

Abstract
Human lung adenocarcinoma A549 cells stably transfected with TPα (A549-TPα) were used to study agonist I-BOP-induced expression of cyclooxygenase-2 (COX-2) and the related mechanisms of induced expression. I-BOP, a TP agonist, induced a time and dose dependent expression of COX-2 in A549-TPα cells as revealed by Western blot and by the synthesis of PGE2 and TXB2 which was totally inhibited by COX-2 inhibitors. The signaling pathways of I-BOP-induced COX-2 expression were elucidated by using various inhibitors of the signaling molecules. The effects of these inhibitors were assessed at three different levels of COX-2 expression: protein, enzyme activity and promoter activity. Within MAPK family, both ERK and p38 MAPK but not JNK/SAPK pathways were involved in the induction. Other pathways such as JAK/Stat3 pathway and β-catenin/TCF/LEF pathway also participated in the induction. The activation of key signaling molecules, ERK, p38 MAPK, CREB and NF-κB, involved in the COX-2 transcription was further studied at the phosphorylation step. Activation of ERK and p38 MAPK appeared to be mediated primarily by transactivation of EGFR, whereas activation of CREB and NF-κB was mediated by PKA, PKC and ERK. The role of CREB and NF-κB in I-BOP-induced COX-2 expression was further explored at the COX-2 promoter level. Studies on promoter fragments of different length and mutation of responsive motifs indicated that CRE and NF-κB sites are critical for the COX-2 induction. Distal NF-κB site is essential for the basal induction of the COX-2 transcription, whereas CRE and proximal NF-κB sites are important for the induced transcription. These results indicate that I-BOP induced COX-2 expression through multiple signaling pathways leading to the activation of CREB and NF-κB transcriptional factors and subsequent COX-2 transcription.
INTRODUCTION
Thromboxane A2 (TXA2) is generated from arachidonic acid by consecutive actions of cyclooxygenase (COX), existing as two isoforms of COX-1 and COX-2, and thromboxane synthase (TXAS) [1]. TXA2 exhibits potent and diverse bioactivities, such as platelet aggregatory activity and smooth muscle constrictive activity [2, 3], which are mediated by the activation of TXA2 receptors (TPs) [4]. Two isoforms of TP, TPα and TPβ, have been identified [5]. They are structurally identical in the first 328 amino acids, but different in the C-terminal tail. Activation of both isoforms of TP leads to stimulation of phospholipase C generating two second messengers, inositol triphosphate and diacylglycerol, which mobilize intracellular Ca2+ and activate protein kinase C (PKC) respectively [6]. Activation of TPα also leads to stimulation of adenylate cyclase generating cyclic AMP which activates protein kinase A (PKA), whereas activation of TPβ results in inhibition of adenylate cyclase [7]. Activation of TPs also initiates signaling cascades leading to activation of various kinases known to be involved in mitogenic responses, such as epidermal growth factor receptor (EGFR) [8], extracellular signal-regulated kinase (ERK) [9], Akt/protein kinase B (PKB) [10] and glycogen synthase kinase (GSK) [11]. Apparently, TP signaling is very much related to mitogenesis of target cells.
Recent reports indicate that TP agonists induce mitogenic and hypertrophic effects in smooth muscle cells [12], and stimulate proliferation of oligodentrocytes [13]. TP agonists also induce endothelial cell migration, angiogenesis and metastasis [14] and tumor metastasis [15]. The role of TPs in tumorigenesis has emerged. Over the last decade, an increased interest in the role of COX-2, a rate-limiting enzyme in PGE2 and TXA2 biosynthesis, in tumorigenesis becomes apparent. This interest was sparked by the fact that COX inhibitors reduced the mortality rate from certain cancer patients [16, 17] and that most of the tumors exhibited over-expression of COX-2, a potential oncogene [18]. One of the major COX-2 derived products, PGE2, has also been shown to induce the growth, migration, and invasiveness of carcinoma cells [19]. In view of the fact that PGE2 up-regulates the expression of COX-2 in several cancer cell lines resulting in positive feedback actions of PGE2 [20, 21], we hypothesize that another COX-2 derived down-stream product with similar mitogenic activity, TXA2, will be also capable of inducing the expression of COX-2.
