

Summary
Proteins destined for export across the cytoplasmic membrane via the post-translational Sec-dependent route have to be maintained in a largely unfolded state within the cytoplasm. In sharp contrast, only proteins that have folded into a native-like state within the cytoplasm are competent for export via the Twin Arginine Translocation (Tat) pathway. Proteins that contain disulfide bonds, such as scFv antibody fragments, can be translocated via Tat only when expressed in E. coli trxB gor mutant strains having an oxidizing cytoplasm. However, export is poor with the majority of the protein accumulating in the cytoplasm and only a fraction exported to the periplasmic space. Using a high throughput fluorescence screen, we isolated a mutant of the anti-digoxin 26-10 scFv from a large library of random mutants which is exported with a higher yield into the periplasm. In vitro refolding experiments revealed that the mutant scFv exhibits a 250% increase in the rate constant of the critical second phase of folding. This result suggests that Tat export competence is related to the protein folding rate and could be exploited for the isolation of faster folding protein mutants.
Keywords: 26-10 scFv, Tat, flow cytometry, refolding kinetics, FACS
The conformation of proteins in the cytoplasm is a critical element in determining their competence for translocation across lipid bilayer membranes. In E. coli, proteins that are exported via the general secretory pathway must be maintained in a partially unfolded conformation so they can be threaded via the narrow pore of the SecYEG translocation apparatus.1–3 For some proteins, this is not an issue because they are translated co-translationally by ribosomes that dock onto the membrane via the interaction of the signal recognition particle with its receptor.4 However, for the majority of proteins that utilize the general secretory pathway, membrane translocation occurs post-translationally. Export competence is ensured by interactions with chaperones such as SecB and by the signal peptide which serves to retard folding.5–8 Proteins that fold too rapidly to benefit from these processes are incompetent for export and remain in the cytoplasm. Consistent with this idea, a recent genetic selection for mutants of the fast folding cytoplasmic protein thioredoxin, which is normally not compatible for export when fused to the signal peptide of a post-translationally exported protein, resulted in the isolation of slower folding variants.9
In sharp contrast to export via the SecYEG pore, proteins that utilize the post-translational Twin Arginine Translocation (Tat) secretion pathway have to be folded in order to be competent for export.10 Misfolded or partially folded proteins cannot be translocated via the Tat pathway, suggesting the existence of a folding quality control mechanism that operates either prior to, or concomitant with, translocation through the Tat pore.11; 12 Not surprisingly, the Tat pathway denies the export of polypeptides prone to aggregation. Recently, Fisher et al. showed that tripartite fusions comprising of: (i) the Tat specific signal peptide ssTorA; (ii) variants of the amyloid peptide Aβ42 and (iii) the reporter protein β-lactamase could be localized in the periplasm and confer resistance to β-lactam antibiotics only if the Aβ42 moiety was soluble.13 Mutations that increased the solubility of the Aβ42 peptide domain of the tripartite fusion allowed better Tat export and therefore resulted in higher resistance to β-lactam antibiotics.
The notion that unfolded proteins cannot be translocated via the Tat pathway is supported by in vivo and in vitro evidence from bacteria and from plant thylakoids.14–19 However, the relationship between the in vitro folding properties of a protein and competence for Tat export has not been investigated. Processes that are dictated by the folding kinetics, such as off-pathway reactions leading to the formation of aggregates or interactions with chaperones, are known to be important for protein translocation.20; 21
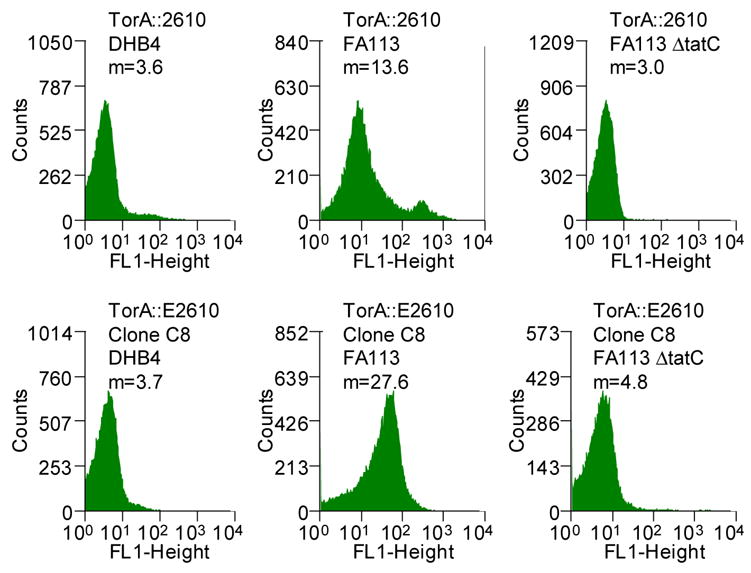
We sought to examine the effect of mutations within the mature protein on the efficiency of export via the Tat apparatus. scFv antibody fragments comprising the VH and the VL immunoglobulin domains linked by a (Gly4Ser)3 are widely used for biotechnology applications, and their folding characteristics have been studied in detail.22–24 The 26-10 scFv antibody fragment binds to digoxin and to other cardiac glycosides with nanomolar affinity.25 Previously, we had reported that the 26-10 scFv can be exported into the periplasmic space by fusion to the Tat specific signal peptide ssTorA from the E. coli trimethylamine N-oxide reductase.11 Export was observed only when the ssTorA-26-10 scFv gene fusion was expressed in E. coli strains that have an oxidizing cytoplasm, such a FA113 (DHB4 trxB gor ahpC*). Expression in trxB gor mutant strains allows the formation of the two disulfide bonds in scFv that are important for the stability of the protein.11; 26; 27 We found that the amount of 26-10 scFv in the periplasm can be monitored by flow cytometry following partial permeabilization of the outer membrane by exposure to hypertonic buffer (5xPBS) and incubation with the fluorescent hapten digoxigenin-BODIPY™ (Figure 1A). Under these conditions, the fluorescent hapten diffuses readily across the outer membrane while the much larger antibody fragment cannot escape from the periplasm. Binding of the hapten by the scFv in the periplasm results in higher cell fluorescence. Figure 1B shows that the cell fluorescence of E. coli FA113 expressing the ssTorA-26-10 scFv fusion was approximately 3 times higher than the background cell autofluorescence in cells that do not contain plasmid. As can be seen in Figure 1B, cells expressing ssTorA-26-10 scFv result in a primary peak corresponding to viable cells with fluorescence considerably higher than the background and a smaller, even more fluorescent secondary peak, corresponding to non-viable cells. Inactivation of the Tat export pathway by deletion of the essential Tat translocon component tatC resulted in low cell fluorescence. Similarly, background fluorescence was detected in the isogenic parental strain E. coli DHB4 (MC4100 phoA(PvuII) phoR malF3) where the oxidative folding of the scFv in the cytoplasm is prevented (Figure 1B).
