

Summary
Glycosylation defects occur in several human diseases. In IgA nephropathy, IgA1 contains O-glycans that are galactose-deficient and consist mostly of core 1 α2,6 sialylated N-acetylgalactosamine, a configuration suspected to prevent β1,3 galactosylation. We confirmed the same aberrancy in IgA1 secreted by the human DAKIKI B cell line. Biochemical assays indicated CMP-NeuAc:GalNAc-IgA1 α2,6-sialyltransferase activity in this cell line. However, a candidate enzyme, ST6-GalNAcI, was not transcribed in DAKIKI cells, B cells isolated from blood, or Epstein-Barr virus (EBV)-immortalized IgA1-producing cells from the blood of IgAN patients and healthy controls. Instead, ST6-GalNAcII transcription was detected at a high level. Expression of the ST6-GalNAcII gene and activity of the CMP-NeuAc:GalNAc-IgA1 α2,6-sialyltransferase were higher in IgA1-producing cell lines from IgAN patients than in such cells from healthy controls. These data are the first evidence that human cells that lack ST6-GalNAcI can sialylate core 1 GalNAc-Ser/Thr.
Keywords: ST6GalNAcII, Dakiki, IgA nephropathy, IgA1 hinge region, sialylation
Introduction
Aberrant glycosylation of immunoglobulins occurs in several diseases, including IgA nephropathy (IgAN). IgAN is the most common primary glomerulonephritis 1; 2 and as many as 20–40% of IgAN patients progress to end-stage renal failure within 20–25 years.3 The pathologic features of IgAN include mesangial deposits of IgA1, often with co-deposits of C3 and IgG, mesangial cell proliferation and matrix expansion. The closely related disease, Henoch-Schoenlein purpura nephritis (HSPN), exhibits pathological features that are indistinguishable from those of IgAN, including aberrant glycosylation of the IgA1.2 The aberrant glycosylation of IgA1 in both the mesangial deposits and in the circulation of patients with IgAN is due to a galactose (Gal) deficiency in the O-linked glycans.4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14 It has been established that the aberrantly glycosylated IgA1 is recognized by naturally occurring antibodies in the circulation of patients with IgAN 13; 15 leading to formation of immune complexes. Deposition of such immune complexes in the glomeruli stimulates mesangial cells to proliferate and secrete matrix proteins.9; 10; 13; 16 Although these lines of evidence suggest a direct causal relationship between aberrant glycosylation and the renal injury in IgAN, the defect underlying the abnormal glycosylation of IgA1 in IgAN has not yet been identified.
IgA1 differs from IgA2 in that it contains a longer hinge region between the first and second heavy-chain constant-region domains.17; 18 Its sequence is unique among the heavy-chain immunoglobulin hinge regions: 19 rich in proline, serine, and threonine and glycosylated with up to six O-linked glycan chains.18; 20; 21; 22; 23 Among other human immunoglobulins, only IgD has a hinge region with attached O-linked glycans.24 O-linked glycans of IgA1 consist of N-acetylgalactosamine (GalNAc) and usually a β1,3-linked Gal. Sialic acid (SA) may be attached to the β1,3-linked Gal through an α2,3 linkage 23; 25; 26; 27; 28 or to the GalNAc by an α2,6 linkage.25; 26; 27 The carbohydrate composition of these O-linked glycans is variable, but the prevailing forms in healthy individuals include the Gal-GalNAc disaccharide, and its mono- and di-sialylated forms. The variant, aberrantly glycosylated form of IgA1 with terminal GalNAc or sialylated GalNAc, is found commonly in sera from IgAN patients, but only rarely in sera from normal individuals.4; 7; 11; 12; 13; 27; 29; 30
O-linked glycans are synthesized in a step-wise manner, beginning with attachment of GalNAc to Ser or Thr. A member of the UDP-GalNAc transferase (GalNAcT) enzyme family catalyzes this first step.31; 32; 33 It has been proposed that all members of this enzyme family can glycosylate a wide range of acceptors, but only one, GalNAcT2, can glycosylate IgA1.34 It has been shown that GalNAcT2 initiates synthesis of all O-linked glycans in the hinge region of IgA1. The O-glycan chain is then extended by sequential attachment of Gal and/or SA residues to the GalNAc. The addition of Gal is mediated by core 1 β1,3-galactosyltransferase (C1β3GalT1) that transfers Gal from UDP-Gal to a GalNAc on Ser/Thr.35 The stability of this enzyme apparently depends on its interaction with the C1β3GalT1-specific molecular chaperone, Cosmc, that assists in the folding of the protein.36; 37; 38 In the absence of Cosmc, the GalT enzyme is degraded rapidly and the O-linked glycans fail to receive the normal complement of Gal residues. The glycan structure is completed by the sialyltransferases, that attach negatively charged SA to the Gal and/or GalNAc residues. If SA is linked to GalNAc prior to attachment of Gal, this “premature” sialylation precludes subsequent attachment of Gal.39
The Gal-deficient IgA1 in IgAN patients is characterized by hinge-region glycans with sialylated GalNAc, rather than terminal GalNAc,29 suggesting that a member of the GalNAc-specific sialyltransferases enzyme family may play a role in the aberrant glycosylation in this disease. We therefore undertook an analysis of the GalNAc-specific sialyltransferases in IgA1-producing human B cells. We found that only one of these sialyltransferases, ST6-GalNAcII, was expressed in human IgA1-producing B cells and that the transcription of the gene encoding this enzyme is higher in the B cells of patients with IgAN than in the B cells of healthy controls.
Results
Analysis of glycosylation of IgA1 secreted by DAKIKI cells
Monosaccharide compositional analysis of IgA1 purified from DAKIKI culture medium showed its Gal deficiency to be similar to that described for IgA1 (Mce) myeloma protein and Gal-deficient IgA1 from patients with IgAN.13; 29 Similar to the other Gal-deficient IgA1 proteins, DAKIKI IgA1 was predominantly in a polymeric form. To further characterize the glycosylation of the DAKIKI IgA1, we used lectin ELISA with lectins specific for O-linked glycans. We also included in this assay IgA1 purified from normal human serum (that was predominantly monomeric) and IgA1 (Mce) myeloma protein (polymeric). Neuraminidase treatment of the purified DAKIKI IgA1 increased its reactivity with Helix aspersa agglutinin (HAA), indicating the presence of GalNAc with terminal SA (Fig. 1). Desialylation of the DAKIKI IgA1 protein also resulted in an increase in its reactivity with peanut agglutinin (PNA) (Fig. 1), indicating that sialylated Gal-β1,3GalNAc structures are also present on the DAKIKI IgA1, i.e., core 1 O-linked glycans. The neuraminidase treatment resulted in a 10-fold increase in the reactivity of the DAKIKI IgA1 with HAA as compared to a 2-fold increase in the reactivity of IgA1 (Mce), suggesting that the glycans are more extensively sialylated on the IgA1 produced by the DAKIKI cells than the myeloma IgA1 protein. Normal serum IgA1 was less sialylated than IgA1 secreted by DAKIKI cells (Fig. 1). Similarly, the neuraminidase treatment resulted in a 5-fold increase in reactivity of the DAKIKI IgA1 with PNA as compared to a 3-fold increase in the reactivity of the IgA1 (Mce), suggesting that more terminal Gal residues are in the O-linked glycans of DAKIKI IgA1 than those of IgA1 (Mce) myeloma protein. These estimates were confirmed by monosaccharide compositional analysis (data not shown). Thus, the DAKIKI cell line secretes Gal-deficient IgA1 and is a suitable model for analysis of the pathways involved in the production of under-galactosylated IgA1 O-linked glycans.