Lung is an organ actively synthesizing TXA2 and PGE2. The roles of TXA2 in pulmonary and cardiovascular functions have been well described [2, 3]. However, its role in lung tumorigenesis is less clear although it induces mitogenesis and hypertrophy of smooth muscle cells and other types of cells as described above. Over-expression of COX-2 in lung tumors has been widely reported [22, 23]. Factors that may induce over-expression of COX-2 in lung tumors are incompletely defined. The aims of this study are to demonstrate that activation of TP induces the expression of COX-2 and to elucidate the signaling pathways leading to the expression of COX-2. We employ human lung adenocarcinoma A549 cells stably over-expressing TPα to investigate these objectives. Our results indicate that activation of TPα induces COX-2 expression through multiple signaling pathways.
MATERIALS AND METHODS
Materials
Culture medium, lipofectamine 2000, heat-inactivated fetal bovine serum (FBS), and restriction enzymes were from Invitrogen (Carlsbad, CA). I-BOP, SQ-29548 (SQ), BM567, and LY294002 (LY) were from Cayman Chemical (Ann Arbor, MI). H89, PKI, wortmannin, PD98059, U0126, AG1478, PD153035, PP2, MG132, Curcumin, SB 203580, SB415286, Celebrex, UK37248, SP600125, JAK inhibitor I and GF109203X (GF) were obtained from Calbiochem (San Diego, CA). Other biochemicals and chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Human lung adenocarcinoma A549 cells and AD293 cells were supplied by the American Type Culture Collection (Manassas, VA). ECL Western blotting detection system was purchased from the Amersham Pharmacia Biotech (Cardiff, UK). pcDNA3 encoding human TPα and rabbit polyclonal antibody specific to N-terminal sequence of TPs were generated as described previously [24]. Rabbit polyclonal antibody specific to COX-2 was from Cayman Chemical (Ann Arbor, MI). Mouse monoclonal antibody specific to pERK was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Rabbit monoclonal antibodies specific to phospho-p38 MAPK, pStat3, phospho-CREB and phospho-NF-κB-p65 (Ser276) were from Cell Signaling Technology Inc. (Beverly, MA). Antibody specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was generated as described [11]. Horseradish peroxidase (HRP)-linked goat anti-mouse and rabbit IgG were supplied by BD Transduction Laboratories (Lexington, KY). Luciferase reporter constructs with the different deletion regions from −918 to +49 bp and site-directed mutation of some consensus elements in COX-2 promoter were prepared as described previously [25, 26].
Cell Culture and Stable Transfectants
A549 cells were cultured as monolayers in RPMI1640 supplement with 10% (v/v) heat inactivated fetal bovine serum (FBS), 100 mg/L streptomycin and 100U/mL of penicillin G at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were plated in 12-well plates for the activation of various kinases and inhibitor studies. Cells were serum-deprived for 24 h before stimulation. For inhibitor study, cells were pretreated with the respective inhibitors at working concentrations or vehicle (0.1% ethanol) for 30 min in serum-free medium prior to stimulation. For stable transfection, cells were allowed to grow in 10% FBS medium for 48 h after transfection using lipofectamine 2000 and then were diluted 10-fold and treated with 1 mg/mL of G418 until the colonies formed as described previously [24]. The western blot detection of the TPα and the [3H] SQ-29548 binding assay were used to monitor the expression level among different colonies. After the colony with a high level of expression of TPα was obtained, it was maintained in RPMI 1640 media supplemented with 10% FBS and 200 μg/mL G418.
Whole Cell Radioligand Binding Assay
Cells were cultured in 10-cm plates and were harvested when 90% confluence was achieved. The cells were washed in ice-cold phosphate-buffered saline three times. Then, 1 × 106 cells were suspended in 95 μL of phosphate-buffered saline using 1.5-mL plastic conical tubes, and the binding assay was conducted in a final volume of 100 μL in the same tubes using [3H] SQ-29548 as a labeled ligand as described previously [24].