Figure 1.
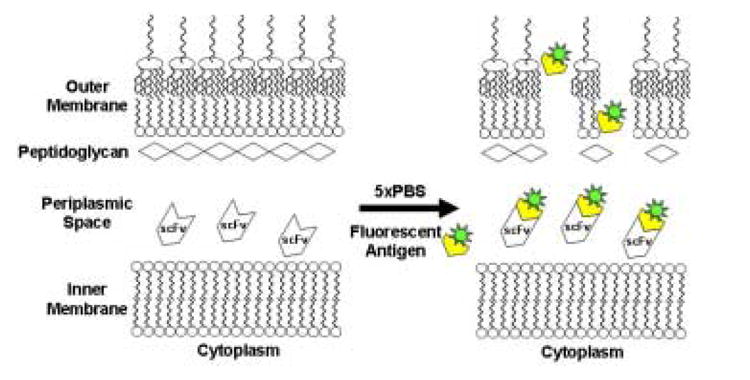
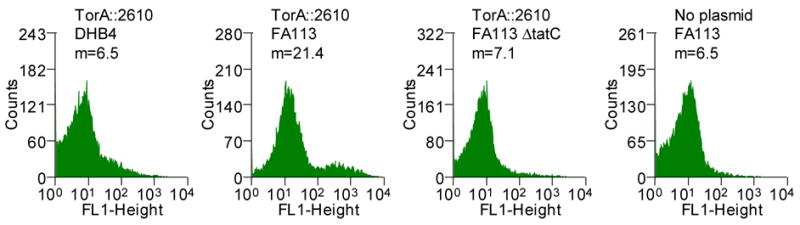
(A) Schematic showing the detection of periplasmic scFv antibodies via binding to fluorescent hapten. (B) Flow cytometric analysis of ssTorA-26-10 in E. coli strains DHB4, FA113 and FA113 ΔtatC. FA113 cells without plasmid is shown as a control. Cells were grown in TB media containing 25 μg/ml chloramphenicol at 37°C to an OD600 of approximately 0.3, and protein synthesis was induced by adding arabinose to a final concentration of 0.2%. Cultures were grown for an additional 4 hours at 30°C at which point 0.1 ml aliquots were mixed with 0.9 ml 5xPBS containing 200 nm digoxigenin-BODIPY™ (Molecular Probes). The samples were incubated at room temperature for one hour with shaking, the cells were pelleted and resuspended in 1 ml of 5xPBS and then analyzed by flow cytometry (Becton-Dickinson FACSort). Cell populations with indicated fluorescence intensities are represented as histograms (m: mean cell fluorescence intensity).
The amount of 26-10 scFv protein in the cytoplasmic and periplasmic fractions was evaluated by cell fractionation using the osmotic shock procedure, followed by Western blot analysis.11; 28 The 26-10 scFv protein was detected using an anti-hexahistidine monoclonal antibody that recognizes the C-terminal His6 tag. Consistent with the FACS analysis, the scFv protein could be detected in the periplasmic fraction from E. coli FA113 cells but not in the periplasm of E. coli DHB4 or FA113 ΔtatC (FA113 tatC::spec). In E. coli FA113 cells, the intensity of the antibody fragment band in the periplasmic fraction was only about 20–25% of the amount found in the cytoplasm indicating that export via Tat was inefficient (see Figure 3A). Accumulation of precursor protein in the cytoplasm is frequently observed when natural Tat substrate proteins or fusions to Tat signal peptides are expressed from multicopy plasmids and this seems to be confirmed here.29–32
Figure 3.
