;35:711–719. doi: 10.1227/00006123-199410000-00018. [DOI] [PubMed] [Google Scholar]
- Pan HL, Khan GM, Alloway KD, Chen SR. Resiniferatoxin induces paradoxical changes in thermal and mechanical sensitivities in rats: mechanism of action. J Neurosci. 2003;23:2911–2919. doi: 10.1523/JNEUROSCI.23-07-02911.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrakis IE, Sciacca V. Spinal cord stimulation in diabetic lower limb critical ischaemia: transcutaneous oxygen measurement as predictor for treatment success. Eur J Vasc Endovasc Surg. 2000;19:587–592. doi: 10.1053/ejvs.1999.1036. [DOI] [PubMed] [Google Scholar]
- Qin C, Farber JP, Miller KE, Foreman RD. Responses of thoracic spinal neurons to activation and desensitization of cardiac TRPV1-containing afferents in rats. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1700–1707. doi: 10.1152/ajpregu.00231.2006. [DOI] [PubMed] [Google Scholar]
- Shealy CN, Mortimer JT, Reswick JB. Electrical inhibition of pain by stimulation of the dorsal columns: preliminary clinical report. Anesth Analg. 1967;46:489–491. [PubMed] [Google Scholar]
- Tanaka S, Barron KW, Chandler MJ, Linderoth B, Foreman RD. Low intensity spinal cord stimulation may induce cutaneous vasodilation via CGRP release. Brain Res. 2001;896:183–187. doi: 10.1016/s0006-8993(01)02144-8. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Barron KW, Chandler MJ, Linderoth B, Foreman RD. Role of primary afferents in spinal cord stimulation-induced vasodilation: characterization of fiber types. Brain Res. 2003a;959:191–198. doi: 10.1016/s0006-8993(02)03740-x. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Barron KW, Chandler MJ, Linderoth B, Foreman RD. Local cooling alters neural mechanisms producing changes in peripheral blood flow by spinal cord stimulation. Auton Neurosci. 2003b;28:117–127. doi: 10.1016/S1566-0702(03)00017-1. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Komori N, Barron KW, Chandler MJ, Linderoth B, Foreman RD. Mechanisms of sustained cutaneous vasodilation induced by spinal cord stimulation. Auton Neurosci. 2004;114:55–60. doi: 10.1016/j.autneu.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Vaishnava P, Wang DH. Capsaicin sensitive-sensory nerves and blood pressure regulation. Curr Med Chem Cardiovasc Hematol Agents. 2003;1:177–188. doi: 10.2174/1568016033477540. [DOI] [PubMed] [Google Scholar]
- Wang DH. The vanilloid receptor and hypertension. Acta Pharmacol Sin. 2005;26:286–294. doi: 10.1111/j.1745-7254.2005.00057.x. [DOI] [PubMed] [Google Scholar]
- Wang LH, Luo M, Wang Y, Galligan JJ, Wang DH. Impaired vasodilation in response to perivascular nerve stimulation in mesenteric arteries of TRPV1-null mutant mice. J Hypertens. 2006;24:2399–2408. doi: 10.1097/01.hjh.0000251900.78051.56. [DOI] [PubMed] [Google Scholar]
- Wu M, Komori N, Qin C, Farber JP, Linderoth B, Foreman RD. Sensory fibers containing vanilloid receptor-1(VR-1) mediate spinal cord stimulation-induced vasodilation. Brain Res. 2006a;1107:177–184. doi: 10.1016/j.brainres.2006.05.087. [DOI] [PubMed] [Google Scholar]
- Wu M, Qin C, Foreman RD, Farber JP. Transient receptor potential vanilloid receptor-1 does not contribute to slowly adapting airway receptor activation by ammonia. Auton Neurosci. 2006b Dec 11; doi: 10.1016/j.autneu.2006.10.007. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Zahner MR, Li DP, Chen SR, Pan HL. Cardiac vanilloid receptor 1-expressing afferent nerves and their role in the cardiogenic sympathetic reflex in rats. J Physiol. 2003;551:515–523. doi: 10.1113/jphysiol.2003.048207. [DOI] [PMC free article] [PubMed] [Google Scholar]