Figure 1. Lectin ELISA analysis of glycosylation of IgA1.
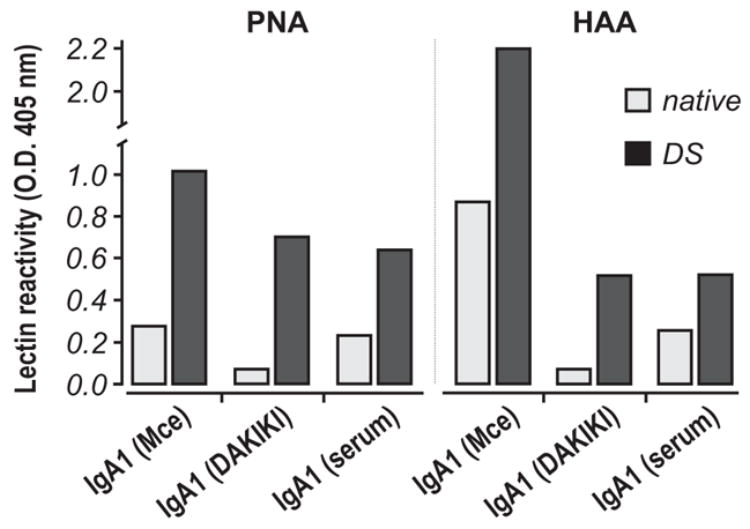
IgA1 proteins isolated from serum of patient with multiple myeloma (Mce), from the supernatants of the DAKIKI cell line, and from normal human serum (polyclonal) were coated onto ELISA wells, treated (dark bars; DS, desialyated) or not-treated (light bars) with neuraminidase, and subsequently reacted with PNA or HAA lectin. Averages of the results from two experiments are shown (SD was about 5% of the average values).
Analysis of sialyltransferase enzyme activity in DAKIKI cells
To test the ability of DAKIKI cells to sialylate terminal GalNAc on Gal-deficient IgA1, we used the Golgi-enriched fraction as the enzyme source, CMP-NeuAc as the SA donor, and desialylated IgA1 (Mce) myeloma protein as the acceptor. The IgA1 was then tested for its reactivity with HAA, such that a reduction in HAA reactivity would indicate the addition of SA to GalNAc on the heavy chains. The sialyltransferase activity in the Golgi-enriched fraction of DAKIKI cells resulted in a reduction in the reactivity of the IgA1 with HAA (Fig. 2a). The specificity was verified by enzymatic desialylation of IgA1 (Mce) protein; neuraminidase treatment restored the level of reactivity with HAA to that observed prior to the sialylation reaction. Coomassie Blue staining of the SDS-PAGE gel (not shown) as well as analysis by western blotting with heavy-chain-specific anti-IgA antibody confirmed equivalent loading of the IgA1 in the different samples (Fig. 2b). These experiments demonstrated that DAKIKI cells have an enzyme(s) capable of sialylating Gal-deficient O-linked GalNAc in the hinge region of IgA1 molecules.
Figure 2. Sialylation of terminal GalNAc in the hinge region of IgA1 (Mce) by DAKIKI cell extracts.
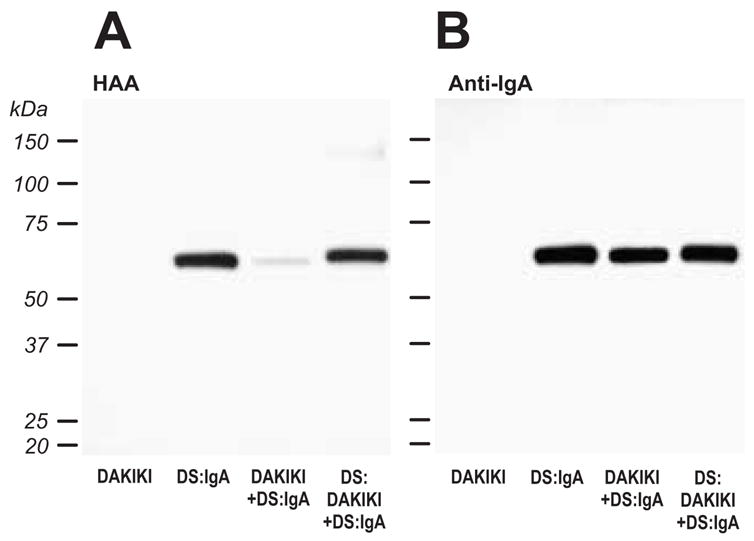
Desialylated IgA1 (Mce) (DS:IgA) was incubated with a Golgi-enriched enzyme preparation from DAKIKI cells (DAKIKI+DS:IgA) and CMP-NeuAc and subsequently desialylated (DS:DAKIKI+DS:IgA). The samples, including DAKIKI Golgi preparations without added IgA1, were separated on SDS-PAGE, western-blotted onto a PVDF membrane and incubated with HAA lectin, that recognizes terminal GalNAc (A). To confirm that equivalent levels of IgA were loaded, the membrane was stripped and re-probed with an IgA-specific antibody (B).
Reverse transcription analysis of ST6-GalNAc activity in DAKIKI cells
Transcription of the sialyltransferase genes was estimated using RealTime RT-PCR with specific primers (Table 1) with the identity of the amplicons being confirmed by sequencing (data not shown). Theoretically, ST6-GalNAcI and ST6-GalNAcII could both sialylate IgA1 in these cells. We did not detect transcription of ST6-GalNAcI, although a parallel analysis of colonic carcinoma HT29 cells and unfractionated peripheral blood mononuclear cells (PBMC) from patients with IgAN and HSPN and from a healthy control indicated the generation of an ST6-GalNAcI-specific amplicon (Figs. 3a and 3b). We discovered that ST6-GalNAcI transcripts in PBMC originated from T cells, but not B cells (Fig 3a). This finding was consistent with the observation that ST6-GalNAcI transcription was not detected in DAKIKI cells from cultures propagated for 1, 2, 21, or 25 weeks (data not shown). Thus, the DAKIKI cells sialylated terminal GalNAc residues of IgA1 although these cells did not express ST6-GalNAcI, the only previously known human sialyltransferase with this activity. Comparative analyses of enzymatic activity of recombinant murine ST6-GalNAcI and ST6-GalNAcII with different acceptors showed that both enzymes can sialylate terminal GalNAc on glycoproteins.40 Consequently, ST6-GalNAcII is likely the enzyme in DAKIKI and other cells without ST6-GalNAcI that sialylates GalNAc on glycoproteins.
Table 1.
Primers used for RealTime RT-PCR.
| Gene | Downstream primer 5′ - 3′ Upstream primer 5′ - 3′ | cDNA [bp] | Genome [bp] | Reference gi|No.: |
|---|---|---|---|---|
| ST6-GalNAcI | GCAACCACAGCCAAGACGCTCATTCCCAA TGTCACGACCTTCTGCACCAAGGAGTAG | 456 | 2059 | 46 |
| ST6-GalNAcII | AAGCTGCTACATCCGGACTTCA GGGACAGATCGTGGTTTGCATA | 247 | 2587 | 60 |
| ST6-GalNAcIV | CTGCAGCTCACCAGGATGTA AACACGATGGGCCTCTTCT | 374 | 3952 | 33989110 |
| ST6-GalNAcVI | GCCACCAGTGTGTGATTGTC TGCTCAACCACGAATGAGAC | 417 | 3461 | 34147672 |
| β-actin | Cat No.: RDP-38-025 | 528 | 528, 969 | R&D |
Figure 3. Transcription of ST6-GalNAc genes in human cells.