Western Blotting
Cells were cultured in 12-well plates to achieve approximately 80% confluence, and then were starved in RPMI 1640 medium without FBS for 24 h. The culture medium was changed and cells were kept for 1 h before the addition of agonist and various inhibitors. After incubation for needed time, cells were harvested and lysed in lysis buffer (1% Nonidet P-40 in 150 mM NaCl, 50 mM HEPES, pH 7.4, 5 mM NaF, 5 mM pyrophosphate, 1 mM sodium orthovanadate, 10 μg/mL aprotinin, 10 μg/mL leupetin, and 1 mM PMSF ) for 1 h on ice. Lysates were cleared by centrifugation at maximum speed on a benchtop centrifuge and then subjected to 12% SDS-PAGE. Proteins were then electrophoretically transferred onto PVDF membrane. The membrane was blocked with 5% nonfat milk in 30 mM Tris-HCl, pH 7.4, containing 120 mm NaCl (TBS) at room temperature for 1 h. It was then incubated for 2 h at room temperature with a primary antibody in TBS with 5% nonfat milk, and then the membrane was washed five times with TBS buffer containing 0.05% Tween 20 (TBST), following incubation with horseradish peroxidase linked goat anti-mouse or rabbit IgG for 1 h at room temperature. Excess antibody was washed with TBST buffer and the immunoreactive bands were detected using ECL Western blotting detection system. All the membranes were stripped using stripping buffer before reprobing with anti-GAPDH polyclonal antibody to ensure equal protein loading.
Transfection and Luciferase Reporter Gene Assay
Cells were transfected with reporter gene constructs by lipofectamine 2000 according to the manufacturer’s instruction. After transfection for 16 h, cells were stimulated with 50 nM of I-BOP for additional 24 h. For inhibitor study, cells were pretreated with the respective inhibitors at working concentrations or vehicle (0.1% ethanol) for 30 min prior to I-BOP stimulation. The luciferase activity in cell lysate was determined as described previously [25, 26].
Measurement of PGE2 and TXB2
Cells were cultured in 12-well culture plates and treated with inhibitors and I-BOP as described above. After treatment, the medium were collected and stored at −80 °C until being assayed. PGE2 and TXB2 were assayed using PGE2 and TXB2 enzyme immunoassay assay kit as described previously [27].
Statistical Analysis
EIA and luciferase assay results were expressed as mean ± S.D. Statistical significance was assessed by Student’s t test. Differences were considered statistically significant when p-values were < 0.05.
RESULTS
Stable Transfection of TPα in A549 Cells and Characterization of Functional Expression of the Receptor
A549 cells were transfected with pcDNA3 encoding TPα cDNA. After screening 40 single clones which had anti-G418 activity, the single clones with highest expression of TPα were selected and designated as A549-TPα. As shown in the previous study [24], Western blotting using an antibody specific to N-terminal peptide of TPs demonstrated that A549-TPα expressed at high level as compared to cells transfected with pcDNA3 vector alone (A549-pcDNA3). A549-TPα cells showed high affinity to the TP antagonist SQ-29548 and its Kd value was 17.4 ± 1.4 nM which was characterized by [3H] SQ-29548 binding assay. The whole cell binding experiment demonstrated that the TPα is functionally located on the surface of the A549-TPα cells, but the A549-pcDNA3 cells express minimal amount of TPs (data not shown). Similarly, AD293-TPα cells were prepared and characterized as described above.
Activation of TPα by I-BOP-Induced Expression of COX-2 and Synthesis of PGE2 and TXB2
A549-pcDNA3 cells and A549-TPα cells both expressed little basal COX-2. Stimulation of control A549-pcDNA3 cells with 50 nM I-BOP for 6 h showed no increased expression of COX-2 (data not shown). However, stimulation of A549-TPα cells with I-BOP exhibited increased expression of COX-2 in a time- and dose-dependent manner (Fig. 1). Time course studies showed that the expression of COX-2 was rapidly induced within 2 h by I-BOP (Fig. 1A), and the maximum induction was achieved following 4–6 h incubation with I-BOP. Therefore, the expression of COX-2 induced by I-BOP was carried out for 6 h in the following studies. As a control, A549-pcDNA3 cells displayed little response to agonist stimulation (data not shown). Dose dependent studies indicated that I-BOP at 0.1 nM clearly led to the expression of COX-2 and the expression reached to a maximum at 50 nM (Fig. 1B). Similarly, levels of PGE2 and TXB2 were also increased in a time- and dose-dependent manner following induction of COX-2 by I-BOP (Fig. 2), but the increase in PGE2 and TXB2 levels occurred slower than that in COX-2 expression suggesting that their production depended on the newly expressed COX-2 (Fig. 2A). The levels of PGE2 and TXB2 in the medium continued to increase since the synthesized prostaglandins were accumulated. I-BOP induced a dose dependent increase in PGE2 and TXB2 levels during a 24 h incubation period (Fig. 2B). PGE2 appeared to be synthesized in two fold higher than TXB2. The maximal increase in the synthesis of PGE2 and TXB2 was found at 50 – 100 nM I-BOP.