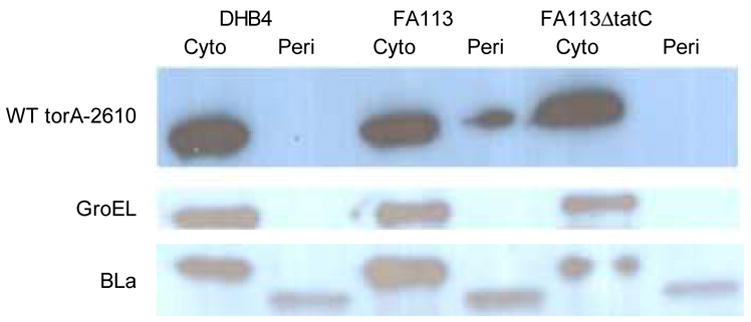
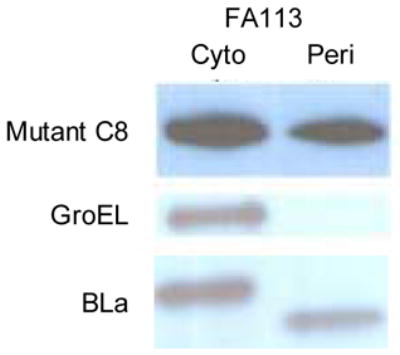
(A) Western blot analysis of cell fractions from E. coli DHB4, FA113 or FA113 ΔtatC expressing ssTorA-26-10 scFv. Cell fractionations were performed by the cold osmotic shock procedure.11 After fractionation, the cell pellet was resuspended in 266 μl of PBS, sonicated for 30 s, and recentrifuged to obtain the soluble cytoplasmic fraction and remove insoluble cell debris. The primary antibody for scFv detection was a monoclonal mouse anti-polyhistidine peroxidase conjugate (Sigma) diluted 1:10,000. Western blots were also probed with rabbit anti-GroEL antibody (Sigma) and rabbit anti-β-Lactamase (Chemicon International) for fractionation controls. Note that, as expected, GroEL and the β-Lactamase precursor are found exclusively in the cytoplasmic fraction and β-Lactamase is found exclusively within the periplasmic fraction. (B) Western blot of cells expressing ssTorA-C8 26-10. Amounts corresponding to equal number of cells were loaded on each lane. Identical results were obtained from duplicate cultures.
The fact that the amount of the 26-10 scFv in the periplasm can be monitored quantitatively by flow cytometry was exploited to screen for mutations that enhance export via Tat. By detecting the digoxigenin-BODIPY™ fluorescence, the amount of periplasmic scFv within each cell is determined directly in the absence of reporter proteins that can otherwise dominate the solubility and folding kinetics of protein fusions.13; 33; 34 The 26-10 scFv gene was subjected to random mutagenesis by error prone PCR35 to give a library of 6×106 independent transformants. Sequencing of six clones picked at random revealed an average of 13 nucleotide substitutions per gene which corresponds to a 1.3% error rate. The library cell population was labeled by incubation with 200 nM digoxigenin-BODIPY™ and fluorescence was analyzed by flow cytometry. As expected, the library displayed a lower mean fluorescence than cells expressing 26-10 scFv since the majority of random mutations are deleterious for function (Figure 2A).36 Clones exhibiting higher cell fluorescence were isolated by FACS. To facilitate the isolation of antibody variants that accumulate at a higher level in the periplasm, rather than mutants exhibiting higher hapten affinity, cell labeling was carried out using 200 nM digoxigenin-BODIPY™, a concentration significantly higher than the 2.3 nM KD of the 26-10 scFv antibody for digoxigenin.37 107 cells were sorted and the 0.3% most fluorescent events were collected. Since incubation in 5xPBS, which is required for outer membrane permeabilization, reduces the cell viability and thus increases the possibility that rare clones might be lost, the collected cells were lysed, and the ssTorA-26-10 scFv genes in that population were recovered by PCR. The PCR product was cloned back to the original vector and electroporated into E. coli XL1-Blue for plasmid propagation. Purified plasmid was obtained from this strain and used to transform E. coli FA113 to give rise to 6×104 transformants. After growth of the pooled transformants in liquid TB media with antibiotic, cells were labeled and subjected to a second round of sorting as above. The cell fluorescence after each round of sorting increased through three rounds of sorting, suggesting successful enrichment of more fluorescent cells (Figure 2A). No additional enrichment was obtained after a fourth round of sorting.
Figure 2.
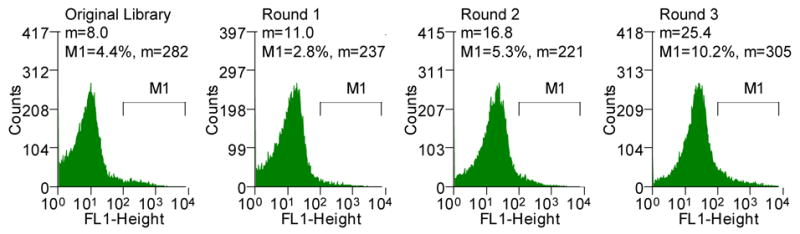
(A) Histograms showing the mean fluorescent intensities of the scFv library and the population of collected cells after each round of sorting (M1, marker indicating the percent of the total population in that region with the corresponding mean fluorescent intensity, m). The torA-2610 scFv fusion was cloned using overlap PCR and primers 1–6 (See Table S1, supplemental information). Primers 3 and 4 changed a NheI restriction site after the torA signal to an XbaI restriction site; primers 5 and 6 removed an XbaI restriction site internal to 26-10 scFv gene with a silent mutation. pTrc99AtorA-2610 (lab collection) was used as the template DNA. The amplified fragment was inserted into pBAD18(Cm). Primers 9 and 10 were used for error-prone PCR35 of the 26-10 scFv gene to create a library with approximately 13 mutations in 26-10 scFv or a 1.3% error rate which was determined by sequencing six clones from the library. The fragments were inserted into the pBAD18(Cm)torA-2610 plasmid using the XbaI and XhoI restriction sites. The ligation mixture was electroporated into the XL1-Blue (Stratagene) strain and plated for growth overnight. Plasmid was isolated from the resulting colonies by QIAprep Spin Miniprep Kit (QIAGEN). The isolated plasmids were then used to transform the library into the E. coli FA113. The library was then sorted on a MoFlo flow cytometer (FC) equipped with an Innova 90C argon laser (Cytomation, Fort Collins, CO). Triggering on side scatter was set to 488 nm light from the argon laser. Detection of digoxigenin-BODIPY binding was monitored by 518 nm emission via a 530/40 band-pass filter (FL-1). By gating for cells showing high FL-1 signal, the library of scFvs were subjected to three rounds of sorting. Between each round of sorting, the collected events were subject to colony PCR using primers 9 and 10 and reinserted into the pBAD18(Cm)torA-2610 construct using the XbaI and XhoI restriction sites. Clones isolated after round 3 PCR and transformation were picked to 96-well plates and checked by flow cytometry. (B) Fluorescent histograms of cells expressing ssTorA-C8 or the ssTorA-26-10 scFv. (C) Fluorescence histograms of E. coli DHB4 expressing ssPelB-C8 or ssPelB-26-10 scFv which are exported via Sec. The PelB signal peptide was cloned onto the pBAD18(cm)torA-2610 construct using the XmaI and XbaI restriction sites.
