

Abstract
Intermittent administration of parathyroid hormone (PTH) stimulates bone formation by increasing osteoblast number, but the molecular and cellular mechanisms underlying this effect are not completely understood. In vitro and in vivo studies have shown that PTH directly activates survival signaling in osteoblasts; and that delay of osteoblast apoptosis is a major contributor to the increased osteoblast number, at least in mice. This effect requires Runx2-dependent expression of anti-apoptotic genes like Bcl-2. PTH also causes exit of replicating progenitors from the cell cycle by decreasing expression of cyclin D and increasing expression of several cyclin-dependent kinase inhibitors. Exit from the cell cycle may set the stage for pro-differentiating and pro-survival effects of locally produced growth factors and cytokines, the level and/or activity of which are known to be influenced by PTH. Observations from genetically modified mice suggest that the anabolic effect of intermittent PTH requires insulin-like growth factor-I (IGF-I), fibroblast growth factor-2 (FGF-2), and perhaps Wnts. Attenuation of the negative effects of PPARγ may also lead to increased osteoblast number. Daily injections of PTH may add to the pro-differentiating and pro-survival effects of locally produced PTH related protein (PTHrP). As a result, osteoblast number increases beyond that needed to replace the bone removed by osteoclasts during bone remodeling. The pleiotropic effects of intermittent PTH, each of which alone may increase osteoblast number, may explain why this therapy reverses bone loss in most osteoporotic individuals regardless of the underlying pathophysiology.
Keywords: parathyroid hormone, bone formation, osteoblasts, differentiation, apoptosis
INTRODUCTION
Daily injections of parathyroid hormone (PTH) amino-terminal peptide 1–34, or the full length protein PTH(1–84), increase bone mass and reduce the incidence of fracture in postmenopausal women, in elderly men, and in women with glucocorticoid-induced osteoporosis (reviewed in [1]). The anabolic effect of intermittent PTH has also been extensively demonstrated in mice and rats [2]. These effects are achieved by repeated transient exposure of the skeleton to PTH as the hormone is cleared from the circulation within 2–3 hours after administration [3–5]. This anabolic response of the skeleton to repeated cycles of systemic PTH elevation is quite distinct from the effect of continuous PTH elevation and results from increased bone formation on the surfaces of cancellous, endocortical and periosteal bone of the appendicular and axial skeleton. Though less studied, intermittent administration of parathyroid hormone related protein (PTHrP) peptide 1–36, an amino-terminal fragment of the other principal ligand of the PTH receptor, is also anabolic [6].
Accretion of bone mass in humans is most rapid during the first 6 to 12 months of PTH administration, and the response tends to wane thereafter [1]. Increased bone formation is manifested as early as 28 days as evidenced by a rise in the level of circulating markers of osteoblast function, and an increase in tetracycline-labeled surface in transileal biopsies [7–9]. By 6 months, indices of bone resorption have also increased [1, 9]. Thus, the increased bone formation occurs without increased bone resorption in the initial stages of the response, whereas anabolism occurs within the context of increased remodeling after approximately 6 months.
Histologic studies have shown that the increase in bone formation is largely due to an increase in the number of matrix-synthesizing osteoblasts [7, 8, 10–13]. Increased osteoblastogenesis, attenuation of osteoblast apoptosis, and activation of quiescent lining cells have been proposed as explanations for this effect of PTH (Fig. 1). This review addresses each of these mechanisms in light of recent advances in the understanding of the control of osteoblast differentiation and survival, the effect of short term exposure to PTH on the behavior of osteoblasts and osteoblast progenitors in vitro and in vivo, and the efficacy of intermittent PTH in genetically modified mice. The emphasis will be placed on regulation of osteoblast number in cancellous bone because information about the effect of PTH on the behavior of osteoblasts has primarily been obtained at this site. When informative, however, findings from periosteal bone will be included.
Figure 1. Proposed cellular mechanisms involved in the anabolic effect of intermittent PTH.
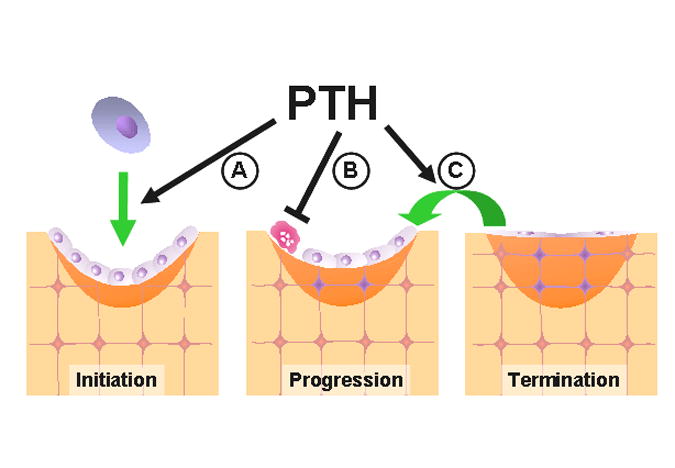
The 3 stages of bone formation are described in the text. Intermittent PTH has been proposed to increase osteoblast number by (A) increasing the development of osteoblasts, (B) inhibiting osteoblast apoptosis, and (C) reactivating lining cells to resume their matrix synthesizing function.
BASIC ASPECTS OF THE CONTROL OF OSTEOBLAST NUMBER
Bone modeling and remodeling
Bone formation in adult cancellous bone takes place only at sites of bone remodeling. During this process, old bone is replaced with new at discrete sites by the basic multicellular unit (BMU), which comprises teams of osteoclasts and osteoblasts. Bone formation also occurs on periosteal surfaces by a process called modeling since it does not require previous resorption.
Remodeling of cancellous bone begins with the retraction of lining cells that cover the bone surface. Osteoclasts, which develop from hematopoietic progenitors, are recruited to the site and excavate the calcified matrix. Then, the cavity is refilled by osteoblasts via a process that occurs in three distinct phases: initiation, progression and termination [14]. During the initiation phase, a team of osteoblasts arising from local mesenchymal stem cells assembles at the bottom of the cavity and bone formation begins. As bone formation progresses, some osteoblasts are entombed within the matrix as osteocytes but the majority die by apoptosis. Bone formation terminates when the cavity has been refilled, at which time the few osteoblasts that remain become the flat lining cells that cover the quiescent surfaces of bone. Once formed, few osteocytes die. Their viability is likely maintained by physiological levels of mechanical stimulation. When mechanical forces are reduced, for example in weightlessness, osteocytes die by apoptosis. This event appears to act as a beacon for osteoclast recruitment and generation of a new BMU, which in turn replaces the old bone containing dead osteocytes with new bone containing viable osteocytes [15].
Unbalanced remodeling with either excess or deficiency of osteoblasts is due to alterations in the commitment, proliferation, and differentiation of osteoblast progenitors, as well as the life span of mature matrix-synthesizing osteoblasts (reviewed in [16] and [17]). Hence, each phase of bone formation represents a potential site at which intermittent PTH could act to increase osteoblast number (Fig. 1).
Histomorphometric measurements made in cancellous bone of animals and humans receiving intermittent PTH have demonstrated a 3–5 fold increase in mineralizing surface, which reflects increased osteoblast number [7, 8, 10–13]. The expected rise in osteoblast number has been quantified in rodents; and it is manifested in mice as early as after 2 daily injections [12]. PTH also increases mineral apposition rate by approximately 50%, which indicates an increase in osteoblast number, osteoblast vigor, or both [14]. Because the magnitude of the increase in mineral apposition rate is smaller compared to the increase in mineralizing surface, increased osteoblast number explains most of the PTH-induced increase in bone formation.
In humans receiving daily injections of PTH for 28 days [7], or for 12–24 months [8], most of the increase in bone formation occurred within pre-existing BMUs. New bone formation also took place on surfaces adjacent to the BMU, perhaps due to spillage of osteoblasts outside the boundary of the bone surface being actively remodeled. Thus, PTH over-rides the mechanisms that normally limit the number of osteoblasts in the BMU to that needed to replace the bone previously excavated by osteoclasts. The increase in bone mass that occurs under these circumstances has been designated as bone anabolism [18]. By contrast, agents like alendronate increase bone mass by halting bone resorption while allowing existing osteoblasts to refill the resorption cavity, and this increase is called anti-catabolism.
The effects of intermittent PTH on the BMU are different from the effects of continuous elevation of the hormone. In mild primary hyperparathyroidism, the rate of remodeling is increased due to an increased number of new BMUs (and therefore increased number of osteoclasts and osteoblasts), but the work achieved within each BMU is balanced or may even slightly favor bone formation, resulting in no loss of bone mass or even gain at trabecular sites [19]. However, in severe primary hyperparathyroidism, or in secondary hyperparathyroidism caused by dietary calcium deficiency, increased remodeling leads to loss of bone mass because within each BMU the work is unbalanced in favor of resorption.
Birth and death as determinants of osteoblast number
The control of osteoblast number is determined by the balance between their production and their death [17]. Multipotential mesenchymal stem cells of the bone marrow give rise to osteoblasts as well as other cell types including marrow adipocytes [20]. Lineage specification is achieved by expression of the transcription factors Runx2 and osterix in the case of osteoblastogenesis, and PPARγ in the case of adipogenesis. Osteoblast number is determined in part by the lineage specification process as shown by the high bone mass phenotype of mice haploinsufficient for PPARγ, as well as mice lacking Alox15, the lipoxygenase responsible for generating oxidized lipid ligands of PPARγ [21, 22]. Thus, PPARγ exerts a tonic suppressive effect on osteoblast differentiation and bone formation, most probably by diverting uncommitted progenitors into the adipocyte lineage at the expense of the osteoblast lineage. In support of this contention, mice given the PPARγ ligand rosiglitazone exhibited increased marrow adipogenesis and decreased osteoblastogenesis associated with decreased bone formation and bone loss [23].
As osteoblast differentiation proceeds, there is a gradual decrease in replication and simultaneous acquisition of differentiated characteristics, culminating in the development of post-mitotic matrix-synthesizing osteoblasts. The replication, differentiation and survival of osteoblast progenitors is controlled by locally produced autocrine/paracrine factors including members of the Wnt, Hedgehog and bone morphogenetic protein (BMP) families, as well as transforming growth factor-β (TGF-β), insulin-like growth factor-I (IGF-I), fibroblast growth factor-2 (FGF-2) and interleukin-6 (IL-6) type cytokines [20, 24, 25]. Moreover, many of these growth factors are deposited into the bone matrix by osteoblasts and are thought to be released in active form during osteoclastic bone resorption.