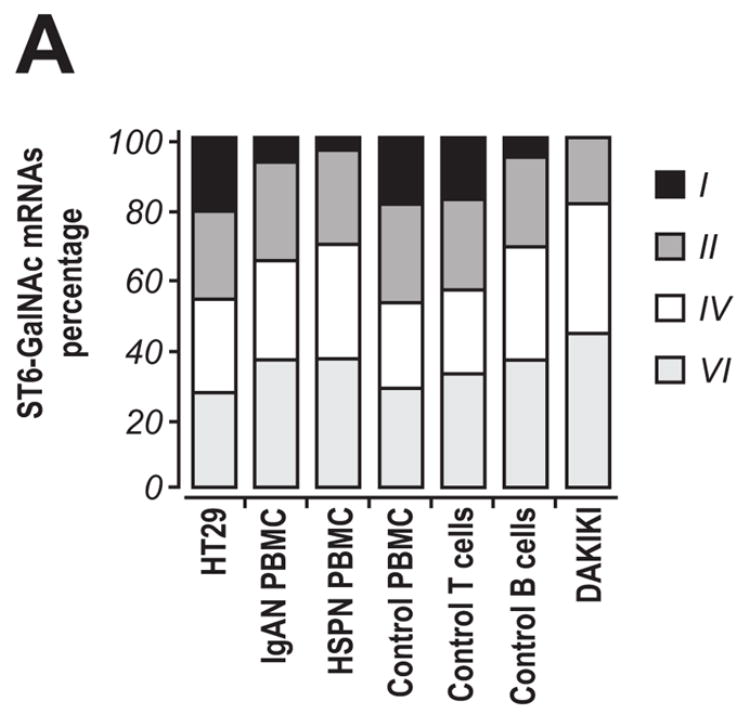
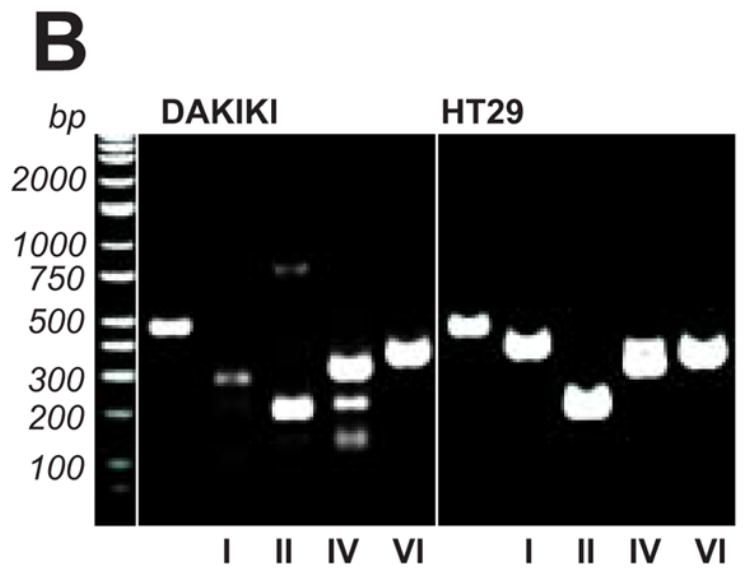
RNA samples from DAKIKI cells were analyzed for transcription of ST6-GalNAc genes. HT29 cells were used as a control and PBMC from patients with IgAN or HSPN, and from a healthy control were also analyzed. In addition, T and B cells isolated by FACS from PBMC of a healthy control were also included. A, The distribution of ST6-GalNAcI, ST6-GalNAcII, ST6-GalNAcIV, and ST6-GalNAcVI mRNA levels detected by RealTime RT-PCR; B, final products of RealTime RT-PCR (total 42 cycles) from DAKIKI cells and HT29 cells, analyzed on 1% TAE agarose gel to confirm specificity of the reactions. Primers used in the reactions (from left to right): β-actin, ST6-GalNAcI, ST6-GalNAcII, ST6-GalNAcIV, and ST6-GalNAcVI.
Reverse transcription analysis of ST6-GalNAc activity in PBMC
ST6-GalNAcI transcripts were detected in unfractionated PBMC from a healthy control, a patient with IgAN, and a patient with HSPN (Fig. 3a). To estimate the levels of ST6-GalNAcI and ST6-GalNAcII in B- and T-cells, we compared using RealTime RT-PCR the ratio of ST6-GalNAcII/ST6-GalNAcI transcripts in freshly isolated and pokeweed mitogen (PWM)-stimulated PBMC, and in the corresponding B- and T-cell enriched fractions (Fig. 4). PBMC were isolated from a patient with IgAN and from two healthy controls. Differences in the ratio were associated mainly with changes in ST6-GalNAcI transcription produced by T cells; the transcription of ST6-GalNAcII was similar among the different cell populations. After depletion of T cells and adherent cells from the PBMC, the remaining B cell population expressed only low amounts of ST6-GalNAcI transcripts originating from contaminating T cells (results not shown). PWM stimulation did not alter the ST6-GalNAcII/ST6-GalNAcI ratio in PBMC from the IgAN patient, but resulted in a small change in the ratio of these transcripts in the cells from healthy individuals. Furthermore, we have shown that B cell populations that did not contain any T cells were devoid of ST6-GalNAcI transcripts and that B cells from the IgAN patients expressed higher levels of ST6-GalNAcII transcript compared to B cells from the two healthy controls. These findings confirmed that B cells did not express ST6-GalNAcI and suggested that the cells from IgAN patients may have upregulated ST6-GalNAcII.
Figure 4. ST6-GalNAcI and ST6-GalNAcII transcription activity in B- and T-cell enriched PBMC.
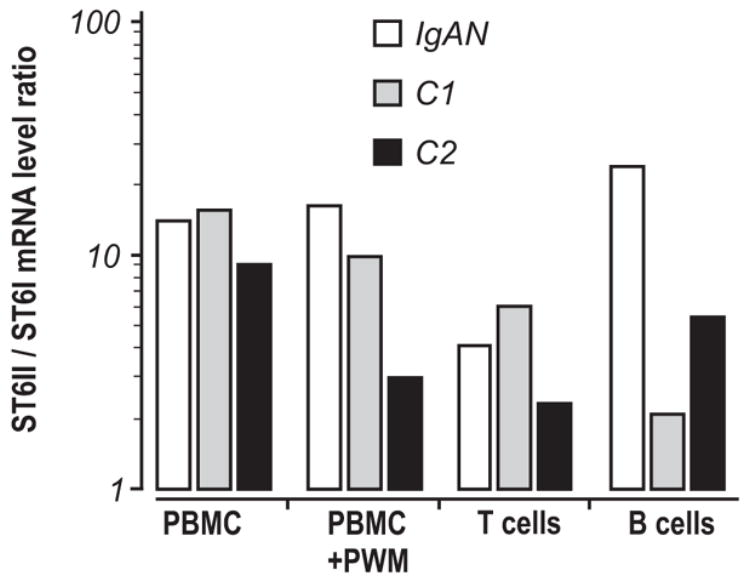
RNA was isolated from PBMC, PWM-stimulated PBMC (PBMC+PWM), a T-cell fraction and an enriched B-cell population from an IgAN patient (IgAN) and two healthy controls (C1 and C2). mRNA levels of specific transcripts were determined by RealTime RT-PCR and ratios of estimated crossing points ST6-GalNAcII / ST6-GalNAcI were calculated.