Clones isolated after transformation with DNA obtained by ligating the PCR product from the third round into vector were grown in 96-well plate format, and the fluorescence of each clonal population was determined. One clone C8, exhibiting the highest increase in fluorescence relative to the parental 26-10 scFv, was analyzed further (Figure 2B). Consistent with the observed 2-fold increase in fluorescence observed with whole cells expressing the C8 scFv, Western blot analysis revealed that, qualitatively about 40% of the C8 scFv protein is found in the periplasm compared to 20–25% for the parental antibody fragment (Figure 3). DNA sequencing revealed that the C8 clone contained 7 amino acid substitutions, 6 in the framework and 1 (L47 Leu→Pro, Kabbat numbering38) in the light chain complementarity determining region 2 (CDR L2) (Figure S1). In immunoglobulins the framework regions act as a scaffold that support each of the three CDR loops found in both the heavy and light chain variable regions. The length and sequence of the CDR loops is largely responsible for antigen specificity. However, L47 does not make contact with the hapten in the crystal structure and therefore the L47 Leu→Pro mutation in C8 is unlikely to affect the binding affinity (see below). In addition to C8, several lower fluorescence clones (about 50% increase in whole cell fluorescence relative to the parental 26-10 scFv) were also isolated and were found to contain one or two amino acid substitutions. No consensus mutations were found among these clones.
The fluorescence of cells expressing the C8 scFv was dependent on an oxidizing cytoplasm and was abolished in a ΔtatC strain background indicating that, as with the parental 26-10 scFv, both disulfide bond formation and a functional Tat pathway are required for export (Figure 2B). Monomeric antibody fragments were purified to >95% as determined by SDS-PAGE from 1 L cultures by immobilized metal chromatography and gel filtration FPLC. The 26-10 and C8 scFvs were produced, respectively at 0.13 mg/L/OD and 0.24 mg/L/OD. The increased yield of the purified C8 scFv is consistent with the approximately 2-fold higher level of periplasmic protein observed by Western blot analysis and with the increased cell fluorescence of C8 scFv-expressing cells (Figures 2 and 3).
We found that the mutations in C8 did not affect the hapten binding kinetics or equilibrium dissociation constant. C8 and the 26-10 scFv both showed identical binding to BSA conjugated digoxigenin by ELISA (data not shown). The hapten binding kinetics were determined quantitatively by surface plasmon resonance on a BIACore 3000 instrument using 15, 5, 1, 0.5 or 0 nM scFv in duplicate. The association rate constant ka, dissociation rate constant kd, and the equilibrium dissociation rate constant KD for C8 were 3.3 ± 0.10 × 105 M−1 s−1, 2.4 ± 0.01 × 10−3 s−1 and 7.2 nM respectively and were statistically indistinguishable from the respective values for the parental 26-10 scFv (2.6 ± 0.05 × 105 M−1 s−1, 2.0 ± 0.006 × 10−3 s−1 and 7.5 nM). The data above show that, consistent with the screening strategy that led to its isolation, the ability of the C8 scFv to confer increased cell fluorescence was not due to a higher hapten affinity but rather, due to enhanced accumulation in the periplasm.
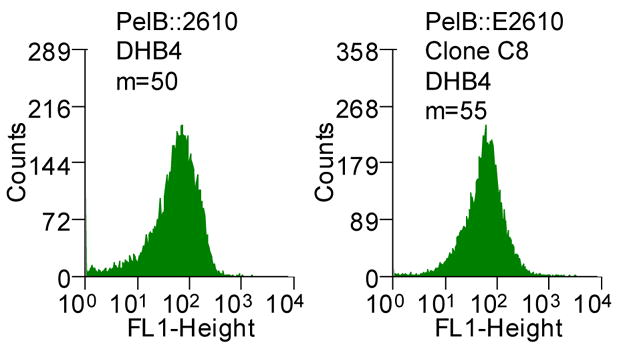
Finally, a higher periplasmic yield of C8 was observed only when the protein was exported via the Tat pathway. Replacement of the ssTorA with the pelB signal sequence directs export through the Sec pathway.25 Precursor proteins competent for Sec export are maintained in a largely unfolded conformation within the cytoplasm in contrast to proteins that are exported through Tat.39 Since the formation of disulfide bonds inhibits Sec export, the accumulation of the scFv in the periplasm was examined in E. coli DHB4 which has a reducing cytoplasm and is isogenic to the oxidizing strain FA113 that had been used for Tat export (Figure 2C). The fluorescence of cells expressing the pelB-C8 scFv fusion was indistinguishable from that of cells expressing the parental pelB-26-10 scFv (Figure 2C). Western blot analysis further revealed identical amounts of C8 and parental 26-10 scFvs in periplasmic fractions (data not shown). Thus, the more efficient periplasmic localization of the C8 scFv occurs only when export is mediated by the Tat pathway.