Figure 1. Time and dose dependent expression of COX-2 in A549-TPα cells upon TP agonist I-BOP stimulation.
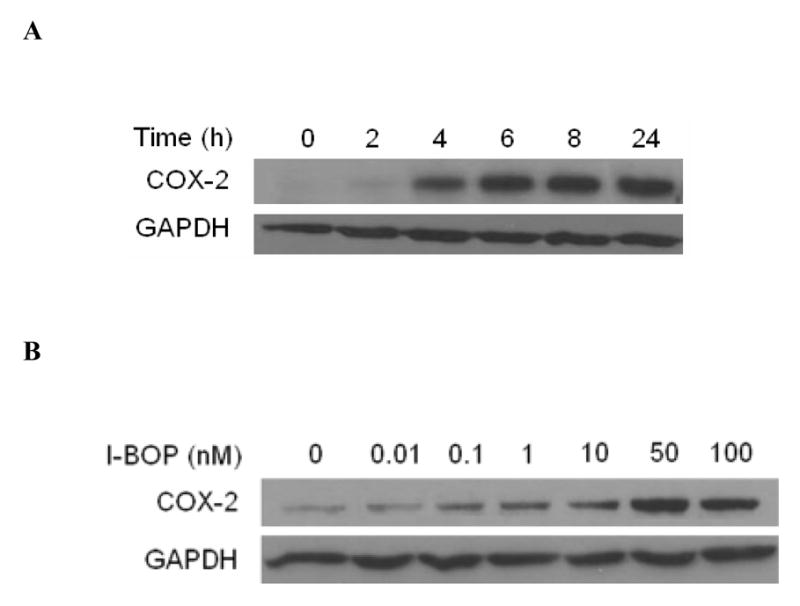
Cells were serum-starved for 16 h and then treated with 50 nM I-BOP or vehicle (0.1% ethanol) for varying length of time as indicated or with I-BOP at different concentrations as indicated for 6 h. The cell lysates were subjected to 12% SDS-PAGE and proteins were transferred to PVDF membranes as described under MATERIALS AND METHODS. The immunoreactive bands were detected with polyclonal antibodies specific for COX-2. The same membrane was probed with GAPDH antibody to ensure equal protein loading. A. Time-dependent effects of I-BOP (50 nM, 2–24 h) on COX-2 expression in A549-TPα cells. B. Dose-dependent effects of I-BOP (0.01–100 nM, 6 h) on COX-2 expression in A549-TPα cells.
Figure 2. Time and dose dependent synthesis of PGE2 and TXB2 in A549-TPα cells upon TP agonist I-BOP stimulation.


Cells were serum-starved for 16 h and then treated with 50 nM I-BOP or vehicle (0.1% ethanol) for varying length of time as indicated or with I-BOP at different concentrations as indicated for 6 h. The TXB2 and PGE2 in the medium were quantified with ELISA as described under MATERIALS AND METHODS. Fold increase (Fold/Basal) in PGE2 and TXB2 were presented as mean ± S.D. (n=3), where the basal levels of PGE2 and TXB2 in the medium of the control cells were assigned a value of 1.0. A. Time dependent effects of I-BOP (50 nM, 1–24 h) on PGE2 and TXB2 synthesis in A549-TPα cells. B. Dose dependent effects of I-BOP (0.1–100 nM, 24 h) on PGE2 and TXB2 synthesis in A549-TPα cells.