It is increasingly appreciated that the Wnt antagonist sclerostin plays an important role in the regulation of bone formation. This protein is made exclusively by osteocytes and prevents binding of Wnt ligands to their receptors, which comprise Frizzled complexed with either LRP5 or LRP6 [26, 27]. Importantly, defects in sclerostin production lead to the high bone mass phenotype of Van Buchem’s disease, and LRP5 mutations associated with high bone mass lead to decreased ability to interact with sclerostin [28]. Thus, sclerostin normally exerts a restraining effect on osteoblast differentiation. Moreover, osteocytes may contribute to regulation of osteoblast number via alterations in sclerostin production.
Apoptosis is also a critical determinant of osteoblast number [16]. However, apoptosis is a fleeting event that leaves no trace after the process has been completed, which makes the phenomenon difficult to recognize and quantify. Nevertheless, cells undergoing apoptosis briefly exhibit unique histologic features before they die. Among these features, fragmented DNA is frequently used to visualize apoptotic cells because it can be conveniently and sensitively detected by deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL). Even though TUNEL stained apoptotic cells are rare in regenerating tissues, it has been calculated that all the cells of an organ would die in 20 days if the prevalence of apoptosis detected by TUNEL was only 0.4% [29].
Parfitt determined that the majority of osteoblasts disappear between the initiation phase and the termination phase of bone formation in human cancellous bone, and suggested that the missing osteoblasts had died by apoptosis [14]. Consistent with this, apoptotic osteoblasts exhibiting TUNEL staining have been detected in murine cancellous bone [16, 30]. Based on estimates of osteoblast life span in mice (10–12 days) and the length of time that apoptotic cells exhibit histologic evidence of DNA fragmentation (which ranges from several hours to 1 day depending on the sensitivity of the reagents used), it was calculated that the majority of osteoblasts in murine cancellous bone die, even though the apoptotic osteoblasts themselves are rarely observed [16, 30]. It is therefore evident that changes in the fraction of osteoblasts that die would cause significant changes in the amount of the work accomplished by osteoblasts within the BMU. Consistent with this notion, the reduced osteoblast number and bone formation rate observed in mice and humans receiving glucocorticoids [31], as well as in aging mice [32], is associated with increased osteoblast apoptosis. Conversely, the high bone mass phenotype of mice expressing an activating mutation of LRP5 is associated with decreased osteoblast apoptosis [33].
In contrast to the situation in murine cancellous bone, apoptotic osteoblasts in transileal biopsies from normal subjects have been difficult to demonstrate [31]. However, this does not mean that fewer osteoblasts die by apoptosis in humans. Rather, the chance to detect dying osteoblasts in human bone specimens must be considerably lower due to a longer osteoblast life span, and a lower bone formation rate in humans as compared to mice (A.M. Parfitt, personal communication). The average osteoblast life span is about 12 days in normal mice [31] and about 150 days in humans [34]. In normal mice, about 1 in 150 osteoblasts are undergoing apoptosis at a given moment as determined by TUNEL in our original study [30]. If the duration of this morphologic feature of apoptosis is similar in the two species, the proportion of apoptotic osteoblasts in human bone would be 1 in 1800. The average bone formation rate in mice is about 55 μm2/μm/y [31] and the corresponding value in humans is about 14 μm2/μm/y [35]. If the relationship between osteoblast number and bone formation rate is similar in the two species, the probability of seeing an apoptotic osteoblast in a histologic section is about 50 times smaller for human than for murine bone. Therefore, the lower prevalence of apoptotic osteoblasts in human as compared to murine bone dictates that 50 times more osteoblasts must be identified and characterized in human specimens before the phenomenon can be accurately quantified.
Apoptotic osteoblast progenitors have not been identified in adult cancellous bone because the progenitors are difficult to detect in an unambiguous fashion. However, identifiable osteoblast progenitors exhibiting TUNEL staining have been observed near the osteogenic front of developing murine calvaria bone [36], and near the newly forming bone that develops during distraction osteogenesis – a process in which long bones are cut and gradually stretched, resulting in de novo bone formation within the expanding gap [37]. Hence, it is very likely that osteoblast progenitors that supply the BMU in adult bone also undergo apoptosis.
MOLECULAR MECHANISM OF PTH ACTION: RECEPTORS, SIGNALING CASCADES AND TRANSCRIPTION FACTORS
General aspects
The actions of PTH and PTHrP are mediated by a G protein coupled receptor, referred to as PTH receptor 1 (PTHR1) [38]. Ligand binding to PTHR1 stimulates Gαs-mediated activation of adenylyl cyclase which stimulates cAMP production, and subsequent activation of protein kinase (PKA). PTHR1 also stimulates Gαq-mediated activation of protein kinase C (PKC). In addition, PTH activates β-arrestin-mediated activation of extracellular regulated kinase (ERK) signaling [39, 40]. β-arrestin is also involved in desensitization of cAMP signaling by PTHR1.
Osteoblast specific actions of PTH signaling involved with anabolism
The differential stimulation of Gs- and Gq-mediated signaling by various amino-terminal fragments of PTH has permitted dissection of signaling pathways required for PTH-induced anabolism. Daily injections of PTH(1–34) or PTH(1–31) produced an equivalent anabolic effect in rats; however, whereas PTH(1–34) activates both cAMP production and PKC, PTH(1–31) only activates cAMP production [41]. On the other hand, PTH(3–38), which activates PKC but not cAMP production, did not cause an anabolic effect [42]. Therefore, PTH-stimulated cAMP production is sufficient for initiation of signaling cascades that increase osteoblast number, but activation of PKC is not.
Intermittent PTH increased osteoblast number in vertebral cancellous bone of mice lacking β-arrestin-2, indicating that β-arrestin-mediated ERK activation is not involved. However, PTH did not increase bone mass in these mice due to an exaggerated osteoclastogenic response [43]. The latter was likely caused by loss of β-arrestin-mediated attenuation of cAMP production, which led to increased production of the pro-osteoclastogenic cytokine receptor activator of NF-kB ligand (RANKL), and an increase in osteoclast number and bone resorption. Interestingly, the anabolic effect of intermittent PTH on periosteal bone in β-arrestin null mice was larger than that in wild type mice. This result suggests that β-arrestin opposes the anabolic actions of PTH in periosteal bone. A similar exaggerated osteoclastogenic response to intermittent PTH may explain why deletion of CREM/ICER, a PTH-induced protein that attenuates cAMP-mediated activation of gene transcription, prevented the anabolic effect of PTH [44]. In that study it was shown that PTH increased osteoblast number and bone formation rate in femoral bone of both wild type and CREM/ICER null mice, but the increase in osteoclasts was much greater in the latter. The findings in these two mouse models of attenuated negative feedback signaling support the notion that the degree of bone anabolism caused by intermittent PTH can be limited by the ability of the hormone to also stimulate bone resorption – a phenomenon that may contribute to the gradual loss of anabolism with continued administration of the hormone [1, 45].
Studies with osteoblastic cell models and cultured rat metatarsal bones indicated that PTH rapidly and transiently increases Runx2 mRNA and protein levels, and also activates a PKA-dependent increase in Runx2 activity [46]. Increased Runx2 levels may be secondary to cAMP-dependent down-regulation of cyclin D1, which besides controlling the cell cycle, also targets Runx2 for proteasomal degradation [47]. PTH may also raise Runx2 activity by increasing the synthesis of the cyclin-dependent kinase inhibitors p27KIP1 and p21Cip1 (which also cell cycle-regulating proteins) in a cAMP-dependent fashion. In this mechanism, a rise in the levels of p27KIP1 and p21Cip1 cause in increase in the activity of retinoblastoma protein, which in turn binds to and enhances Runx2 transactivation capacity [48, 49]. As discussed below, the PTH-activated signaling cascades involving cAMP-dependent activation of PKA, and changes in the expression of cyclins, cyclin dependent kinase inhibitors and Runx2, may play an important role in both the pro-survival and pro-differentiating effects of PTH on cells of the osteoblast lineage.
HOW DOES INTERMITTENT PTH INCREASE OSTEOBLAST NUMBER?
Advantages and limitations of in vivo and in vitro approaches to the study the actions of intermittent PTH
Important information on the mechanisms by which intermittent PTH increases osteoblast number has been obtained by studying the response in remodeling cancellous bone of rodents, in which at least some aspects of the birth and death of osteoblasts can be quantified. However, in vivo studies of effects of PTH on osteoblast progenitors have been limited by the difficulty in identifying them, particularly in adults. Moreover, the complexity of bone tissue makes it difficult to obtain detailed molecular information in a cell-specific manner. These problems can be overcome with the use of primary cultures of osteoblast progenitors established from fetal or neonatal calvaria or from the marrow of long bones of adult animals, as well as osteoblast-like cell lines, but these in vitro systems cannot reproduce the architectural and cellular complexity of bone tissue. Also, the only way to ensure that in vitro responses to PTH are relevant to the in vivo effects caused by transient exposure to injected PTH is to study short term effects of the hormone, or to expose cells to the hormone for a few hours per day during longer term studies. This is because, unlike the in vivo situation, PTH is not substantially degraded after addition to cultured osteoblastic cells, and remains fully active for at least 72 hours [12, 50]. Although PTHR1 is desensitized and internalized within minutes after addition of PTH, this phenomenon does not model the effects of transient exposure to the hormone. Indeed, some type of continued PTHR1 signaling in the presence of the hormone seems likely in view of the dramatic differences between the effects of continuous and intermittent PTH elevation on the skeleton, as exemplified by the response of genes like RANKL to the two forms of hormone administration [51]. Recycling of the PTHR1 to the membrane surface following internalization may be one explanation for continued signaling in the presence of the ligand [52].
Effect of intermittent PTH on osteoblast apoptosis
Studies from our laboratory showed that daily administration of 3 to 300 ng/g/d of PTH for 28 days to adult mice caused a dose dependent increase in the bone mineral density of the spine and hindlimb that was associated with a reduction in osteoblast apoptosis at both skeletal sites [12]. Histomorphometric measurements made in the secondary spongiosa of the distal femur indicated that the same doses of PTH that inhibited osteoblast apoptosis also increased osteoblast number, bone formation rate, and the amount of cancellous bone. Moreover, the prevalence of osteoblast apoptosis (% of osteoblasts with TUNEL labeling) exhibited a strong inverse correlation with circulating osteocalcin, bone formation rate and osteoblast number. A decline in the number of apoptotic osteoblasts was detected after only 2 injections. After 4 injections, there was a 50% reduction in the prevalence of apoptotic osteoblasts, and osteoblast number increased by 2-fold. These results strongly argue that increased survival is a major contributor to the increase in osteoblast number caused by intermittent PTH.