Reverse transcription analysis of ST6-GalNAc activity and enzyme ST6-GalNAc activity in EBV-immortalized IgA1-producing cells
IgA1-producing cells were subcloned from four IgAN patients and four healthy controls. ST6-GalNAcI was not detectable using RealTime RT-PCR in any of these cell lines (Table 2), whereas all of the cell lines expressed ST6-GalNAcII. The transcription of ST6-GalNAcII was greater in IgA1-producing cell lines subcloned from IgAN patients than in the cell lines subcloned from healthy controls (Table 2). The increased transcription correlated with higher enzyme activity: the cell lines from IgAN patients showed 156.9±32.5 U/mg protein compared to 54.2±32.9 U/mg protein in the cell lines from healthy controls. Lectin ELISA confirmed that the cell lines from the IgAN patients secreted predominantly Gal-deficient IgA1, while most of the IgA1 secreted by the cells from the healthy controls displayed a normal Gal content. Moreover, the Gal-deficient IgA1 secreted by the cells from IgAN patients contained mostly sialylated GalNAc rather than terminal GalNAc.
Table 2.
Expression of ST6GalNAcI and II mRNA in EBV-immortalized B cells measured by RealTime RT-PCR.
| IgAN patients* | Healthy controls | |
|---|---|---|
| ST6-GalNAcI | N/D | N/D |
| ST6-GalNAcII | 2.68 ± 0.11 | 1.97 ± 0.44 |
Data are presented as mean ± SD of crossing points normalized to the crossing points of β-actin, measured by RealTime RT-PCR. IgA1-producing B cells were generated from 4 patients with IgAN (total of 7 different clones) and 4 healthy controls (total of 7 different clones). The difference in the expression of ST6GalNAcII in the cells from IgAN patients and healthy controls was statistically significant, P < 0.01. N/D, not detected.
Discussion
Patients with IgAN and HSPN exhibit elevated levels of aberrantly glycosylated IgA1 in their circulation.4; 13; 29; 30; 41; 42 Furthermore, most of the mesangial IgA1 is similarly Gal-deficient.11; 12 Differences between patients with IgAN/HSPN and healthy individuals in the glycosylation of circulatory IgA1 are quantitative. The glycosylation aberrancy, Gal deficiency of O-linked glycans, affects a very small proportion of the molecules in healthy individuals, but a larger fraction in IgAN patients.13; 29 Furthermore, Gal-deficient IgA1 molecules are not completely devoid of Gal, but rather contain both Gal-deficient and Gal-containing GalNAc.11; 28; 43 This feature is reflected by the binding of these proteins to GalNAc- and GalNAc-Gal-binding lectins, such as HAA and PNA, respectively.
It is well established that the Gal-deficient hinge-region O-glycans of IgA1 contain increased amounts of sialylated GalNAc,13; 16; 29 but the exact enzymatic mechanism(s) for this sialylation remains a matter of debate. In some cells and tissues, ST6-GalNAcI adds sialic acid to GalNAc in an α2,6 position to generate a disaccharide.44; 45; 46 It has been postulated that another enzyme of the same family, ST6-GalNAcII, adds SA to GalNAc of GalNAc-Gal and GalNAc-Gal-SA, but not to terminal GalNAc.46 More recently, it was shown that murine ST6-GalNAcII can also add SA to terminal GalNAc.40 In the current studies, we have shown for the first time that human B cells without ST6-GalNAcI can express ST6Gal-NAcII. This enzyme may contribute to the pathogenesis of IgAN and HSPN by enhancing the synthesis of aberrantly glycosylated O-linked glycans on circulatory IgA1. This hypothesis is based on the observation that sialylated GalNAc cannot be galactosylated or otherwise modified by additional glycan residues.39
Synthesis of Gal-deficient IgA1 is apparently restricted to a relatively few IgA1-secreting B cells and the glycosylation defect does not affect other O-glycosylated circulatory proteins, such as C1 inhibitor or IgD.30; 47 These observations suggest that during late differentiation of B cells, a clone(s) that exhibits abnormal O-glycosylation of its secreted IgA1 molecules is selected.
The mechanisms responsible for the O-glycosylation aberrancy in IgAN are not known, but may include an abnormal expression or activity of the enzymes that add individual monosaccharides to the IgA1 hinge region. For example, reduced activity of C1GalT1 (or its chaperone, Cosmc) or enhanced activity of ST6-GalNAcII would lead to an excess of GalNAc-SA.9; 48 Because the Gal-deficient O-linked glycans on IgA1 from IgAN patients contained mostly sialylated GalNAc,29 we had hypothesized that another mechanism may be premature sialylation of the GalNAc residue.9 We based this hypothesis on the observation that addition of SA to GalNAc prevents attachment of Gal to GalNAc 39 and thus precludes further growth of the glycan. However, it is possible that both mechanisms may be involved.
Investigation of the molecular mechanisms involved in the synthesis of Gal-deficient IgA1 in IgAN required an appropriate model. We had established earlier that DAKIKI, an IgA1-secreting EBV-immortalized B cell line, secrete polymeric IgA1 with some of its O-glycans Gal-deficient 49 and that these glycans contain SA-GalNAc. In the current study, we confirmed these findings and furthermore established that the Golgi-enriched fractions of these cells possess ST6-GalNAc enzymatic activity. The enzyme reaction was conducted with desialylated IgA1 (Mce) myeloma protein as the acceptor. Gal deficiency and removal of SA from the acceptor IgA1 was verified by gas-liquid chromatography and western blotting with lectins (HAA, PNA). O-glycans of this IgA1 had been previously characterized in detail.23 Because CMP-NeuAc but no UDP-Gal was provided in the reaction, only sialylation without galactosylation could have occurred. The sialyltransferase reaction was assessed indirectly by western blotting as a loss of HAA reactivity. In addition, under controlled conditions for total amount of IgA (anti-IgA antibody blotting) we assessed the reappearance of HAA reactivity after treatment with neuraminidase that removed alpha2,6-bound SA from GalNAc. These controls showed conclusively that we measured sialylation of GalNAc on IgA1 glycans. As the candidate enzyme for this function, ST6-GalNAcI, was not transcribed in these cells, we concluded that another enzyme must display such activity. Using RealTime RT-PCR, we determined that DAKIKI cells express ST6-GalNAcII, the other enzyme that can add sialic acid to terminal GalNAc.40
Analysis of the transcription of the ST6-GalNAc genes in IgA1-secreting EBV-immortalized cells from IgAN patients and healthy controls indicated that none of the cell lines transcribed ST6-GalNAcI, but that all expressed ST6-GalNAcII. Furthermore, the EBV-immortalized cells from the patients with IgAN expressed more ST6-GalNAcII than did cells from healthy controls. The activity of the ST6-GalNAc enzyme was 2-fold to 3-fold higher in the cells from IgAN patients compared to the cells from healthy controls. These data are consistent with glycosylation of IgA1 proteins secreted by the corresponding cell lines and the frequency of aberrantly glycosylated IgA1 hinge-region glycans in the circulation, i.e., while normal individuals have some Gal-deficient IgA1, the amount is significantly higher in patients with IgAN or HSPN.4; 13; 29; 30; 42
Analysis of the transcription of the ST6-GalNAc genes in B cell-enriched and T cell-enriched subpopulations from PBMC confirmed lower levels of transcription of ST6-GalNAcI and higher levels of transcription of ST6-GalNAcII in the B cell population of PBMC from patients with IgAN than in such cells from healthy controls. The respective populations of B cell-enriched and T cell-enriched subpopulations had about 95% purity. This impurity explains why the B cell subpopulation from PBMC has some ST6-GalNAcI transcripts. When the subcloned IgA1-secreting cell lines were examined for transcripts of ST6-GalNAcI and ST6-GalNAcII, only the latter was detected. We also studied EBV-immortalized IgG-secreting cell lines and primary B cells and lymphoblasts and determined that ST6-GalNAcII but not ST6-GalNAcI was expressed. Taken together, our data from the studies with DAKIKI and EBV-transformed peripheral blood cells are unlikely to represent an artifact of EBV transformation, but rather to accurately reflect the situation in the subpopulation of IgA1-producing cells. Interestingly, the levels of transcription of ST6-GalNAcI and ST6-GalNAcII in T cells enriched from the PBMC of patients with IgAN were equivalent to those of healthy controls. This finding is consistent with the lack of aberrant glycosylation of proteins other than IgA1 in patients with IgAN.30; 47 Furthermore, EBV-immortalized B cells from patients with IgAN produced IgA1 with aberrant O-glycans while B cells from healthy controls produced normally galactosylated O-glycans.50 This observation indicated that the glycosylation aberrancy is not caused by the EBV transformation, but rather is an inherent characteristic of the differentiated IgA1-producing cells in patients with IgAN or HSPN.