As shown in Figure 3, the increased amount of C8 scFv in the periplasm was accompanied by a reduction in the amount of protein that remained in the cytoplasm. The observed increase in the yield of periplasmic scFv can be due to one or more of the following factors: (i) increased stability of the C8 protein to degradation in the periplasm; (ii) more efficient targeting of the precursor polypeptide to the TatABC translocon; (iii) greater export competence. The finding that export of the C8 and the 26-10 scFvs via Sec resulted in equal periplasmic yields suggests that the two proteins have similar in vivo stability. Targeting of the scFvs to the Tat translocon is mediated by the signal peptide. Since the mutations in the C8 scFv are all located in the mature protein, they are less likely to influence the efficiency of translocon targeting.40 The higher ratio of periplasmic to cytoplasmic C8 scFv compared to the parental 26-10 scFv is consistent with the third possibility discussed above, namely more efficient export. Unfortunately, as is the case with many Tat precursor proteins expressed from multicopy plasmids, processing by leader peptidase in the periplasm is inefficient and therefore the kinetics of export cannot be measured using pulse-chase techniques.
As was discussed above, for proteins translocated via Sec export competence depends on the folding kinetics of the precursor within the cytoplasm. To investigate whether there is an analogous relationship between folding and the improved export of the C8 scFv via Tat, we examined the guanidine-HCl (GdnHCl) induced equilibrium denaturation, folding kinetics and aggregation propensities of the mutant the parental proteins. The GdnHCl-induced denaturation transition was determined both for oxidized and for completely reduced scFv proteins (Figure 4A). Fluorescence spectroscopy (excitation wavelength 280 nm) revealed that increasing concentrations of GdnHCl caused a shift in the fluorescence emission maxima from 325 nm to 360 nm for the fully denatured proteins. Normalized transition curves were prepared based on the change in emission intensity at 360 nm. For the reduced C8 and 26-10 scFvs, the transition curves were essentially superimposable with midpoints at 2.75 M and 2.65 M, respectively. The transition midpoints for the oxidized scFvs exhibited a similar difference (3.60 M for C8 compared 3.45 M for 26-10). These differences represent around 4% of the midpoint concentrations and are clearly within the experimental error of the measurements. Therefore, the higher export efficiency is not correlated to a change in the stability of the mutant scFv to GdnHCl denaturation.
Figure 4.
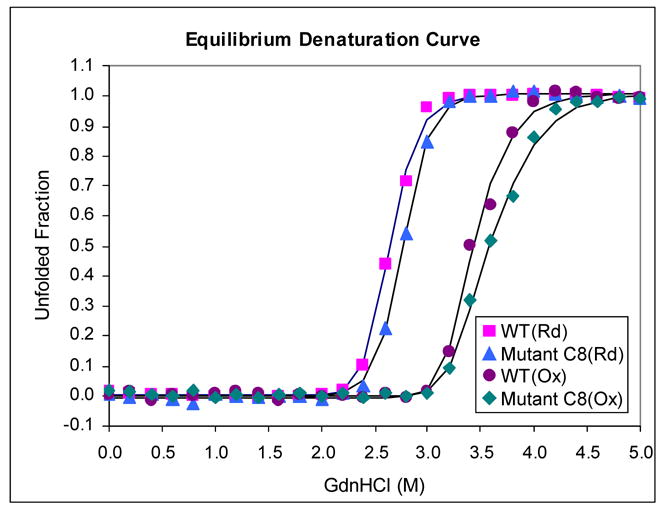
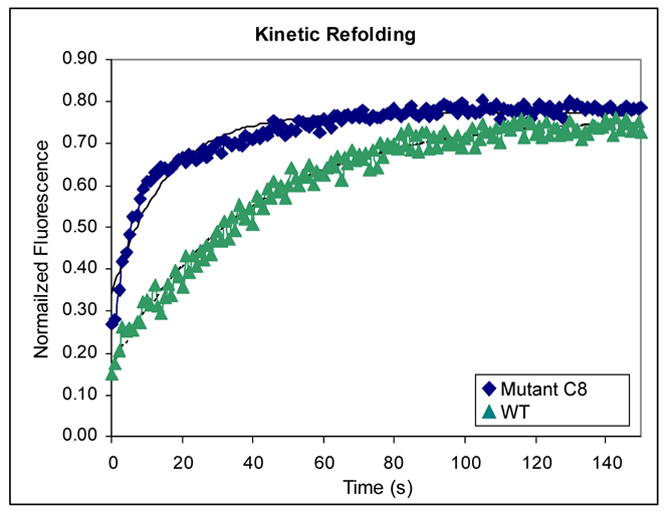
(A) GdnHCl-induced denaturation equilibrium curves. Denaturation equilibrium experiments were carried out in degassed and sterile-filtered BBS (50 mM sodium borate, 150 mM sodium chloride) at 20 °C at a final protein concentration of 15 μg/ml. 15 mM dithiothreitol (DTT) was added to keep the protein reduced when necessary. Excitation wavelength was 280 nm, and the emission spectrum of the protein from three identical samples were scanned in parallel from 320 nm to 370 nm using an AMINCO-Bowman Series 2 Luminescence Spectrometer. The three spectra were averaged, and the fraction of unfolded protein was calculated as described previously by using the maxima at 360 nm.45 Fitted curves are provided only as a visual aid. Rd-reduced, Ox-oxidized. (B) Renaturation under reducing conditions. Proteins were first denatured overnight at 20 °C in BBS containing 80 mM DTT and 5.0 M GdnHCl. The samples were then diluted to 0.5 M GdnHCl using BBS to a final protein concentration of 15 μg/ml, and the time-course for folding was measured by following the increased in fluorescence at 325 nm with excitation at 275 nm. Data points represented the average of four separate experiments. Fitted curves were used to determine the macroscopic rate constants.