In contrast to the evidence for an anti-apoptotic effect of intermittent PTH on osteoblasts in the remodeling secondary spongiosa of adult mice, Stanislaus et al reported a 50% increase in apoptotic osteoblasts in the primary spongiosa of the distal femur of 1 month old rats after 7 and 21 days, but not after 28 days, of PTH injections [53]. The percentage of osteoblasts exhibiting TUNEL staining was not reported, and the phenomenon was not examined in the remodeling secondary spongiosa. Therefore, it is unknown if the number of apoptotic osteoblasts in primary spongiosa is similar to that reported in secondary spongiosa of murine bone [12]. Interestingly however, these investigators found that the activity of caspases 2, 3, and 7 (several of the proteases that mediate apoptosis) was dramatically decreased in extracts of the femoral metaphyseal bone, which included both primary and secondary spongiosa. This result is consistent with an anti-apoptotic effect of daily hormone injections on osteoblasts in the secondary spongiosa, and/or the pre-osteoblasts that are prevalent in the metaphysis during growth [54].
Recent evidence from our laboratory indicates that the prevalence of apoptotic osteoblasts on the surface of murine vertebral periosteal bone was very low, as compared to osteoblasts on cancellous bone [55]. Assuming an equivalent life span at the two sites, this finding indicates that only a small proportion of osteoblasts die during bone formation on periosteal bone. Moreover, ablation of osteoblast progenitors blocked the increase in osteoblast number caused by intermittent PTH on the surface of periosteal bone. Thus, unlike the situation in cancellous bone, inhibition of apoptosis of matrix-synthesizing osteoblasts does not appear to substantially contribute to the increase in osteoblast number caused by intermittent PTH in murine periosteal bone.
Lindsey et al recently reported that 28 days of intermittent PTH increased the number of apoptotic osteoblasts (expressed as the number of TUNEL-labeled cells per mm of osteoblast surface) in cancellous bone of transileal biopsies in postmenopausal women [56]. These investigators also found a positive correlation between osteoblast apoptosis and bone formation rate, unlike the negative correlation seen in mice receiving intermittent PTH [12]. The opposite effects of PTH on osteoblast apoptosis in mice versus humans could be due to a species difference in the survival and death signaling activated by the hormone. Another possibility is increased background staining related to osteoblasts but not to osteoblast apoptosis. Indeed, experience has shown that TUNEL staining is prone to artifacts due to lack of standardization of the staining procedure and the use of samples with only a limited number of cells available for inspection [57]. Finally, because the number of osteoblasts per mm of bone surface is 2–3 fold higher at the initiation bone formation than at the termination of this process [14], enumeration of the number of TUNEL-stained osteoblasts per mm of osteoblast surface may not accurately reflect the percentage of osteoblasts that are apoptotic during PTH-induced anabolism. These issues, together with the ascertainment difficulties caused by a 50-fold difference in the prevalence of osteoblast apoptosis between mice and humans, mandates more extensive studies of the effect of PTH on osteoblast survival in humans.
Consistent with the in vivo findings in mice, in vitro studies by us and others have demonstrated that short term exposure to PTH or PTHrP inhibits the pro-apoptotic effects of the topoisomerase inhibitor etoposide, serum withdrawal and dexamethasone in a variety of rat, murine and human osteoblastic cell preparations [12, 30, 58]. The only in vitro study reporting a PTH-induced increase in apoptosis of osteoblastic cells required overnight exposure of post-confluent C3H10T1/2 or MC3T3-E1cells to PTH [58]. Thus, this observation does not seem to be relevant to the effects of transient hormone exposure on osteoblast survival.
Mechanistic studies using cultured osteoblastic cells showed that PTH rapidly activated anti-apoptotic signaling pathways that involve cAMP-mediated activation of PKA and subsequent phosphorylation and inactivation of the pro-apoptotic protein Bad, as well as increased transcription of survival genes like Bcl-2 [12] (Fig. 2). Moreover, the increased synthesis of survival genes required cAMP response element-binding protein (CREB) and Runx2. Hence, the ability of PTH to transiently increase the level and activity of Runx2 [46] may not only promote differentiation but also enhance survival signaling in osteoblasts and perhaps osteoblast progenitors. The PTH-stimulated increase in p21Cip1 that contributes to the increase in Runx2 may also have a separate pro-survival effect as p21Cip1 can prevent activation of caspases and block the pro-apoptotic effects of apoptosis signal-regulating kinase 1 [59]. Indeed, p21Cip1 mediates the pro-differentiating and pro-survival effects of IL-6 type cytokines on osteoblastic cells [60].
Figure 2. PTH-stimulated survival signaling in osteoblasts.
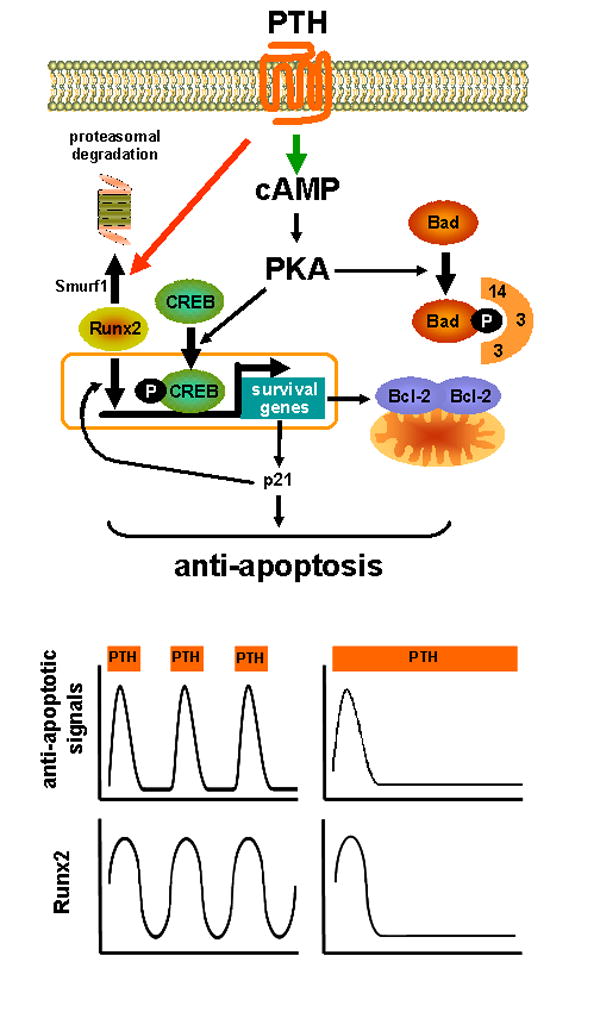
The upper panel depicts PTH-activated survival pathways, and the negative feedback on these pathways caused by stimulation of Runx2 degradation. The lower panel depicts the transient anti-apoptotic signaling and the changes in Runx2 that occur upon each injection of PTH. The net result is a temporary delay in osteoblast apoptosis. As injected PTH disappears from the circulation, Runx2 returns to basal levels and the cells are positioned to generate another episode of anti-apoptotic signaling in response to a subsequent injection. With sustained PTH elevation, on the other hand, the negative feedback pathway remains active, and further anti-apoptosis signaling is abrogated.
The anti-apoptotic effect of PTH lasts only about 6 hours when examined in vitro [12]. This is due to Smurf1-activated proteasomal degradation of Runx2 as indicated by evidence that survival signaling in OB-6 cells was prolonged to at least 24 hours by a proteasome inhibitor, a dominant negative mutant of Smurf1 that targets Runx2 for degradation, or a mutant Runx2 lacking the Smurf1 recognition site. Consistent with short-lived PTH-induced survival signaling, osteoblast apoptosis was not affected by 6 days of continuous elevation of PTH in mice by infusion, or by 2 days of hormone elevation caused by feeding a calcium deficient diet [12].
These findings have led us to propose that intermittent PTH activates short bursts of survival signaling resulting in repeated delays of apoptosis leading to a reduction in the prevalence of dying osteoblasts and a corresponding increase in osteoblast number (Figure 2). On the other hand, continuous PTH does not affect osteoblast life span because it causes sustained reduction in Runx2 below the threshold needed for survival signaling. Unlike the situation in adult mice with continuous PTH elevation, 2 week old mice expressing a constitutively active PTHR1 in cells of the osteoblast lineage exhibited decreased apoptosis of osteoblasts, and this was associated with increased proliferation of osteoblast progenitors [61]. Unlike the short-term studies mentioned above, the long-term PTHR1 signaling in these mice may have increased the level of growth factors that exert their own proliferative and pro-survival effects, which could override the deleterious effects of any reduction in Runx2 induced by the constitutively active PTHR1.
Effects of intermittent PTH on the replication and differentiation of osteoblast progenitors
The impact of intermittent PTH on the replication of osteoblast progenitors has been studied in 16 month old rats following administration of 3H-thymidine to label dividing cells [62]. Seven daily injections of PTH increased osteoblast number in cancellous bone, but the percentage of labeled osteoblasts that developed from the labeled progenitors was not affected. Therefore, at least in the early stages of PTH-induced anabolism, the increase in osteoblast number in cancellous bone is not due to stimulation of the proliferation of osteoblast progenitors. Daily administration of PTH to young, adult, or aged mice for 4 weeks did not affect the number of mesenchymal stem cells as measured by the number colony forming unit-osteoblasts (CFU-OB) present in femoral marrow isolates [13, 63, 64]. Moreover, SAMP6 mice, a strain with low CFU-OB number, gave an anabolic response to PTH that was equivalent to that of control mice [13], suggesting that anabolism does not depend on actions of the hormone on these uncommitted progenitors.
It is commonly accepted that differentiation requires exit from the cell cycle, and numerous in vitro studies have shown that proliferation decreases as osteoblast differentiation proceeds. Consistent with the possibility that PTH increases osteoblast differentiation by suppressing proliferation, in vivo and in vitro studies have shown that short term exposure to PTH causes exit of osteoblast progenitors from the cell cycle. Thus, intermittent PTH reduced the expression of histone H4, a marker of the cell cycle [65], as well as expression of the cell cycle inhibitors p27KIP1 and p21Cip1 [49], in metaphyseal bone of young rats, a site rich in replicating osteoblast progenitors [54].
Short term exposure to PTH has also been shown to exert rapid suppressive effects on the replication of osteoblastic cell lines [49] and osteoblastic cells isolated from the primary spongiosa of growing rats [54]. Mechanistic studies have shown that the anti-mitotic effect of PTH is due to decreased expression of cyclin D1, which is required for cell cycle progression, as well as increased expression of cyclin-dependent kinase inhibitors p27KIP1, p21Cip1 and p16, which inhibit cell cycle progression [49, 66]. Taken together with the ability of PTH to rapidly and transiently increase Runx2 levels and activity, PTH-induced exit from the cell cycle may be part of a mechanism that promotes osteoblast differentiation (Fig. 3). Locally produced autocrine/paracrine growth factors, or growth factors released during bone resorption, are probably required to complete any pro-differentiating events initiated by PTH. Consistent with a PTH-induced increase in the number of differentiating osteoblast progenitors, marrow cell cultures established from young ovariectomized rats receiving intermittent PTH exhibited an increased ability to form a mineralized matrix compared to vehicle controls [67]. Moreover, intermittent PTH accelerated the development of ossicles formed by murine bone marrow-derived osteoblast progenitors following transplantation into immunocompromised mice [68].