In conclusion, ST6-GalNAcII may be the enzyme that is primarily responsible for the synthesis of sialylated-GalNAc. Additional studies are necessary to determine whether overexpression of this enzyme is a major factor in the generation of Gal-deficient IgA1 in patients with IgAN or HSPN.
Materials and methods
Reagents
All chemicals, unless otherwise specified, were from Sigma (St. Louis, MO, USA). Tissue culture media and reagents were purchased from Gibco (Invitrogen, Carlsbad, CA, USA).
Cell lines and primary cells
DAKIKI (ATCC TIB-206), a surface-IgA1-positive human lymphocyte cell line derived from lymphocytes of an African patient with nasopharyngeal carcinoma,51 were obtained from ATCC (Manassas, VA, USA). The HT29 human colorectal adenocarcinoma cell line was used as a positive control for ST6-GalNAcI, ST6-GalNAcII, ST6-GalNAcIV, and ST6-GalNAcVI (ATCC; HTB-38).
The study included cells from patients with biopsy-proven IgAN and HSPN and healthy controls. The Institutional Review Boards at the University of Alabama at Birmingham and the University of Tennessee Health Sciences Center approved this study. Informed written consent was obtained from all adults and from a parent or legally authorized representative for children; children age 8 years or older provided signed assent. PBMC were isolated using a Ficoll-Hypaque density gradient from heparinized peripheral blood of healthy controls and patients with IgAN or HSPN and then stimulated with 3 μg/ml PWM (Sigma) for 3–5 days.52 Enriched B-cell population was derived from PBMC by removal of adherent cells through incubation in a plastic tissue-culture flask for 1 h at 37°C, and removal of T cells by CD3 (panT) Dynabeads, according to the manufacturer’s instructions (Dynal ASA, Oslo, Norway). An alternative protocol for isolation of B cells (CD19 surface-positive) and T cells (CD3 surface-positive) included immunofluorescence surface staining followed by FACS sorting. A fraction of PBMC was immortalized with EBV 53 in the General Clinical Research Center Laboratory Core Facility at the University of Alabama at Birmingham. Then, we subcloned several IgA1-secreting EBV-immortalized B cell lines by limiting dilution in RPMI 1640 supplemented with L-glutamine, 20% FCS, penicillin, and streptomycin. We generated seven clones from four IgAN patients and seven clones from four healthy controls that secreted only IgA1.
IgA1 isolation and lectin ELISA
IgA1 was purified using a combination of affinity- and size-exclusion chromatography from culture supernatants of DAKIKI cells 20; 23; 54; 55 and from serum of a healthy control. IgA1 purified from the plasma of a patient with IgA1 multiple myeloma 13; 54 was used as the standard for Gal-deficient IgA1. The purity and molecular form of the IgA1 preparations was assessed by size-exclusion chromatography and SDS-PAGE.
Lectin ELISA was used to characterize the O-linked glycans in the IgA1 hinge region. HAA lectin reacts with terminal GalNAc, but not with sialylated GalNAc or GalNAc-Gal disaccharide, whereas PNA lectin reacts with GalNAc-Gal, but not with the sialylated disaccharide, sialylated GalNAc or terminal GalNAc. The ELISA plates were coated with 100 μl/well of purified DAKIKI IgA1, IgA1 isolated from serum of a healthy control, or IgA1 (Mce) myeloma protein (5 μg/ml protein). Some of the coated plates were treated with 1 mU/well neuraminidase (Roche Diagnostic Corp., Indianapolis, IN, USA) in 0.01 M acetate buffer, pH 5.0 for 3 h at 37°C to remove sialic acid 29. The coated wells were then reacted with biotin-labeled HAA (Sigma), a GalNAc-specific lectin, or with biotin-labeled PNA (Sigma), a GalNAc+Gal-specific lectin, for 3 h at 37°C. Subsequently, alkaline phosphatase-streptavidin conjugate was added and the plates were incubated at 37°C. After addition of the substrate for colorimetric detection,13 the absorbance at 405 nm was measured using an EL312 Bio-Kinetics microplate reader (Bio-Tek Instruments Inc., Winooski, VT, USA).
Monosaccharide compositional analysis
The monosaccharides from purified IgA1 secreted by the DAKIKI cell line were determined as trifluoroacetates of methylglycosides by gas chromatography.13 The analyses were performed with a gas chromatograph (Model 5890, Hewlett-Packard, Sacramento, CA, USA equipped with a 25-m fused silica (0.22-mm inner diameter) OV-1701 WCOT column (Chrompack, Bridgewater, NJ, USA) and electron capture detector. About 20 μg of each purified IgA1 protein (DAKIKI and IgA1 [Mce] myeloma protein) was used for the analysis. IgA1 (Mce) myeloma protein has naturally Gal-deficient O-linked glycans and its glycosylation thus resembles the aberrant glycosylation of the IgA1 in patients with IgAN.13
Preparation of Desialylated-IgA1
IgA1 (Mce) myeloma protein (10 μg) was desialylated with 3 mU neuraminidase (Roche) in 40 μl SB buffer (50 mM sodium acetate buffer pH 5.5, 4 mM CaCl2, 100 μg/ml BSA) at 37°C for 12 h. The desialylated Mce IgA1 was recovered by incubation with jacalin-agarose (Pierce, Rockford, IL, USA) in Dulbecco’s PBS (DPBS) at 22°C for 3 h.29 After washing, the jacalin-agarose-Mce IgA1 was resuspended in DPBS to achieve a final concentration 1 μg IgA1 per 2 μl of the agarose suspension. For determination of ST6-GalNAc enzyme activity, the desialylated Mce IgA1 was recovered by size-exclusion chromatography.13
Preparation of the Golgi-enriched fraction from IgA1-producing cells
Golgi enzyme extraction was performed using a modification of published methods.56 Briefly, cells (1 × 107) were harvested and homogenized on ice in a Potter-Elvehjem homogenizer in 2 ml 25 mM MES buffer (pH 6.5) containing 75 mM NaCl and 1x Complete Mini EDTA-free protease inhibitor mix (Roche). The homogenate was centrifuged at 1,000 x g for 10 min. The supernatant was re-centrifuged at 27,000 x g for 1 h and the pellet was resuspended in 25 mM MES buffer, pH 6.5 containing 75 mM NaCl, 10 mM MnCl2, 10% glycerol (v/v), 1% Triton-X100, and 1x Complete Mini EDTA-free protease inhibitor mix (Roche). After 1 h, the suspension was centrifuged at 100,000 x g for 1 h and the supernatant, designated as the Golgi-enriched fraction, was used as the source of the sialyltransferase enzyme. All purification steps were performed at 4°C.