The folding kinetics were determined by diluting completely reduced and denatured proteins to a final concentration of 0.5 M GdnHCl while following the change in fluorescence at 325 nm. Consistent with previous studies,27 the folding reaction exhibited three distinct phases. The first phase was completed during manual mixing (~10 sec) and accounted for 15–30% of the total amplitude of the fluorescence increase. The second phase followed first order kinetics and was completed within 180 seconds. Finally, a third slow phase was also observed. The third, slow phase has been shown to arise from peptidyl proline isomerization and exhibits a macroscopic rate <10−3 s−1.24; 41 Differences in the folding kinetics of the C8 and 26-10 scFvs were observed during the second phase (Figure 4B). The fluorescence data could be fit to a single exponential equation resulting in a rate constant of 0.063 s−1 for C8 and 0.025 s−1 for 26-10. In other words, the second critical phase in the folding of the C8 scFv occurs at a rate that is approximately 2.5 times higher than that of the parental antibody fragment, 26-10. It should be noted that cysteine oxidation in E. coli trxB gor strains is relatively slow,26 and therefore the formation of the disulfide bonds in scFvs occurs at a late step, most likely prior to or concomitant with cis-trans proline isomerization.
Finally, we determined the aggregation propensity of the scFv fragments by monitoring the increase in light scattering following prolonged incubation (16 hrs) in different concentrations of GdnHCl (data not shown). An increase in light scattering was observed following prolonged incubation in GdnHCl concentrations near the midpoint of the denaturation transition of both the reduced and the oxidized proteins. In 2.5–3.0 M GdnHCl, the reduced C8 scFv exhibited approximately 30% lower light scattering than the 26-10 scFv; a smaller difference was observed for the oxidized proteins, which showed a lower degree of aggregation.
CONCLUSIONS
Export of the anti-digoxin 26-10 scFv antibody fragment via the Tat pathway by means of the ssTorA signal peptide could only occur in E. coli strains having an oxidizing cytoplasm that allows the formation of its two disulfide bonds. However, we found that the yield of Tat-exported 26-10 scFv in the periplasm was only 0.12 mg/L/OD which is considerably less than the yield obtained when export is mediated by a Sec signal peptide (approximately 1 mg/L/OD, unpublished data). A large fraction of the ssTorA-26-10 scFv is localized in the cytoplasm and is incompatible for export (Figure 3). Accumulation of Tat precursor proteins in the cytoplasm is routinely observed not only with heterologous proteins but also with many native Tat substrates expressed at non-physiological levels.29–32 We took advantage of a quantitative assay for monitoring the amount of periplasmic 26-10 scFv at the single cell level42 to search for mutations in the mature protein that increase the export yield. A single clone, C8, that exhibited a 2-fold higher yield of periplasmic scFv and a proportional reduction in the amount of protein that remained in the cytoplasm was isolated from a library of 6×106 random mutants. Increased periplasmic accumulation was only observed when the protein was exported via the Tat pathway indicating that the mutations in the C8 scFv specifically affected a step important in protein translocation. Comparison of the folding properties of the mutant and the parental scFvs revealed that the former was only marginally more stable to GdnHCl denaturation under either reducing or oxidizing conditions. However, kinetic experiments showed that for the reduced C8 scFv, the critical second phase in refolding is accelerated substantially. In addition, the C8 scFv exhibited a reduction in the aggregation of a folding intermediate populated at denaturant concentrations near the transition midpoint. The folding properties of the C8 scFv bear many similarities to those of the anti-β-galactosidase 13R4 scFv that was isolated earlier based on its ability to fold into an active conformation within the reducing environment of the E. coli cytoplasm.27; 43 Similar to what we found for the anti-digoxigenin scFv mutant in the present study, the reduced 13R4 scFv exhibited a 3-fold acceleration in the intermediate refolding phase and a somewhat lower propensity to aggregate compared to its parental protein. Martineau and Betton suggested that the increase in the folding rate of the 13R4 scFv helps prevent off-pathway interactions that lead to aggregation.27 A reduction in protein aggregation can also play a role in Tat export competence.13 However, the export of scFvs is a complex process that also depends on disulfide bond formation, since only the oxidized protein can be translocated, and ultimately, on the features that are recognized by the folding quality control mechanism of the Tat pathway.
Regardless of the precise mechanism, our data indicate that the rate of folding plays an important role in the Tat export competence of scFv antibodies and possibly of other proteins. Along these lines, we note that thioredoxin-1, a protein that exhibits very fast folding kinetics9 when fused to ssTorA, is exported with very high efficiency even when its expression is driven by very strong promoters (Masip and Georgiou, in preparation). In contrast, when thioredoxin is fused to a signal peptide that directs post-translational export via Sec, it is localized in the periplasm very inefficiently.9; 44 Thus, it is tempting to speculate that Tat export hinges on folding properties that are diametrically opposite to those required for post-translational export through the Sec translocon. If that is the case, genetic screens for improved Tat export could be useful for selecting mutants of soluble proteins exhibiting faster folding. Apart from its practical ramifications for protein expression, the isolation of faster folding proteins can provide insights into the nature of the rate limiting steps in folding.