Figure 3. Actions of intermittent PTH on osteoblast progenitors.
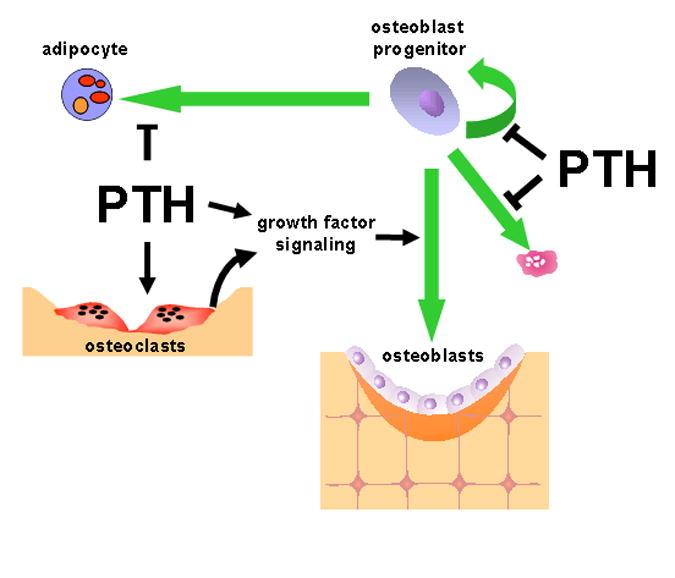
PTH exerts anti-mitotic effects on replicating osteoblast progenitors, and may also inhibit their apoptosis. The anti-mitotic effects may be necessary for differentiation in response to locally produced autocrine/paracrine growth factors regulated by PTH, as well as factors released from the bone matrix during bone resorption. PTH may also increase the number of osteoblast progenitors by preventing the differentiation of adipocytes from multipotential progenitors.
In contrast to the in vivo and in vitro evidence for a direct anti-mitotic action of short term exposure of osteoblast progenitors to PTH, osteogenic cell replication was stimulated by 6 hours of exposure to PTH or PTHrP in cultures established from the bone marrow [69] or calvaria [70] of young rats. In addition, treatment of embryonic chick periosteal cells with PTH for 5–30 minutes stimulated proliferation [71]. Although pro-mitotic actions of PTH have also been reported in cultures of osteoblastic cells maintained for 24 hours or longer in the presence of PTH [72, 73], these responses do not appear to be relevant to the anabolism caused by repeated transient PTH exposure. In view of the available in vivo evidence against a substantial contribution of increased osteoblast progenitor proliferation in response to intermittent PTH [62], it seems reasonable to conclude that the proliferative effects of short term PTH exposure seen in vitro reflects either a phenomenon that is highly favored in the tissue culture environment, or the response of a progenitor that makes a comparatively small contribution to the increase in osteoblast number in remodeling cancellous bone. That proliferative effects of PTH may be important in other settings, however, is suggested by the finding that 4 daily injections of the hormone increased proliferation of mesenchymal progenitors, and the development of soft callus, in the early stages of fracture repair in the rat [74].
Attenuation of adipogenesis
Intermittent PTH may also increase osteoblast number by reducing the tonic negative effects of PPARγ on osteoblast differentiation (Fig. 3). In vitro studies have shown that activation of the PTH receptor in pre-adipocytic cells with PTHrP caused phosphorylation of PPARγ and inhibition of its transactivation activity [75]. Further, culture of human mesenchymal stem cells with PTH for 1 hour per day prevented the ability of a cocktail of dexamethasone, insulin, isobutyl-methylxanthine and troglitazone to stimulate the development of adipocytes; and this inhibitory effect of the hormone was mediated by cAMP-activated PKA [50]. Interestingly, PTH inhibition of adipogenesis was not seen when cells were maintained in the continuous presence of PTH in this study. More to the point, the anabolic effect of intermittent PTH in monkeys was associated with a reduction in the number of adipocytes in the bone marrow [76].
Effect of intermittent PTH on lining cell activation
Leaffer et al reported that the increase in osteoblasts covering the surface of cancellous bone of rats following intermittent PTH administration was accompanied by a reciprocal reduction in the number of the flat lining cells that cover the surface of quiescent bone [77]. Following cessation of hormone administration, osteoblast number declined while lining cells increased. Based on this evidence, these investigators proposed that intermittent PTH caused lining cells to revert to their osteoblast phenotype. The appeal of this hypothesis is that it provides a potential explanation for the rapid appearance of osteoblasts on previously quiescent bone surfaces in response to PTH. However, there are too few lining cells to account for the osteoblasts needed to cover the expanded cancellous bone surface and the increased osteocyte number and density observed in mice receiving daily PTH injections [13]. Moreover, the reciprocal changes in osteoblast and lining cell number could also be explained if lining cells were replaced by osteoblasts that overfill the resorption cavity and migrate onto adjacent quiescent surfaces as suggested by the evidence from recent humans studies [7, 8]. Lining cell activation has also been invoked to explain the PTH-induced increase in osteoblasts when remodeling is low or absent, but the ability of the hormone to stimulate osteoblast differentiation could accomplish the same result. A better understanding of the biosynthetic properties of lining cells, and their response to PTH or to factors affected by PTH, is needed before the role of these cells in PTH-induced anabolism can be established.
THE ROLE OF AUTOCRINE/PARACRINE REGULATORS OF THE GENESIS AND SURVIVAL OF OSTEOBLASTS
Besides direct activation of pro-differentiating and pro-survival signaling in osteoblastic cells, PTH also affects the synthesis of many osteogenic growth factors and cytokines [78–81], as well as their antagonists and proteolytic activators [4, 81–83]. Hence, PTH may indirectly stimulate the differentiation and survival of pre-osteoblasts and osteoblasts via regulation of these autocrine/paracrine factors; and this response could complete the pro-differentiating effects caused by exit of replicating progenitors from the cell cycle. Results from studies with genetically modified mice have indicated a role for several of these factors in PTH-induced bone anabolism.
IGF-I
PTH stimulated the synthesis of IGF-I via a cAMP-dependent mechanism, and this factor exerts pro-differentiating and pro-survival effects on osteoblasts [78, 84]. In rats, 4 weeks of daily PTH injections increased IGF-I transcripts in osteoblasts as determined by in situ hybridization [85]. Interestingly, however, mRNA levels were not affected by short term exposure of cultured osteoblastic cells to PTH [86]. Therefore, the increase in IGF-I seen in vivo after 4 weeks of PTH administration may be mediated by another PTH-regulated factor; or it may reflect increased synthesis of IGF-I with repeated exposure to PTH as has been seen for p21Cip1 as well as several other genes [49, 87].
Consistent with a role of IGF-I in the anabolic effects of intermittent PTH, daily hormone injections for 14 days to IGF-I null mice failed to stimulate periosteal bone formation or to increase cortical thickness at the tibiofibular junction, whereas wild type mice exhibited the expected anabolic response [88]. For reasons that are unclear, intermittent PTH caused loss of cancellous bone in proximal tibia in wild type mice in this experiment, but this response did not occur in the IGF-I null mice. In a separate study, it was found that 5 week old IGF-I null mice failed to exhibit an anabolic response to 10 days of PTH injections at the distal femoral metaphysis, whereas wild type mice gave a 40% increase in bone mass at this site [89]. Further evidence for the involvement of IGF-I in PTH-induced anabolism was provided by the observation that 10 week old mice lacking insulin receptor substrate adapting molecule-1, which transmits IGF-I receptor signaling, failed to exhibit an anabolic response to 4 weeks of intermittent PTH administration [90]. Finally, deletion of the IGF-I gene from the liver did not affect PTH-induced anabolism in cancellous bone, suggesting that locally produced IGF-I mediates the response at this site [91]. Interestingly, however, the anabolic response was reduced in cortical bone of these mice suggesting a site specific role of liver-derived IGF-I.
FGF-2
PTH rapidly increased FGF-2 transcripts in cultured murine osteoblastic cells via increased cAMP production, and this factor exerts both proliferative and anti-apoptotic effects on osteoblast progenitors [80]. FGF-2 may contribute to the anabolic effect of intermittent PTH because the increase in cancellous bone volume was strongly attenuated in either 2 or 10 month old FGF-2 null mice [92]. Daily injections of FGF-2 also induced bone anabolism, but unlike intermittent PTH which thickened existing trabeculae, FGF-2 increased trabecular number and connectivity [93]. The latter may result from the proliferative effect of FGF-2 on osteoblast progenitors. Since the impact of intermittent FGF-2 on trabecular architecture did not mimic the effect of intermittent PTH, either the increase in FGF-2 induced by intermittent PTH was not big enough to increase trabecular number and connectivity, or the cellular response of osteoblasts and osteoblast progenitors was modified by other growth factors affected by PTH, or perhaps by the anti-mitotic effect of PTH itself.
Sclerostin and Wnt signaling
Administration of PTH to mice or rats caused a transient reduction in the level of sclerostin mRNA, the product of the Sost gene that is expressed exclusively in osteocytes [4, 94]. The effect was rapid, consistent with direct activation of the PTHR1 that is known to be expressed in osteocytes [95]. PTH also rapidly decreased the expression of the Wnt signaling antagonist secreted frizzled related protein-2 in UMR-106 osteoblastic cells [81]. Hence, intermittent PTH could increase osteoblast number via bursts of Wnt signaling, which would lead to increased differentiation and survival of osteoblasts and osteoblast progenitors [25, 96].
Three independent studies in mice lacking the Wnt co-receptor LRP5 have shown that the PTH-stimulated increase in femoral cortical bone mass does not require Wnt signaling [97–99]. However, it might be involved in the anabolic response in cancellous bone. In one of the aforementioned studies [99], administration of PTH every other day for 6 weeks increased osteoblast number in vertebral bone of males and females of both wild type and LRP5 null mice. However, whereas bone formation rate was elevated in females in both mutant and wild type mice, this effect was sharply attenuated in LRP5 null males. PTH failed to increase vertebral cancellous bone in either wild type or LRP5 null mice in this study, perhaps due to the less frequent administration of PTH combined with the fact that manifestation of the anabolic response is comparatively slow in murine vertebral bone [10, 12]. Nevertheless, another group of investigators reported that 30 days of daily PTH injections increased vertebral cancellous bone in male wild type mice [97], but not in male LRP5 null mice [97]. It is possible that Wnt signaling is not completely lost in LRP5 null mice because they still express LRP6, which can also be activated by Wnts. Overall, these studies support the notion that LRP5-mediated Wnt signaling may contribute to the anabolic effect of intermittent PTH in cancellous bone, at least in male mice.