Sialyltransferase assay
The sialyltransferase activity was determined using desialylated IgA1 (Mce) myeloma protein as the acceptor and CMP-Neu5Ac (Sigma) as the donor substrate. A 6 μl aliquot of the desialylated IgA1 (Mce) bound to jacalin-agarose was incubated with 10 μl Golgi-enriched fraction for 16 h at 37°C in 50 μl reaction buffer (50 mM MES buffer, pH 6.5; 2 mM CaCl2, 2 mM MnCl2, 10 mM MgCl2, 0.6% Triton-X100, 100 mM CMP-NeuAc, 1 mM 2,3-dehydro-2-deoxy-Neu5Ac [sialidase inhibitor, Sigma]; 1x Complete Mini EDTA-free protease inhibitor mix). To assess the extent of IgA1 protein sialylation, 25 μl of the reaction mixture containing jacalin-agarose-Mce IgA1 (Mce) myeloma protein was washed three times with DPBS and the pellet resuspended in 15 μl SB buffer containing 1 mU neuraminidase (Roche). After incubation for another 12 h at 37°C, the suspension was centrifuged for 5 min at 10,000 x g and the supernatant was collected.
Quantitative assay for measurement of ST6-GalNAc-IgA1 enzyme activity
To measure enzymatic activity of ST6-GalNAc, we used a modified protocol of a previous method.57 Briefly, the sialyltransferase activity was determined using desialylated IgA1 (Mce) myeloma protein as the acceptor and CMP-Neu5Ac (Sigma) as the donor substrate. Two μg desialylated IgA1 (Mce) myeloma protein was incubated with 10 μl Golgi-enriched fraction for 4 h at 37°C in 30 μl reaction buffer (50 mM MES buffer, pH 6.5; 2 mM CaCl2, 2 mM MnCl2, 10 mM MgCl2, 0.4% Triton-X100, 10 mM CMP-NeuAc, 1 mM 2,3-dehydro-2-deoxy-Neu5Ac [sialidase inhibitor, Sigma]; 1x Complete Mini EDTA-free protease inhibitor mix). The reaction was stopped by snap-freezing the samples at −80°C. Sialylation of GalNAc residues on IgA1 was determined as a decrease in the HAA lectin binding to IgA1 in the reaction mixture by ELISA 58 without neuraminidase treatment. Specificity of the assay was verified in a parallel reaction of HAA with the IgA1 in the reaction mixture that was treated with neuraminidase after capture on the ELISA plate. One unit of the enzyme activity was defined as a decrease of HAA binding to IgA1 by 1.0 OD in 4 h. The enzyme activity was normalized per mg protein in the Golgi-enriched preparation in the reaction mixture.
SDS-PAGE and western Blot
IgA1 proteins and cellular extracts were separated by SDS-PAGE under reducing conditions and gels were stained for protein by Coomassie Blue or blotted on PVDF membrane and probed with IgA-specific antibody (Sigma) or HAA lectin (Sigma).16 Enhanced chemiluminescence (Pierce) was used for detection.59
Reverse transcription (RT) RealTime PCR
Total RNA was isolated using RNAStat60.54 The mRNA was purified using the Oligotex mRNA isolation kit (Qiagen, Valencia, CA, USA); mRNA was then converted to cDNA with SuperScriptII reverse transcriptase (Invitrogen). RealTime PCR was performed using LightCycler FastStart DNA Master SYBR Green I chemistry as suggested by the manufacturer (Roche) with MgCl2 adjusted to a final concentration of 2.6 mM.60 Primers for detection of ST6-GalNAcI, ST6-GalNAcII, ST6-GalNAcIV, and ST6-GalNAcVI were as described in Table 1. Primers for β-actin amplification were purchased from R&D Systems (Minneapolis, MN, USA). RealTime PCR was performed for 42 cycles: denaturation at 95°C, annealing at 64°C, and extension at 72°C. For confirmation of the specificity of each reaction, the PCR product was analyzed using 1.5% agarose Tris-Acetate-EDTA (TAE) electrophoresis.54
Acknowledgments
This work was supported by grants DK61525, DK078244, DK71802, DK64400, and DE13694 from the National Institutes of Health and by General Clinical Research Centers of the University of Alabama at Birmingham (M01 RR00032) and the University of Tennessee Health Sciences Center (M01 RR00211) and by KONTAKT ME875 (Czech Republic). The authors express their appreciation to Catherine Barker and Sue Woodford (University of Alabama at Birmingham), and Dr. Kimberly Fisher and Sandra Grimes (University of Tennessee) for collection of the clinical samples.
The abbreviations used are
- C1β3GalT1
core 1 β1,3-galactosyltransferase
- C1β3GalT1
specific molecular chaperone
- C3
third component of complement cascade
- CMP-Neu5Ac
CMP- 5 N-Acetylneuraminic acid
- Cosmc (C1β3GalT2)
DPBS, Dulbecco’s phosphate buffered saline
- EBV
Epstein-Barr virus
- FCS
fetal calf serum
- Gal
galactose
- GalNAc
N-acetylgalactosamine
- GalT
β1,3-galactosyltransferase
- HAA
Helix aspersa agglutinin
- HSPN
Henoch-Schoenlein purpura nephritis
- IgAN
IgA nephropathy
- MES
4-morpholinethanesulfonic acid
- NeuAc
N-acetyl neuraminic acid
- PBMC
peripheral blood mononuclear cells
- PNA
peanut agglutinin
- PWM
pokeweed mitogen
- SA
sialic acid
- SDS-PAGE
sodiumdodecylsulfate polyacrylamide gel electrophoresis
- ST6-GalNAcI (SIAT7A), ST6-GalNAcII (SIAT7B), ST6-GalNAcIV (SIAT7D), ST6-GalNAcVI (ST6-GalNAcVI)
-
CMP-NeuAc:R-GalNAc-α1-O-Ser/Thr α2,6-sialyltransferase
- TAE
Tris-acetate-EDTA buffer
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Julian BA, Waldo FB, Rifai A, Mestecky J. IgA nephropathy, the most common glomerulonephritis worldwide. A neglected disease in the United States? Am J Med. 1988;84:129–132. doi: 10.1016/0002-9343(88)90019-8. [DOI] [PubMed] [Google Scholar]
- 2.Emancipator SN. IgA nephropathy and Henoch-Schönlein syndrome. In: Jennette JC, Olson JL, Schwartz MM, Silva FG, editors. Heptinstall’s Pathology of the Kidney. Lippincott-Raven Publishers; Philadelphia: 1998. pp. 479–539. [Google Scholar]
- 3.Emancipator SN, Mestecky J, Lamm ME. IgA nephropathy and related diseases. In: Ogra PL, Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, editors. Mucosal Immunology. Academic Press; San Diego: 1999. pp. 1365–1380. [Google Scholar]
- 4.Mestecky J, Tomana M, Crowley-Nowick PA, Moldoveanu Z, Julian BA, Jackson S. Defective galactosylation and clearance of IgA1 molecules as a possible etiopathogenic factor in IgA nephropathy. Contrib Nephrol. 1993;104:172–182. doi: 10.1159/000422410. [DOI] [PubMed] [Google Scholar]
- 5.Couser WG. Glomerulonephritis. Lancet. 1999;353:1509–1515. doi: 10.1016/S0140-6736(98)06195-9. [DOI] [PubMed] [Google Scholar]
- 6.Floege J, Feehally J. IgA nephropathy: recent developments. J Am Soc Nephrol. 2000;11:2395–2403. doi: 10.1681/ASN.V11122395. [DOI] [PubMed] [Google Scholar]