Supplementary Material
Acknowledgments
We are grateful to Christian Cobaugh and Mark Gebhard for help with BiaCore experiments and Clint Leysath for help with protein purification and to Lluis Masip, Eva-Maria Strauch and Jamie Link for reading the manuscript. This wok was supported by NIH RO1 GM069872 and by a grant from the Foundation of Research.
Abbreviations
- scFv
single chain fragment variable
- Tat
Twin Arginine Translocation
- ssTorA
TorA signal peptide
- TorA
trimethylamine N-oxide reductase
- ssPelB
PelB signal peptide
- FACS
fluorescence activated cell sorting
- PBS
phosphate buffered saline
- GdnHCl
guanidine HCl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271:1519–26. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 2.Kim J, Kendall DA. Sec-dependent protein export and the involvement of the molecular chaperone SecB. Cell Stress Chaperones. 2000;5:267–75. doi: 10.1379/1466-1268(2000)005<0267:sdpeat>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breyton C, Haase W, Rapoport TA, Kuhlbrandt W, Collinson I. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 2002;418:662–5. doi: 10.1038/nature00827. [DOI] [PubMed] [Google Scholar]
- 4.Connolly T, Gilmore R. The signal recognition particle receptor mediates the GTP-dependent displacement of SRP from the signal sequence of the nascent polypeptide. Cell. 1989;57:599–610. doi: 10.1016/0092-8674(89)90129-3. [DOI] [PubMed] [Google Scholar]
- 5.Nakonechny WS, Teschke CM. GroEL and GroES control of substrate flux in the in vivo folding pathway of phage P22 coat protein. J Biol Chem. 1998;273:27236–44. doi: 10.1074/jbc.273.42.27236. [DOI] [PubMed] [Google Scholar]
- 6.Doyle SM, Anderson E, Zhu D, Braswell EH, Teschke CM. Rapid unfolding of a domain populates an aggregation-prone intermediate that can be recognized by GroEL. J Mol Biol. 2003;332:937–51. doi: 10.1016/s0022-2836(03)00955-0. [DOI] [PubMed] [Google Scholar]
- 7.Randall LL, Crane JM, Liu G, Hardy SJ. Sites of interaction between SecA and the chaperone SecB, two proteins involved in export. Protein Sci. 2004;13:1124–33. doi: 10.1110/ps.03410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Randall LL, Hardy SJ. SecB, one small chaperone in the complex milieu of the cell. Cell Mol Life Sci. 2002;59:1617–23. doi: 10.1007/PL00012488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huber D, Cha MI, Debarbieux L, Planson AG, Cruz N, Lopez G, Tasayco ML, Chaffotte A, Beckwith J. A selection for mutants that interfere with folding of Escherichia coli thioredoxin-1 in vivo. Proc Natl Acad Sci U S A. 2005;102:18872–7. doi: 10.1073/pnas.0509583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee PA, Tullman-Ercek D, Georgiou G. The Bacterial Twin-Arginine Translocation Pathway. Annu Rev Microbiol. 2006 doi: 10.1146/annurev.micro.60.080805.142212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLisa MP, Tullman D, Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A. 2003;100:6115–20. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richter S, Bruser T. Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J Biol Chem. 2005;280:42723–30. doi: 10.1074/jbc.M509570200. [DOI] [PubMed] [Google Scholar]
- 13.Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 2006;15:449–58. doi: 10.1110/ps.051902606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halbig D, Wiegert T, Blaudeck N, Freudl R, Sprenger GA. The efficient export of NADP-containing glucose-fructose oxidoreductase to the periplasm of Zymomonas mobilis depends both on an intact twin-arginine motif in the signal peptide and on the generation of a structural export signal induced by cofactor binding. Eur J Biochem. 1999;263:543–51. doi: 10.1046/j.1432-1327.1999.00536.x. [DOI] [PubMed] [Google Scholar]
- 15.Rodrigue A, Chanal A, Beck K, Muller M, Wu LF. Co-translocation of a periplasmic enzyme complex by a hitchhiker mechanism through the bacterial tat pathway. J Biol Chem. 1999;274:13223–8. doi: 10.1074/jbc.274.19.13223. [DOI] [PubMed] [Google Scholar]
- 16.Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- 17.Clark SA, Theg SM. A folded protein can be transported across the chloroplast envelope and thylakoid membranes. Mol Biol Cell. 1997;8:923–34. doi: 10.1091/mbc.8.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson C, Bolhuis A. Protein targeting by the twin-arginine translocation pathway. Nat Rev Mol Cell Biol. 2001;2:350–6. doi: 10.1038/35073038. [DOI] [PubMed] [Google Scholar]
- 19.Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem. 2001;276:8159–64. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- 20.Li SY, Chang BY, Lin SC. Coexpression of TorD enhances the transport of GFP via the TAT pathway. J Biotechnol. 2006;122:412–21. doi: 10.1016/j.jbiotec.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 21.Hatzixanthis K, Clarke TA, Oubrie A, Richardson DJ, Turner RJ, Sargent F. Signal peptide-chaperone interactions on the twin-arginine protein transport pathway. Proc Natl Acad Sci U S A. 2005;102:8460–5. doi: 10.1073/pnas.0500737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holt LJ, Herring C, Jespers LS, Woolven BP, Tomlinson IM. Domain antibodies: proteins for therapy. Trends Biotechnol. 2003;21:484–90. doi: 10.1016/j.tibtech.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Hoyer W, Ramm K, Pluckthun A. A kinetic trap is an intrinsic feature in the folding pathway of single-chain Fv fragments. Biophys Chem. 2002;96:273–84. doi: 10.1016/s0301-4622(02)00022-4. [DOI] [PubMed] [Google Scholar]