The involvement of osteocytes, the primary mechanosensory cell of bone, in the anabolic effect of intermittent PTH may explain why weight bearing and intermittent PTH exert synergistic effects on bone formation [63, 100–102]; and why the anabolic response is greater in tibiae, which experience high mechanical strain, than in the vertebra, which experience less strain [103]. Moreover, like PTH, mechanical loading of murine bone causes a fall in the level of sclerostin [104]. The potential involvement of osteocytes in PTH-induced anabolism is strongly supported by evidence obtained by our group demonstrating a dramatic increase in bone mass in mice expressing a constitutively active PTHR1 in osteocytes [105].
Osteoclast-derived factors
Intermittent PTH provoked a transient increase in RANKL [51] that may result in a transient increase in osteoclastic resorptive activity. The resulting release of osteoblastogenic growth factors from the bone matrix may be an additional source of osteogenic signals that contribute to PTH-induced anabolism (Figure 3). RANKL may also stimulate the secretion of an osteoblastogenic factor from osteoclasts [106]. Evidence that bisphosphonates attenuated the anabolic effect of intermittent PTH in animal models and humans is consistent with the involvement of osteoclasts in the anabolic effect [107–111], but such inhibition was not seen in other studies [45, 112–114]. Indeed, both OPG and alendronate prolonged the duration of the anabolic effect in ovariectomized mice, apparently by preventing the increased bone resorption that occurs in the later stages of treatment [45]. Thus, the available evidence suggests that the stimulatory effect of PTH on bone resorption may represent a two-edged sword in the setting of anabolic therapy. Intermittent PTH may increase release of osteogenic growth factors from the bone matrix or from osteoclasts, but this effect may be counterbalanced by increased resorption, particularly in later stages of the therapy. However, the later may be overcome administering PTH for short periods of time in a cyclic fashion to maximize the bone forming effects and minimize the pro-resorptive effects [113, 115].
DOES INTERMITTENT PTH INTENSIFY THE OSTEOGENIC EFFECTS OF ENDOGENOUS PTHRP?
The anabolic effect of intermittent PTH has been puzzling because there seems to be no a priori reason why a hormone so intimately involved in calcium homeostasis and bone remodeling should also be anabolic, but only when given in an intermittent fashion. However, recent evidence demonstrates that PTHrP, produced by early osteoblast progenitors [116] as well as osteocytes [117], is an important member of the network of cytokines and growth factors that regulate the differentiation and survival of osteoblasts [118, 119]. Specific deletion of PTHrP in cells of the osteoblast lineage caused a reduction in bone mass that was associated with decreased osteoblast progenitors, decreased osteoblast number and increased osteoblast apoptosis [119]. In contrast, studies with mice lacking PTH indicated that the hormone is mainly involved in the regulation of bone resorption, and thereby the rate of bone remodeling [119].
Sustained elevation of PTHrP, as occurs in malignancy, causes increased osteoclastogenesis and bone resorption via activation of RANKL synthesis. Therefore, under normal circumstances, the exposure of bone cells to PTHrP must be intermittent in a way that ensures that activation of PTHR1 results in osteogenic rather than catabolic effects [120]. In view of the pro-differentiating and pro-survival role of locally produced PTHrP on osteoblasts and osteoblast progenitors, and the similar effects of intermittent PTH as outlined in this review, it is tempting to speculate that daily injections of PTH simply add to existing osteogenic signals arising from PTHrP-induced activation of PTHR1.
THE THERAPEUTIC EFFICACY OF INTERMITTENT PTH ON THE OSTEOPOROTIC SKELETON
Intermittent PTH activates several mechanisms that promote the development and survival of osteoblasts. The pleiotropic effects of PTH, each of which alone might be sufficient to increase osteoblast number, may explain the potent ability of intermittent PTH to increase bone mass in most subjects with osteoporosis regardless of the underlying pathophysiology. Even if the nature of the disease process prevents activation, or reduces the importance, of one of the mechanisms by which PTH increases osteoblast number, other mechanisms may remain which are sufficient for anabolism. Furthermore, some of the pathways activated by the hormone may antagonize pathologic mechanisms leading to bone loss.
In glucocorticoid-induced osteoporosis, osteoblast number is reduced due to deceased genesis and increased apoptosis [31]. Yet, clinical studies have established that intermittent PTH causes a dramatic increase in bone mass in established disease [121], albeit the anabolic response is blunted in mice given prednisolone simultaneously with PTH [122]. In mice with osteoblast-specific expression of 11β-hydroxysteroid dehydrogenase, an enzyme that inactivates glucocorticoids, osteoblasts were protected against the pro-apoptotic effects of glucocorticoids and the negative effects of glucocorticoids on bone formation were ameliorated [123]. These findings indicate that attenuation of osteoblast apoptosis alone is a powerful mechanism for increasing bone formation in the presence of glucocorticoids. Therefore, the prevention of glucocorticoid-induced osteoblast apoptosis by intermittent PTH could contribute strongly to the ability of this therapy PTH to reverse glucocorticoid-induced bone loss. However, the contribution of increased osteoblast survival alone would be small at the beginning of therapy because osteoblast number is low. The ability of PTH to reverse the anti-osteoblastogenic effects of glucocorticoids has not been demonstrated, but such effects may also contribute to the anabolic effects of PTH in the presence of excess glucocorticoids.
Interestingly, intermittent PTH increased osteoblast number and added more bone to the vertebra of 18 month old mice with diminished bone mass than in 3 month old mice that are at or near peak bone mass [64]. And, it was recently reported that the increase in vertebral bone in humans receiving intermittent PTH was slightly greater in older subjects, albeit the reduction in fracture risk was independent of age [124]. As mentioned earlier, osteoblast apoptosis increases with advancing age in mice [32]. Osteoblastogenesis also declines with age, perhaps due to diversion of the common progenitor toward the adipocyte lineage [23, 125]. We have obtained preliminary evidence that the tonic suppression of bone formation by PPARγ increases with advancing age in mice as indicated by increased expression of Alox15 and PPARγ, as well as increased levels of oxidized lipid ligands of PPARγ [126]. Recent evidence from our lab also indicates that the increase in ROS that occurs with advancing age may cause attenuation of Wnt signaling [127]. Thus, the increased efficacy of intermittent PTH to reverse age-related bone loss at least in mice may be due to the pro-differentiating and pro-survival effects of the hormone combined with a reduction in the negative effects of PPARγ on osteoblastogenesis.
CONCLUDING REMARKS
In vitro and in vivo evidence indicates that transient activation of the PTHR1 activates multiple interconnected pathways leading to increased survival signaling, decreased replication of osteoblast progenitors, increased differentiation, and the production and/or activation of osteogenic growth factors. The result of these actions is an increase in osteoblast number beyond that needed to refill the resorption cavity created by osteoclasts. Nevertheless, the new knowledge raises several issues that need to be addressed. The relative importance of pro-differentiating and pro-survival pathways to the ability of intermittent PTH to increase osteoblast number remains to be determined in both cancellous and periosteal bone, as does the potential contribution of pro-survival signaling in osteoblast progenitors, in both animals and humans. In addition, conflicting evidence from animal versus human studies, particularly regarding the role of PTH-induced survival signaling, needs to be resolved. Finally, ways must be found to determine the potential contribution of lining cell activation and increased differentiation of osteoblasts to the anabolic effect of intermittent PTH, particularly under conditions when osteoblast number is low due to the underlying pathologic mechanism.