- 7.Coppo R, Amore A. Aberrant glycosylation in IgA nephropathy (IgAN) Kidney Int. 2004;65:1544–7. doi: 10.1111/j.1523-1755.2004.05407.x. [DOI] [PubMed] [Google Scholar]
- 8.Barratt J, Feehally J. IgA Nephropathy. J Am Soc Nephrol. 2005;16:2988–2097. doi: 10.1681/ASN.2005020134. [DOI] [PubMed] [Google Scholar]
- 9.Novak J, Julian BA, Tomana M, Mestecky J. Progress in molecular and genetic studies of IgA nephropathy. J Clin Immunol. 2001;21:310–327. doi: 10.1023/a:1012284402054. [DOI] [PubMed] [Google Scholar]
- 10.Julian BA, Novak J. IgA nephropathy: an update. Current Opin Nephrol Hypertens. 2004;13:171–179. doi: 10.1097/00041552-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 11.Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, Shinzato T, Kobayashi Y, Maeda K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59:1077–1085. doi: 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 12.Allen AC, Bailey EM, Brenchley PEC, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001;60:969–973. doi: 10.1046/j.1523-1755.2001.060003969.x. [DOI] [PubMed] [Google Scholar]
- 13.Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Novak J, Coward L, DeSilva T, Kirk M, Novak L, Vu HL, Cook WJ, Kim H, Barnes S, Julian BA, Mestecky J, Tomana M. Galactose (Gal)-deficient IgA1-containing circulatory immune complexes from IgA nephropathy (IgAN) patients bind to cultured human mesangial cells and induce cell activation. J Am Soc Nephrol. 2001;12:688A. [Google Scholar]
- 15.Kokubo T, Hashizume K, Iwase H, Arai K, Tanaka A, Toma K, Hotta K, Kobayashi Y. Humoral immunity against the proline-rich peptide epitope of the IgA1 hinge region in IgA nephropathy. Nephrol Dial Transplant. 2000;15:28–33. doi: 10.1093/ndt/15.1.28. [DOI] [PubMed] [Google Scholar]
- 16.Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, Julian BA, Wyatt RJ, Mestecky J. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–513. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 17.Frangione B, Wolfenstein-Todel C. Partial duplication in the “hinge” region of IgA1 myeloma proteins. Proc Natl Acad Sci USA. 1972;69:3673–3676. doi: 10.1073/pnas.69.12.3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Putnam FW. Structure of the human IgA subslasses and allotypes. Protides Biol Fluids. 1989;36:27–37. [Google Scholar]
- 19.Mestecky J, Moro I, Kerr MA, Woof JM. Mucosal immunoglobulins. In: Mestecky J, Bienenstock J, Lamm ME, Mayer L, McGhee JR, Strober W, editors. Mucosal Immunology. 3. Vol. 1. Elsevier Academic Press; Amsterdam: 2005. pp. 153–181. [Google Scholar]
- 20.Mestecky J, Kilian M. Immunoglobulin A (IgA) Methods Enzymol. 1985;116:37–75. doi: 10.1016/s0076-6879(85)16005-2. [DOI] [PubMed] [Google Scholar]
- 21.Mestecky J, Russell MW. IgA subclasses. Monogr Allergy. 1986;19:277–301. [PubMed] [Google Scholar]
- 22.Tarelli E, Smith AC, Hendry BM, Challacombe SJ, Pouria S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr Res. 2004;339:2329–35. doi: 10.1016/j.carres.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Renfrow MB, Cooper HJ, Tomana M, Kulhavy R, Hiki Y, Toma K, Emmett MR, Mestecky J, Marshall AG, Novak J. Determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation Fourier transform-ion cyclotron resonance mass spectrometry. J Biol Chem. 2005;280:19136–19145. doi: 10.1074/jbc.M411368200. [DOI] [PubMed] [Google Scholar]
- 24.Mellis SJ, Baenziger JU. Structures of the O-glycosidically linked oligosaccharides of human IgD. J Biol Chem. 1983;258:11557–11563. [PubMed] [Google Scholar]
- 25.Baenziger J, Kornfeld S. Structure of the carbohydrate units of IgA1 immunoglobulin II. Structure of the O-glycosidically linked oligosaccharide units. J Biol Chem. 1974;249:7270–7281. [PubMed] [Google Scholar]
- 26.Field MC, Dwek RA, Edge CJ, Rademacher TW. O-linked oligosaccharides from human serum immunoglobulin A1. Biochem Soc Trans. 1989;17:1034–1035. doi: 10.1042/bst0171034. [DOI] [PubMed] [Google Scholar]
- 27.Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, Rudd PM, Woof JM, Dwek RA. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J Biol Chem. 1998;273:2260–2272. doi: 10.1074/jbc.273.4.2260. [DOI] [PubMed] [Google Scholar]
- 28.Novak J, Tomana M, Kilian M, Coward L, Kulhavy R, Barnes S, Mestecky J. Heterogeneity of O-glycosylation in the hinge region of human IgA1. Mol Immunol. 2000;37:1047–1056. doi: 10.1016/s0161-5890(01)00019-0. [DOI] [PubMed] [Google Scholar]
- 29.Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997;52:509–516. doi: 10.1038/ki.1997.361. [DOI] [PubMed] [Google Scholar]
- 30.Allen AC, Harper SJ, Feehally J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin Exp Immunol. 1995;100:470–474. doi: 10.1111/j.1365-2249.1995.tb03724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulson JC, Colley KJ. Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J Biol Chem. 1989;246:17615–17618. [PubMed] [Google Scholar]
- 32.Schachter H, Roseman S. Mammalian glycosyltransferases. In: Lennarz WJ, editor. The Biochemistry of Glycoproteins and Proteoglycans. Plenum Press; New York: 1980. pp. 85–160. [Google Scholar]
- 33.Piller V, Piller F, Fukuda M. Biosynthesis of truncated O-glycans in the T cell line Jurkat. Localization of O-glycan initiation. J Biol Chem. 1990;265:9264–9271. [PubMed] [Google Scholar]
- 34.Iwasaki H, Zhang Y, Tachibana K, Gotoh M, Kikuchi N, Kwon YD, Togayachi A, Kudo T, Kubota T, Narimatsu H. Initiation of O-glycan synthesis in IgA1 hinge region is determined by a single enzyme, UDP-N-acetyl-alpha-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2. J Biol Chem. 2003;278:5613–21. doi: 10.1074/jbc.M211097200. [DOI] [PubMed] [Google Scholar]
- 35.Ju T, Brewer K, D’Souza A, Cummings RD, Canfield WM. Cloning and expression of human core 1 β1,3-galactosyltransferase. J Biol Chem. 2002;277:178–86. doi: 10.1074/jbc.M109060200. [DOI] [PubMed] [Google Scholar]
- 36.Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 β3-galactosyltransferase. Proc Natl Acad Sci U S A. 2002;99:16613–8. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kudo T, Iwai T, Kubota T, Iwasaki H, Takayma Y, Hiruma T, Inaba N, Zhang Y, Gotoh M, Togayachi A, Narimatsu H. Molecular cloning and characterization of a novel UDP-Gal:GalNAcα peptide β1,3-galactosyltransferase (C1Gal-T2), an enzyme synthesizing a core 1 structure of O-glycan. J Biol Chem. 2002;277:47724–31. doi: 10.1074/jbc.M205839200. [DOI] [PubMed] [Google Scholar]