- 24.Jager M, Pluckthun A. Folding and assembly of an antibody Fv fragment, a heterodimer stabilized by antigen. J Mol Biol. 1999;285:2005–19. doi: 10.1006/jmbi.1998.2425. [DOI] [PubMed] [Google Scholar]
- 25.Daugherty PS, Chen G, Olsen MJ, Iverson BL, Georgiou G. Antibody affinity maturation using bacterial surface display. Protein Eng. 1998;11:825–32. doi: 10.1093/protein/11.9.825. [DOI] [PubMed] [Google Scholar]
- 26.Bessette PH, Aslund F, Beckwith J, Georgiou G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci U S A. 1999;96:13703–8. doi: 10.1073/pnas.96.24.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martineau P, Betton JM. In vitro folding and thermodynamic stability of an antibody fragment selected in vivo for high expression levels in Escherichia coli cytoplasm. J Mol Biol. 1999;292:921–9. doi: 10.1006/jmbi.1999.3105. [DOI] [PubMed] [Google Scholar]
- 28.Levy R, Weiss R, Chen G, Iverson BL, Georgiou G. Production of correctly folded Fab antibody fragment in the cytoplasm of Escherichia coli trxB gor mutants via the coexpression of molecular chaperones. Protein Expr Purif. 2001;23:338–47. doi: 10.1006/prep.2001.1520. [DOI] [PubMed] [Google Scholar]
- 29.Chanal A, Santini CL, Wu LF. Specific inhibition of the translocation of a subset of Escherichia coli TAT substrates by the TorA signal peptide. J Mol Biol. 2003;327:563–70. doi: 10.1016/s0022-2836(03)00170-0. [DOI] [PubMed] [Google Scholar]
- 30.DeLisa MP, Lee P, Palmer T, Georgiou G. Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J Bacteriol. 2004;186:366–73. doi: 10.1128/JB.186.2.366-373.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikhaleva NI, Santini CL, Giordano G, Nesmeyanova MA, Wu LF. Requirement for phospholipids of the translocation of the trimethylamine N-oxide reductase through the Tat pathway in Escherichia coli. FEBS Lett. 1999;463:331–5. doi: 10.1016/s0014-5793(99)01661-0. [DOI] [PubMed] [Google Scholar]
- 32.Stanley NR, Sargent F, Buchanan G, Shi J, Stewart V, Palmer T, Berks BC. Behaviour of topological marker proteins targeted to the Tat protein transport pathway. Mol Microbiol. 2002;43:1005–21. doi: 10.1046/j.1365-2958.2002.02797.x. [DOI] [PubMed] [Google Scholar]
- 33.Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, Rogers GK. Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc Natl Acad Sci U S A. 2004;101:9193–8. doi: 10.1073/pnas.0400187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cabantous S, Pedelacq JD, Mark BL, Naranjo C, Terwilliger TC, Waldo GS. Recent advances in GFP folding reporter and split-GFP solubility reporter technologies. Application to improving the folding and solubility of recalcitrant proteins from Mycobacterium tuberculosis. J Struct Funct Genomics. 2005;6:113–9. doi: 10.1007/s10969-005-5247-5. [DOI] [PubMed] [Google Scholar]
- 35.Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal Biochem. 1995;224:347–53. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 36.Daugherty PS, Chen G, Iverson BL, Georgiou G. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain Fv antibodies. Proc Natl Acad Sci U S A. 2000;97:2029–34. doi: 10.1073/pnas.030527597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen G, Dubrawsky I, Mendez P, Georgiou G, Iverson BL. In vitro scanning saturation mutagenesis of all the specificity determining residues in an antibody binding site. Protein Eng. 1999;12:349–56. doi: 10.1093/protein/12.4.349. [DOI] [PubMed] [Google Scholar]
- 38.Martin A. Accessing the Kabat Antibody Sequence Database by Computer. PROTEINS: Structure, Function and Genetics. 1996;25:130–133. doi: 10.1002/(SICI)1097-0134(199605)25:1<130::AID-PROT11>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Knoblauch NT, Rudiger S, Schonfeld HJ, Driessen AJ, Schneider-Mergener J, Bukau B. Substrate specificity of the SecB chaperone. J Biol Chem. 1999;274:34219–25. doi: 10.1074/jbc.274.48.34219. [DOI] [PubMed] [Google Scholar]
- 40.Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Muller M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell. 2003;12:937–46. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- 41.Jager M, Gehrig P, Pluckthun A. The scFv fragment of the antibody hu4D5–8: evidence for early premature domain interaction in refolding. J Mol Biol. 2001;305:1111–29. doi: 10.1006/jmbi.2000.4342. [DOI] [PubMed] [Google Scholar]
- 42.Chen G, Hayhurst A, Thomas JG, Harvey BR, Iverson BL, Georgiou G. Isolation of high-affinity ligand-binding proteins by periplasmic expression with cytometric screening (PECS) Nat Biotechnol. 2001;19:537–42. doi: 10.1038/89281. [DOI] [PubMed] [Google Scholar]
- 43.Martineau P, Jones P, Winter G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 1998;280:117–27. doi: 10.1006/jmbi.1998.1840. [DOI] [PubMed] [Google Scholar]
- 44.Schierle CF, Berkmen M, Huber D, Kumamoto C, Boyd D, Beckwith J. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol. 2003;185:5706–13. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pace CN. Measuring and increasing protein stability. Trends Biotechnol. 1990;8:93–8. doi: 10.1016/0167-7799(90)90146-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.