Acknowledgments
The author thanks Maria Almeida, Teresita Bellido, Charles A. O’Brien, and Robert S. Weinstein for their comments and suggestions, Stavros C. Manolagas for his critical pre-review of earlier versions of this article, and the NIH (R01 AR46823, P01 AG13918) and the Dept. of Veterans Affairs (Merit Review) for their support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Hodsman AB, Bauer DC, Dempster D, Dian L, Hanley DA, Harris ST, et al. Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev. 2005;26:688–703. doi: 10.1210/er.2004-0006. [DOI] [PubMed] [Google Scholar]
- 2.Hodsman AB, Hanley DA, Watson PH, Fraher LJ. Parathyroid hormone. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principals of Bone Biology. San Diego: Academic Press; 2002. pp. 1305–24. [Google Scholar]
- 3.Frolik CA, Black EC, Cain RL, Satterwhite JH, Brown-Augsburger PL, Sato M, et al. Anabolic and catabolic bone effects of human parathyroid hormone (1–34) are predicted by duration of hormone exposure. Bone. 2003;33:372–9. doi: 10.1016/s8756-3282(03)00202-3. [DOI] [PubMed] [Google Scholar]
- 4.Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, et al. Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology. 2005;146:4577–83. doi: 10.1210/en.2005-0239. [DOI] [PubMed] [Google Scholar]
- 5.Lindsay R, Nieves J, Henneman E, Shen V, Cosman F. Subcutaneous administration of the amino-terminal fragment of human parathyroid hormone-(1–34): kinetics and biochemical response in estrogenized osteoporotic patients. J Clin Endocrinol Metab. 1993;77:1535–9. doi: 10.1210/jcem.77.6.8263137. [DOI] [PubMed] [Google Scholar]
- 6.Horwitz MJ, Tedesco MB, Gundberg C, Garcia-Ocana A, Stewart AF. Short-term, high-dose parathyroid hormone-related protein as a skeletal anabolic agent for the treatment of postmenopausal osteoporosis. J Clin Endocrinol Metab. 2003;88:569–75. doi: 10.1210/jc.2002-021122. [DOI] [PubMed] [Google Scholar]
- 7.Lindsay R, Cosman F, Zhou H, Bostrom MP, Shen VW, Cruz JD, et al. A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J Bone Miner Res. 2006;21:366–73. doi: 10.1359/JBMR.051109. [DOI] [PubMed] [Google Scholar]
- 8.Ma YL, Zeng Q, Donley DW, Ste-Marie LG, Gallagher JC, Dalsky GP, et al. Teriparatide increases bone formation in modeling and remodeling osteons and enhances IGF-II immunoreactivity in postmenopausal women with osteoporosis. J Bone Miner Res. 2006;21:855–64. doi: 10.1359/jbmr.060314. [DOI] [PubMed] [Google Scholar]
- 9.Arlot M, Meunier PJ, Boivin G, Haddock L, Tamayo J, Correa-Rotter R, et al. Differential Effects of Teriparatide and Alendronate on Bone Remodeling in Postmenopausal Women Assessed by Histomorphometric Parameters. J Bone Miner Res. 2005;20:1244–53. doi: 10.1359/JBMR.050309. [DOI] [PubMed] [Google Scholar]
- 10.Iida-Klein A, Zhou H, Lu SS, Levine LR, Ducayen-Knowles M, Dempster DW, et al. Anabolic action of parathyroid hormone is skeletal site specific at the tissue and cellular levels in mice. J Bone Miner Res. 2002;17:808–16. doi: 10.1359/jbmr.2002.17.5.808. [DOI] [PubMed] [Google Scholar]
- 11.Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138:4607–12. doi: 10.1210/endo.138.11.5505. [DOI] [PubMed] [Google Scholar]
- 12.Bellido T, Ali AA, Plotkin LI, Fu Q, Gubrij I, Roberson PK, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts: a putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem. 2003;278:50259–72. doi: 10.1074/jbc.M307444200. [DOI] [PubMed] [Google Scholar]
- 13.Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis with parathyroid hormone. J Clin Invest. 1999;104:439–46. doi: 10.1172/JCI6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parfitt AM. Bone-forming cells in clinical conditions. In: Hall BK, editor. Bone, The Osteoblast and Osteocyte. Vol. 1. Boca Raton, FL: Telford Press and CRC Press; 1990. pp. 351–429. [Google Scholar]
- 15.Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J Bone Miner Res. 2006;21:605–15. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 16.Boyce BF, Xing L, Jilka RL, Bellido T, Weinstein RS, Parfitt AM, et al. Apoptosis in Bone Cells. In: Bilezikian JP, Raisz LG, Rodan G, editors. Principles of Bone Biology. 2. San Diego: Academic Press; 2002. pp. 151–68. [Google Scholar]
- 17.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–37. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 18.Riggs BL, Parfitt AM. Drugs used to treat osteoporosis: the critical need for a uniform nomenclature based on their action on bone remodeling. J Bone Miner Res. 2005;20:177–84. doi: 10.1359/JBMR.041114. [DOI] [PubMed] [Google Scholar]
- 19.Dempster DW, Parisien M, Silverberg SJ, Liang XG, Schnitzer M, Shen V, et al. On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism. J Clin Endocrinol Metab. 1999;84:1562–6. doi: 10.1210/jcem.84.5.5652. [DOI] [PubMed] [Google Scholar]
- 20.Aubin JE, Triffitt JT. Mesenchymal stem cells and osteoblast differentiation. In: Bilezikian JP, Raisz LG, Rodan G, editors. Principles of Bone Biology. 2. San Diego: Academic Press; 2002. pp. 59–81. [Google Scholar]
- 21.Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung Ui, Kubota N, et al. PPARγ insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–55. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klein RF, Allard J, Avnur Z, Nikolcheva T, Rotstein D, Carlos AS, et al. Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science. 2004;303:229–32. doi: 10.1126/science.1090985. [DOI] [PubMed] [Google Scholar]
- 23.Ali AA, Weinstein RS, Stewart SA, Parfitt AM, Manolagas SC, Jilka RL. Rosiglitazone causes bone loss in mice by suppressing osteoblast differentiation and bone formation. Endocrinology. 2005;146:1226–35. doi: 10.1210/en.2004-0735. [DOI] [PubMed] [Google Scholar]
- 24.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 25.Krishnan V, Bryant HU, MacDougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116:1202–9. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Bezooijen RL, Roelen BAJ, Visser A, van der Wee-Pals L, de Wilt E, Karperien M, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. The Journal of Experimental Medicine. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 28.Semenov MV, He X. LRP5 Mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006;281:38276–84.i. doi: 10.1074/jbc.M609509200. [DOI] [PubMed] [Google Scholar]
- 29.Tidball JG, Albrecht DE. Regulation of apoptosis by cellular interactions with the extracellular matrix. In: Lockshin RA, Zakeri Z, Tilly JL, editors. When Cells Die. New York: Wiley-Liss; 1998. pp. 411–26. [Google Scholar]
- 30.Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- 31.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–82. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manolagas SC, Almeida M, Han L, Weinstein R, Jilka R, Bellido T, et al. Decreased defense against reactive oxygen species: a common pathogenetic mechanism of the effects of aging and estrogen deficiency on bone. J Bone Miner Res. 2005;20:S94. [Google Scholar]
- 33.Babij P, Zhao W, Small C, Kharode Y, Yaworsky PJ, Bouxsein ML, et al. High bone mass in mice expressing a mutant LRP5 gene. J Bone Miner Res. 2003;18:960–74. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 34.Parfitt AM, Han ZH, Palnitkar S, Rao DS, Shih MS, Nelson D. Effects of ethnicity and age or menopause on osteoblast function, bone mineralization, and osteoid accumulation in iliac bone. J Bone Miner Res. 1997;12:1864–73. doi: 10.1359/jbmr.1997.12.11.1864. [DOI] [PubMed] [Google Scholar]
- 35.Han ZH, Palnitkar S, Rao DS, Nelson D, Parfitt AM. Effects of ethnicity and age or menopause on the remodeling and turnover of iliac bone: Implications for mechanisms of bone loss. J Bone Miner Res. 1997;12:498–508. doi: 10.1359/jbmr.1997.12.4.498. [DOI] [PubMed] [Google Scholar]
- 36.Rice DP, Kim HJ, Thesleff I. Apoptosis in murine calvarial bone and suture development. Eur J Oral Sci. 1999;107:265–75. doi: 10.1046/j.0909-8836.1999.eos107406.x. [DOI] [PubMed] [Google Scholar]
- 37.Li G, Dickson GR, Marsh DR, Simpson H. Rapid new bone tissue remodeling during distraction osteogenesis is associated with apoptosis. J Orthop Res. 2003;21:28–35. doi: 10.1016/S0736-0266(02)00097-9. [DOI] [PubMed] [Google Scholar]
- 38.Potts JT. Parathyroid hormone: past and present. J Endocrinol. 2005;187:311–25. doi: 10.1677/joe.1.06057. [DOI] [PubMed] [Google Scholar]
- 39.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–64. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- 40.Gensure RC, Gardella TJ, Juppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328:666–78. doi: 10.1016/j.bbrc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 41.Whitfield JF, Morley P, Willick GE, Ross V, Barbier JR, Isaacs RJ, et al. Stimulation of the growth of femoral trabecular bone in ovariectomized rats by the novel parathyroid hormone fragment, hPTH-(1–31)NH2 (Ostabolin) Calcif Tissue Int. 1996;58:81–7. doi: 10.1007/BF02529728. [DOI] [PubMed] [Google Scholar]
- 42.Armento-Villareal R, Ziambaras K, Abbasi-Jarhomi SH, Dimarogonas A, Halstead L, Fausto A, et al. An intact N terminus is required for the anabolic action of parathyroid hormone on adult female rats. J Bone Miner Res. 1997;12:384–92. doi: 10.1359/jbmr.1997.12.3.384. [DOI] [PubMed] [Google Scholar]
- 43.Ferrari SL, Pierroz DD, Glatt V, Goddard DS, Bianchi EN, Lin FT, et al. Bone response to intermittent parathyroid hormone is altered in mice null for β-arrestin2. Endocrinology. 2005;146:1854–62. doi: 10.1210/en.2004-1282. [DOI] [PubMed] [Google Scholar]
- 44.Liu F, Lee SK, Adams DJ, Gronowicz GA, Kream BE. CREM deficiency in mice alters the response of bone to intermittent parathyroid hormone treatment. Bone. 2006 doi: 10.1016/j.bone.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samadfam R, Xia Q, Goltzman D. Cotreatment of parathyroid hormone with osteoprotegerin or alendronate increases its anabolic effect on the skeleton of oophorectomized mice. J Bone Miner Res. 2007;22:55–63. doi: 10.1359/jbmr.060915. [DOI] [PubMed] [Google Scholar]
- 46.Krishnan V, Moore TL, Ma YL, Helvering LM, Frolik CA, Valasek KM, et al. PTH bone anabolic action requires Cbfa1/Runx2-dependent signaling. Mol Endocrinol. 2003;17:423–35. doi: 10.1210/me.2002-0225. [DOI] [PubMed] [Google Scholar]