- 38.Ju T, Cummings RD. Protein glycosylation: chaperone mutation in Tn syndrome. Nature. 2005;437:1252. doi: 10.1038/4371252a. [DOI] [PubMed] [Google Scholar]
- 39.Schachter H, McGuire EJ, Roseman S. Sialic acids. XIII A uridine diphosphate D-galactose: mucin galactosyltransferase from porcine submaxillary gland. J Biol Chem. 1971;246:5321–5328. [PubMed] [Google Scholar]
- 40.Kono M, Tsuda T, Ogata S, Takashima S, Liu H, Hamamoto T, Itzkowitz SH, Nishimura S, Tsuji S. Redefined substrate specificity of ST6GalNAc II: a second candidate sialyl-Tn synthase. Biochem Biophys Res Commun. 2000;272:94–97. doi: 10.1006/bbrc.2000.2745. [DOI] [PubMed] [Google Scholar]
- 41.Hiki Y, Horii A, Iwase H, Tanaka A, Toda Y, Hotta K, Kobayashi Y. O-linked oligosaccharide on IgA1 hinge region in IgA nephropathy. Fundamental study for precise structure and possible role. Contrib Nephrol. 1995;111:73–84. [PubMed] [Google Scholar]
- 42.Allen AC, Willis FR, Beattie TJ, Feehally J. Abnormal IgA glycosylation in Henoch-Schönlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant. 1998;13:930–934. doi: 10.1093/ndt/13.4.930. [DOI] [PubMed] [Google Scholar]
- 43.Renfrow MB, Hall S, Brown R, Tomana M, Julian BA, Mestecky J, Emmett MR, Novak J, AG M. Human IgA1 hinge region O-glycan profiling by FT-ICR MS. 53rd Annual Conference of the American Society for Mass Spectrometry; San Antonio, Texas. June 5–9.2005. [Google Scholar]
- 44.Ikehara Y, Kojima N, Kurosawa N, Kudo T, Kono M, Nishihara S, Issiki S, Morozumi K, Itzkowitz S, Tsuda T, Nishimura SI, Tsuji S, Narimatsu H. Cloning and expression of a human gene encoding an N-acetylgalactosamine-α2,6-sialyltransferase (ST6GalNAc I): a candidate for synthesis of cancer-associated sialyl-Tn antigens. Glycobiology. 1999;9:1213–1224. doi: 10.1093/glycob/9.11.1213. [DOI] [PubMed] [Google Scholar]
- 45.Marcos NT, Cruz A, Silva F, Almeida R, David L, Mandel U, Clausen H, Von Mensdorff-Pouilly S, Reis CA. Polypeptide GalNAc-transferases, ST6GalNAc-transferase I, and ST3Gal-transferase I expression in gastric carcinoma cell lines. J Histochem Cytochem. 2003;51:761–771. doi: 10.1177/002215540305100607. [DOI] [PubMed] [Google Scholar]
- 46.Marcos NT, Pinho S, Grandela C, Cruz A, Samyn-Petit B, Harduin-Lepers A, Almeida R, Silva F, Morais V, Costa J, Kihlberg J, Clausen H, Reis CA. Role of the human ST6GalNAc-I and ST6GalNAc-II in the synthesis of the cancer-associated sialyl-Tn antigen. Cancer Res. 2004;64:7050–7057. doi: 10.1158/0008-5472.CAN-04-1921. [DOI] [PubMed] [Google Scholar]
- 47.Smith AC, de Wolff JF, Molyneux K, Feehally J, Barratt J. O-Glycosylation of serum IgD in IgA nephropathy. J Am Soc Nephrol. 2006;17:1192–9. doi: 10.1681/ASN.2005101115. [DOI] [PubMed] [Google Scholar]
- 48.Allen AC, Topham PS, Harper SJ, Feehally J. Leucocyte β1,3 galactosyltransferase activity in IgA nephropathy. Nephrol Dial Transplant. 1997;12:701–706. doi: 10.1093/ndt/12.4.701. [DOI] [PubMed] [Google Scholar]
- 49.Novak J, Raska M, Tomana M, Hall S, Moldoveanu Z, Brown R, Sarabu N, Novak L, Julian BA, Mestecky J. Transcriptional regulation by cytokines of O-glycan-specific glycosyltransferases in human IgA1-producing cells: Implications for IgA nephropathy (IgAN) J Am Soc Nephrol. 2003;14:632A. [Google Scholar]
- 50.Suzuki H, Moldoveanu Z, Hall S, Brown R, Vu HL, Novak L, Julian BA, Wyatt RJ, Tomino Y, Mestecky J, Novak J. Epstein-Barr virus (EBV)-immortalized B cells from patients with IgA nephropathy (IgAN) secrete IgA1 with galactose (Gal)-deficient O-linked glycans. J Am Soc Nephrol. 2006;17:351–352A. [Google Scholar]
- 51.Steinitz M, Klein G. EBV-transformation of surface IgA-positive human lymphocytes. J Immunol. 1980;125:194–196. [PubMed] [Google Scholar]
- 52.Moldoveanu Z, Egan ML, Mestecky J. Cellular origins of human polymeric and monomeric IgA: intracellular and secreted forms of IgA. J Immunol. 1984;133:3156–62. [PubMed] [Google Scholar]
- 53.Hajdu I, Moldoveanu Z, Cooper MD, Mestecky J. Ultrastructural studies of human lymphoid cells. μ and J chain expression as a function of B cell differentiation. J Exp Med. 1983;158:1993–2006. doi: 10.1084/jem.158.6.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Novak J, Vu HL, Novak L, Julian BA, Mestecky J, Tomana M. Interactions of human mesangial cells with IgA and IgA-containing circulating immune complexes. Kidney Int. 2002;62:465–475. doi: 10.1046/j.1523-1755.2002.00477.x. [DOI] [PubMed] [Google Scholar]
- 55.Sandin C, Linse S, Areschoug T, Woof JM, Reinholdt J, Lindahl G. Isolation and detection of human IgA using a streptococcal IgA-binding peptide. J Immunol. 2002;169:1357–64. doi: 10.4049/jimmunol.169.3.1357. [DOI] [PubMed] [Google Scholar]
- 56.Preuss U, Gu X, Gu T, Yu RK. Purification and characterization of CMP-N-acetylneuraminic acid:lactosylceramide (alpha 2-3) sialyltransferase (GM3-synthase) from rat brain. J Biol Chem. 1993;268:26273–8. [PubMed] [Google Scholar]
- 57.Basset C, Durand V, Mimassi N, Pennec YL, Youinou P, Dueymes M. Enhanced sialyltransferase activity in B lymphocytes from patients with primary Sjogren’s syndrome. Scand J Immunol. 2000;51:307–11. doi: 10.1046/j.1365-3083.2000.00692.x. [DOI] [PubMed] [Google Scholar]
- 58.Moore JS, Kulhavy R, Tomana M, Moldoveanu Z, Suzuki H, Brown R, Hall S, Kilian M, Poulsen K, Mestecky J, Julian BA, Novak J. Reactivities of N-acetylgalactosamine-specific lectins with human IgA1 proteins. Mol Immunol. 2007;44:2598–604. doi: 10.1016/j.molimm.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore JS, Wu X, Kulhavy R, Tomana M, Novak J, Moldoveanu Z, Brown R, Goepfert PA, Mestecky J. Increased levels of galactose-deficient IgG in sera of HIV-1-infected individuals. AIDS. 2005;19:381–389. doi: 10.1097/01.aids.0000161767.21405.68. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen HH, Broker TR, Chow LT, Alvarez RD, Vu HL, Andrasi J, Brewer LR, Jin G, Mestecky J. Immune responses to human papillomavirus in genital tract of women with cervical cancer. Gynecol Oncol. 2005;96:452–61. doi: 10.1016/j.ygyno.2004.10.019. [DOI] [PubMed] [Google Scholar]