- 47.Shen R, Wang X, Drissi H, Liu F, O’Keefe RJ, Chen D. Cyclin D1-Cdk4 induce Runx2 ubiquitination and degradation. J Biol Chem. 2006;281:16347–53. doi: 10.1074/jbc.M603439200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas DM, Johnson SA, Sims NA, Trivett MK, Slavin JL, Rubin BP, et al. Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol. 2004;167:925–34. doi: 10.1083/jcb.200409187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin L, Li X, Ko JK, Partridge NC. Parathyroid hormone uses multiple mechanisms to arrest the cell cycle progression of osteoblastic cells from G1 to S phase. J Biol Chem. 2005;280:3104–11. doi: 10.1074/jbc.M409846200. [DOI] [PubMed] [Google Scholar]
- 50.Rickard DJ, Wang FL, Rodriguez-Rojas AM, Wu Z, Trice WJ, Hoffman SJ, et al. Intermittent treatment with parathyroid hormone (PTH) as well as a non-peptide small molecule agonist of the PTH1 receptor inhibits adipocyte differentiation in human bone marrow stromal cells. Bone. 2006;39:1361–72. doi: 10.1016/j.bone.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 51.Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, et al. Catabolic effects of continuous human PTH (1–38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology. 2001;142:4047–54. doi: 10.1210/endo.142.9.8356. [DOI] [PubMed] [Google Scholar]
- 52.Vilardaga JP, Krasel C, Chauvin S, Bambino T, Lohse MJ, Nissenson RA. Internalization determinants of the parathyroid hormone receptor differentially regulate β-arrestin/receptor association. J Biol Chem. 2002;277:8121–9. doi: 10.1074/jbc.M110433200. [DOI] [PubMed] [Google Scholar]
- 53.Stanislaus D, Yang X, Liang JD, Wolfe J, Cain RL, Onyia JE, et al. In vivo regulation of apoptosis in metaphyseal trabecular bone of young rats by synthetic human parathyroid hormone (1–34) fragment. Bone. 2000;27:209–18. doi: 10.1016/s8756-3282(00)00309-4. [DOI] [PubMed] [Google Scholar]
- 54.Onyia JE, Miller B, Hulman J, Liang J, Galvin R, Frolik C, et al. Proliferating cells in the primary spongiosa express osteoblastic phenotype in vitro. Bone. 1997;20:93–100. doi: 10.1016/s8756-3282(96)00350-x. [DOI] [PubMed] [Google Scholar]
- 55.Ali AA, O’Brien CA, Gubrij I, Wynne RA, Parfitt AM, Weinstein RS, et al. Evidence that the cellular mechanisms responsible for the anabolic effects of intermittent PTH are different in murine cancellous and periosteal bone. J Bone Min Res. 2005;20:S412. [Google Scholar]
- 56.Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB. Effects of a one-month treatment with parathyroid hormone (1–34) on bone formation on cancellous, endocortical and periosteal surfaces of the human ilium. J Bone Miner Res. 2007 doi: 10.1359/jbmr.070103. [DOI] [PubMed] [Google Scholar]
- 57.Labat-Moleur F, Guillermet C, Lorimier P, Robert C, Lantuejoul S, Brambilla E, et al. TUNEL apoptotic cell detection in tissue sections: Critical evaluation and improvement. J Histochem Cytochem. 1998;46:327–34. doi: 10.1177/002215549804600306. [DOI] [PubMed] [Google Scholar]
- 58.Chen HL, Demiralp B, Schneider A, Koh AJ, Silve C, Wang CY, et al. Parathyroid hormone and parathyroid hormone-related protein exert both pro- and anti-apoptotic effects in mesenchymal cells. J Biol Chem. 2002;277:19374–81. doi: 10.1074/jbc.M108913200. [DOI] [PubMed] [Google Scholar]
- 59.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–49. [PubMed] [Google Scholar]
- 60.Bellido T, O’Brien CA, Roberson PK, Manolagas SC. Transcriptional Activation of the p21WAF1, CIP1, SDI1 gene by interleukin-6 type cytokines. A prerequisite for their pro-differentiating and anti-apoptotic effects on human osteoblastic cells. J Biol Chem. 1998;273:21137–44. doi: 10.1074/jbc.273.33.21137. [DOI] [PubMed] [Google Scholar]
- 61.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–86. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dobnig H, Turner RT. Evidence that intermittent treatment with parathyroid hormone increases bone formation in adult rats by activation of bone lining cells. Endocrinology. 1995;136:3632–8. doi: 10.1210/endo.136.8.7628403. [DOI] [PubMed] [Google Scholar]
- 63.Sakai A, Sakata T, Ikeda S, Uchida S, Okazaki R, Norimura T, et al. Intermittent administration of human parathyroid hormone(1–34) prevents immobilization-related bone loss by regulating bone marrow capacity for bone cells in ddY mice. J Bone Miner Res. 1999;14:1691–9. doi: 10.1359/jbmr.1999.14.10.1691. [DOI] [PubMed] [Google Scholar]
- 64.Knopp E, Troiano N, Bouxsein M, Sun Bh, Lostritto K, Gundberg C, et al. The effect of aging on the skeletal response to intermittent treatment with parathyroid hormone. Endocrinology. 2005;146:1983–90. doi: 10.1210/en.2004-0770. [DOI] [PubMed] [Google Scholar]
- 65.Onyia JE, Bidwell J, Herring J, Hulman J, Hock JM. In vivo, human parathyroid hormone fragment (hPTH 1–34) transiently stimulates immediate early response gene expression, but not proliferation, in trabecular bone cells of young rats. Bone. 1995;17:479–84. doi: 10.1016/8756-3282(95)00332-2. [DOI] [PubMed] [Google Scholar]
- 66.Datta NS, Chen C, Berry JE, McCauley LK. PTHrP signaling targets cyclin D1 and induces osteoblastic cell growth arrest. J Bone Miner Res. 2005;20:1051–64. doi: 10.1359/JBMR.050106. [DOI] [PubMed] [Google Scholar]
- 67.Kostenuik PJ, Harris J, Halloran BP, Turner RT, Morey-Holton ER, Bikle DD. Skeletal unloading causes resistance of osteoprogenitor cells to parathyroid hormone and to insulin-like growth factor-I. J Bone Miner Res. 1999;14:21–31. doi: 10.1359/jbmr.1999.14.1.21. [DOI] [PubMed] [Google Scholar]
- 68.Pettway GJ, Schneider A, Koh AJ, Widjaja E, Morris MD, Meganck JA, et al. Anabolic actions of PTH (1–34): use of a novel tissue engineering model to investigate temporal effects on bone. Bone. 2005;36:959–70. doi: 10.1016/j.bone.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 69.Miao D, Tong XK, Chan GK, Panda D, McPherson PS, Goltzman D. Parathyroid hormone-related peptide stimulates osteogenic cell proliferation through protein kinase C activation of the ras/mitogen-activated protein kinase signaling pathway. J Biol Chem. 2001;276:32204–13. doi: 10.1074/jbc.M101084200. [DOI] [PubMed] [Google Scholar]
- 70.Ishizuya T, Yokose S, Hori M, Noda T, Suda T, Yoshiki S, et al. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J Clin Invest. 1997;99:2961–70. doi: 10.1172/JCI119491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scutt A, Duvos C, Lauber J, Mayer H. Time-dependent effects of parathyroid hormone and prostaglandin E2 on DNA synthesis by periosteal cells from embryonic chick calvaria. Calcif Tissue Int. 1994;55:208–15. doi: 10.1007/BF00425877. [DOI] [PubMed] [Google Scholar]
- 72.Swarthout JT, Doggett TA, Lemker JL, Partridge NC. Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C- dependent. J Biol Chem. 2001;276:7586–92. doi: 10.1074/jbc.M007400200. [DOI] [PubMed] [Google Scholar]
- 73.MacDonald BR, Gallagher JA, Russell RG. Parathyroid hormone stimulates the proliferation of cells derived from human bone. Endocrinology. 1986;118:2445–9. doi: 10.1210/endo-118-6-2445. [DOI] [PubMed] [Google Scholar]
- 74.Nakazawa T, Nakajima A, Shiomi K, Moriya H, Einhorn TA, Yamazaki M. Effects of low-dose, intermittent treatment with recombinant human parathyroid hormone (1–34) on chondrogenesis in a model of experimental fracture healing. Bone. 2005;37:711–9. doi: 10.1016/j.bone.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 75.Chan GK, Deckelbaum RA, Bolivar I, Goltzman D, Karaplis AC. PTHrP inhibits adipocyte differentiation by down-regulating PPARγ activity via a MAPK-dependent pathway. Endocrinology. 2001;142:4900–9. doi: 10.1210/endo.142.11.8515. [DOI] [PubMed] [Google Scholar]
- 76.Sato M, Westmore M, Ma YL, Schmidt A, Zeng QQ, Glass EV, et al. Teriparatide [PTH(1–34)] strengthens the proximal femur of ovariectomized nonhuman primates despite increasing porosity. J Bone Miner Res. 2004;19:623–9. doi: 10.1359/JBMR.040112. [DOI] [PubMed] [Google Scholar]
- 77.Leaffer D, Sweeney M, Kellerman LA, Avnur Z, Krstenansky JL, Vickery BH, et al. Modulation of osteogenic cell ultrastructure by RS-23581, an analog of human parathyroid hormone (PTH)-related peptide- (1–34), and bovine PTH-(1–34) Endocrinology. 1995;136:3624–31. doi: 10.1210/endo.136.8.7628402. [DOI] [PubMed] [Google Scholar]
- 78.Conover CA. Insulin-like growth factors and the skeleton. In: Canalis E, editor. Skeletal Growth Factors. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 101–16. [Google Scholar]
- 79.Nakchbandi IA, Grey A, Masiukiewicz U, Mitnick M, Insogna K. Cytokines in primary hyperparathyroidism. In: Bilezikian JP, Marcus R, Levine MA, editors. The Parathyroids Basic and Clinical Concepts. San Diego: Academic Press; 2001. pp. 411–21. [Google Scholar]
- 80.Hurley MM, Tetradis S, Huang YF, Hock J, Kream BE, Raisz LG, et al. Parathyroid hormone regulates the expression of fibroblast growth factor-2 mRNA and fibroblast growth factor receptor mRNA in osteoblastic cells. J Bone Miner Res. 1999;14:776–83. doi: 10.1359/jbmr.1999.14.5.776. [DOI] [PubMed] [Google Scholar]
- 81.Qin L, Qiu P, Wang L, Li X, Swarthout JT, Soteropoulos P, et al. Gene expression profiles and transcription factors involved in parathyroid hormone signaling in osteoblasts revealed by microarray and bioinformatics. J Biol Chem. 2003;278:19723–31. doi: 10.1074/jbc.M212226200. [DOI] [PubMed] [Google Scholar]
- 82.McClelland P, Onyia JE, Miles RR, Tu Y, Liang J, Harvey AK, et al. Intermittent administration of parathyroid hormone (1–34) stimulates matrix metalloproteinase-9 (MMP-9) expression in rat long bone. J Cell Biochem. 1998;70:391–401. [PubMed] [Google Scholar]
- 83.Miles RR, Sluka JP, Halladay DL, Santerre RF, Hale LV, Bloem L, et al. ADAMTS-1: A cellular disintegrin and metalloprotease with thrombospondin motifs is a target for parathyroid hormone in bone. Endocrinology. 2000;141:4533–42. doi: 10.1210/endo.141.12.7817. [DOI] [PubMed] [Google Scholar]
- 84.Grey A, Chen Q, Xu X, Callon K, Cornish J. Parallel phosphatidylinositol-3 kinase and p42/44 mitogen-activated protein kinase signaling pathways subserve the mitogenic and antiapoptotic actions of insulin-like growth factor I in osteoblastic cells. Endocrinology. 2003;144:4886–93. doi: 10.1210/en.2003-0350. [DOI] [PubMed] [Google Scholar]
- 85.Watson P, Lazowski D, Han V, Fraher L, Steer B, Hodsman A. Parathyroid hormone restores bone mass and enhances osteoblast insulin-like growth factor I gene expression in ovariectomized rats. Bone. 1995;16:357–65. doi: 10.1016/8756-3282(94)00051-4. [DOI] [PubMed] [Google Scholar]
- 86.Locklin RM, Khosla S, Turner RT, Riggs BL. Mediators of the biphasic responses of bone to intermittent and continuously administered parathyroid hormone. J Cell Biochem. 2003;89:180–90. doi: 10.1002/jcb.10490. [DOI] [PubMed] [Google Scholar]
- 87.Chen X, Stewart SA, Roberson PK, Wynne R, Manolagas SC, Jilka RL. Gene expression patterns in response to daily injections of PTH to mice: evidence for priming by the initial dose. J Bone Miner Res. 2006;21:S322. [Google Scholar]
- 88.Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, et al. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17:1570–8. doi: 10.1359/jbmr.2002.17.9.1570. [DOI] [PubMed] [Google Scholar]
- 89.Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142:4349–56. doi: 10.1210/endo.142.10.8436. [DOI] [PubMed] [Google Scholar]
- 90.Yamaguchi M, Ogata N, Shinoda Y, Akune T, Kamekura S, Terauchi Y, et al. Insulin receptor substrate-1 is required for bone anabolic function of parathyroid hormone in mice. Endocrinology. 2005;146:2620–8. doi: 10.1210/en.2004-1511. [DOI] [PubMed] [Google Scholar]
- 91.Yakar S, Bouxsein ML, Canalis E, Sun H, Glatt V, Gundberg C, et al. The ternary IGF complex influences postnatal bone acquisition and the skeletal response to intermittent parathyroid hormone. J Endocrinol. 2006;189:289–99. doi: 10.1677/joe.1.06657. [DOI] [PubMed] [Google Scholar]
- 92.Hurley MM, Okada Y, Xiao L, Tanaka Y, Ito M, Okimoto N, et al. Impaired bone anabolic response to parathyroid hormone in Fgf2−/− and Fgf2+/− mice. Biochem Biophys Res Commun. 2006;341:989–94. doi: 10.1016/j.bbrc.2006.01.044. [DOI] [PubMed] [Google Scholar]
- 93.Lane NE, Yao W, Kinney JH, Modin G, Balooch M, Wronski TJ. Both hPTH(1–34) and bFGF increase trabecular bone mass in osteopenic rats but they have different effects on trabecular bone architecture. J Bone Miner Res. 2003;18:2105–15. doi: 10.1359/jbmr.2003.18.12.2105. [DOI] [PubMed] [Google Scholar]
- 94.Keller H, Kneissel M. SOST is a target gene for PTH in bone. Bone. 2005;37:148–58. doi: 10.1016/j.bone.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 95.Fermor B, Skerry TM. PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J Bone Miner Res. 1995;10:1935–43. doi: 10.1002/jbmr.5650101213. [DOI] [PubMed] [Google Scholar]
- 96.Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by β-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem. 2005;280:41342–51. doi: 10.1074/jbc.M502168200. [DOI] [PubMed] [Google Scholar]
- 97.Kharode YP, Sharp MC, Milligan CL, Pirrello JM, Selim SF, Bodine PVN, et al. Selective involvement of WNT signaling pathway in bone anabolic action of parathyroid hormone. J Bone Miner Res. 2006;21:S114. [Google Scholar]
- 98.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006;281:23698–711. doi: 10.1074/jbc.M601000200. [DOI] [PubMed] [Google Scholar]
- 99.Iwaniec UT, Wronski TJ, Liu J, Rivera MF, Arzaga RR, Hansen G, et al. PTH stimulates bone formation in mice deficient in Lrp5. J Bone Miner Res. 2007;22:394–402. doi: 10.1359/jbmr.061118. [DOI] [PubMed] [Google Scholar]
- 100.Kim CH, Takai E, Zhou H, von Stechow D, Muller R, Dempster DW, et al. Trabecular bone response to mechanical and parathyroid hormone stimulation: the role of mechanical microenvironment. J Bone Miner Res. 2003;18:2116–25. doi: 10.1359/jbmr.2003.18.12.2116. [DOI] [PubMed] [Google Scholar]
- 101.Turner RT, Lotinun S, Hefferan TE, Morey-Holton E. Disuse in adult male rats attenuates the bone anabolic response to a therapeutic dose of parathyroid hormone. J Appl Physiol. 2006;101:881–6. doi: 10.1152/japplphysiol.01622.2005. [DOI] [PubMed] [Google Scholar]
- 102.Zhou H, Iida-Klein A, Lu SS, Ducayen-Knowles M, Levine LR, Dempster DW, et al. Anabolic action of parathyroid hormone on cortical and cancellous bone differs between axial and appendicular skeletal sites in mice. Bone. 2003;32:513–20. doi: 10.1016/s8756-3282(03)00057-7. [DOI] [PubMed] [Google Scholar]
- 103.Ma Y, Jee WS, Yuan Z, Wei W, Chen H, Pun S, et al. Parathyroid hormone and mechanical usage have a synergistic effect in rat tibial diaphyseal cortical bone. J Bone Miner Res. 1999;14:439–48. doi: 10.1359/jbmr.1999.14.3.439. [DOI] [PubMed] [Google Scholar]
- 104.Robling AG, Bellido TM, Turner CH. Mechanical loading reduces osteocyte expression of sclerostin protein. J Bone Miner Res. 2006;21:S72. [PubMed] [Google Scholar]
- 105.O’Brien CA, Plotkin LI, Vyas K, Cazer PE, Gortazar AR, Goellner JJ, et al. Activation of PTH receptor 1 specifically in osteocytes suppresses Sost expression and increases bone mass in transgenic mice. J Bone Miner Res. 2006;20:S4. [Google Scholar]
- 106.Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 107.Black DM, Greenspan SL, Ensrud KE, Palermo L, McGowan JA, Lang TF, et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–15. doi: 10.1056/NEJMoa031975. [DOI] [PubMed] [Google Scholar]
- 108.Finkelstein JS, Hayes A, Hunzelman JL, Wyland JJ, Lee H, Neer RM. The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med. 2003;349:1216–26. doi: 10.1056/NEJMoa035725. [DOI] [PubMed] [Google Scholar]
- 109.Ettinger B, San Martin J, Crans G, Pavo I. Differential Effects of Teriparatide on BMD After Treatment With Raloxifene or Alendronate. J Bone Miner Res. 2004;19:745–51. doi: 10.1359/JBMR.040117. [DOI] [PubMed] [Google Scholar]
- 110.Delmas PD, Vergnaud P, Arlot ME, Pastoureau P, Meunier PJ, Nilssen MHL. The anabolic effect of human PTH (1–34) on bone formation is blunted when bone resorption is inhibited by the bisphosphonate tiludronate - Is activated resorption a prerequisite for the in vivo effect of PTH on formation in a remodeling system. Bone. 1995;16:603–10. doi: 10.1016/8756-3282(95)00113-r. [DOI] [PubMed] [Google Scholar]
- 111.Wronski TJ, Yen C-F, Qi H, Dann LM. Parathyroid hormone is more effective than estrogen or bisphosphonates for restoration of lost bone mass in ovariectomized rats. Endocrinology. 1993;132:823–31. doi: 10.1210/endo.132.2.8425497. [DOI] [PubMed] [Google Scholar]
- 112.Cosman F, Nieves J, Woelfert L, Shen V, Lindsay R. Alendronate does not block the anabolic effect of PTH in postmenopausal osteoporotic women. J Bone Miner Res. 1998;13:1051–5. doi: 10.1359/jbmr.1998.13.6.1051. [DOI] [PubMed] [Google Scholar]
- 113.Cosman F, Nieves J, Zion M, Woelfert L, Luckey M, Lindsay R. Daily and Cyclic Parathyroid Hormone in Women Receiving Alendronate. N Engl J Med. 2005;353:566–75. doi: 10.1056/NEJMoa050157. [DOI] [PubMed] [Google Scholar]
- 114.Ma YL, Bryant HU, Zeng Q, Schmidt A, Hoover J, Cole HW, et al. New bone formation with teriparatide [human parathyroid hormone-(1–34)] is not retarded by long-term pretreatment with alendronate, estrogen, or raloxifene in ovariectomized rats. Endocrinology. 2003;144:2008–15. doi: 10.1210/en.2002-221061. [DOI] [PubMed] [Google Scholar]
- 115.Iida-Klein A, Lu SS, Cosman F, Lindsay R, Dempster DW. Effects of cyclic vs. daily treatment with human parathyroid hormone (1–34) on murine bone structure and cellular activity. Bone. 2007;40:391–8. doi: 10.1016/j.bone.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 116.Walsh CA, Birch MA, Fraser WD, Lawton R, Dorgan J, Walsh S, et al. Expression and secretion of parathyroid hormone-related protein by human bone-derived cells in vitro: effects of glucocorticoids. J Bone Miner Res. 1995;10:25. doi: 10.1002/jbmr.5650100106. [DOI] [PubMed] [Google Scholar]
- 117.Chen X, Macica CM, Ng KW, Broadus AE. Stretch-induced PTH-related protein gene expression in osteoblasts. J Bone Miner Res. 2005;20:1454–61. doi: 10.1359/jbmr.2005.20.8.1454. [DOI] [PubMed] [Google Scholar]
- 118.Miao D, Li J, Xue Y, Su H, Karaplis AC, Goltzman D. Parathyroid hormone-related peptide is required for increased trabecular bone volume in parathyroid hormone-null mice. Endocrinology. 2004;145:3554–62. doi: 10.1210/en.2003-1695. [DOI] [PubMed] [Google Scholar]
- 119.Miao D, He B, Jiang Y, Kobayashi T, Soroceanu MA, Zhao J, et al. Osteoblast-derived PTHrP is a potent endogenous bone anabolic agent that modifies the therapeutic efficacy of administered PTH 1–34. J Clin Invest. 2005;115:2402–11. doi: 10.1172/JCI24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martin TJ. Osteoblast-derived PTHrP is a physiological regulator of bone formation. J Clin Invest. 2005;115:2322–4. doi: 10.1172/JCI26239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud C. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. J Clin Invest. 1998;102:1627–33. doi: 10.1172/JCI3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oxlund H, Ortoft G, Thomsen JS, Danielsen CC, Ejersted C, Andreassen TT. The anabolic effect of PTH on bone is attenuated by simultaneous glucocorticoid treatment. Bone. 2006;39:244–52. doi: 10.1016/j.bone.2006.01.142. [DOI] [PubMed] [Google Scholar]
- 123.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–41. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 124.Marcus R, Wang O, Satterwhite J, Mitlak B. The skeletal response to teriparatide is largely independent of age, initial bone mineral density, and prevalent vertebral fractures in postmenopausal women with osteoporosis. J Bone Miner Res. 2003;18:18–23. doi: 10.1359/jbmr.2003.18.1.18. [DOI] [PubMed] [Google Scholar]
- 125.Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2:165–71. doi: 10.1023/a:1011513223894. [DOI] [PubMed] [Google Scholar]
- 126.Jilka RL, Wynne RA, Chen X, Stewart S, Xu L, Thaden JA, et al. The pathogenesis of age-related bone loss in mice involves increased production of PPARγ-activating oxidized lipids derived from the lipoxygenase Alox15. J Bone Min Res. 2005;20:S36. [Google Scholar]
- 127.Almeida M, Han L, Lowe V, Warren A, Kousteni S, O’Brien CA, et al. Reactive oxygen species antagonize the skeletal effects of Wnt/β-catenin in vitro and aging mice by diverting β-catenin to FOXO-mediated transcription. J Bone Miner Res. 2006;21:S26. [Google Scholar]