Several Key Signal Transduction Pathways Are Involved in Agonist-Induced COX-2 Expression and PGE2 and TXB2 Synthesis
Based on the results described above, we investigated signal transduction pathways that led to the expression of COX-2 and the synthesis of PGE2 and TXB2 with their specific inhibitors. TPs antagonists, SQ29548 and BM567, totally blocked I-BOP-induced expression of COX-2 providing evidence that activation of TPα mediated COX-2 expression (Fig. 3A). We studied further the involvement of various kinases in the signaling pathways in I-BOP-induced COX-2 expression. As shown in Fig. 3A, I-BOP-induced COX-2 expression was significantly inhibited by PKA inhibitors H89 and PKI, and by PKC inhibitor GF109203X indicating that both PKA and PKC are involved in the TP-mediated induction of COX-2. However, the expression was not significantly affected by PI-3K/Akt/PKB inhibitors wortmannin and LY294002. The roles of other kinases were further examined. I-BOP-induced COX-2 expression was greatly diminished by EGFR kinase inhibitors AG1478 and PD153035, and Src kinase inhibitor PP2. These results suggest that EGFR kinase and Src kinase are significantly involved in I-BOP-induced COX-2 expression.
Figure 3. Effect of various inhibitors on I-BOP-induced COX-2 expression and TXB2 and PGE2 synthesis in A549-TPα cells.


The cells were serum-starved for 16 h and then treated with 5 μM of SQ29548 or BM567, 20 μM of H-89 and PKI, 5 μM U0126, 20 μM PD98059, 10 μM SB203580, 25 μM SB415286, 10 μM MG132, 20 μM curcumin, 5 μM of celebrex, 5 μM of DuP 697, 5 μM UK37248, 30 μM SP600125, 250 nM GF109203X, 2 μM AG1478, 2 μM PD153035, 5 μM PP2, 200 nM wortmannin, 25 μM LY294002, or vehicle (0.1% ethanol )for 30 min followed by stimulation with 50 nM I-BOP for 6 h. Cells were lysed and the lysates were subjected to SDS/PAGE and Western blotting analysis as described in the MATERIALS AND METHODS. The COX-2 was detected by Western blotting as described in Figure 1. GAPDH was used to ensure equal protein loading. TXB2 and PGE2 in the medium were quantified with ELISA as described under MATERIALS AND METHODS. Fold increase (Fold/Basal) in PGE2 and TXB2 were presented as mean ± S.D. (n=3), where the basal levels of PGE2 and TXB2 in the medium of the control cells were assigned a value of 1.0. A and B Effects of various inhibitors on I-BOP-induced COX-2 expression in A549-TPα cells. C. Effects of various inhibitors on I-BOP-induced PGE2 and TXB2 synthesis in A549-TPα cells. * and ** are P values less than 0.05 and 0.01 respectively when compared with I-BOP stimulated level of PGE2 or TXB2.
We also examined the contribution of several downstream signaling molecules in their signal transduction pathways and found that I-BOP-induced COX-2 expression was significantly attenuated by MEK inhibitors U0126 and PD98059, p38 MAPK inhibitor SB203580, JAK inhibitor I, and by NF-κB phosphorylation inhibitor curcumin, and proteasome inhibitor MG132 (inhibiting IκB degradation), but was not significantly inhibited by JNK inhibitor SP600125, and GSK inhibitor SB415286 (Fig. 3B). In fact, SB415286 increased slightly COX-2 expression. The results indicate that ERK, p38 MAPK, JAK and NF-κB are involved in the signal transduction pathways that lead to COX-2 expression following I-BOP stimulation. Fig. 3B showed that COX-2 inhibitor celebrex, and TXA2 synthase inhibitor UK37248 did not affect COX-2 expression suggesting that the increase in COX-2 expression depended on I-BOP induction rather than by the newly produced PGE2 and TXA2 in this period and that these inhibitors suppress the respective enzyme activities but not the expression of COX-2 protein.
Alterations in COX-2 expression were further corroborated with changes in the synthesis of PGE2 and TXB2 following I-BOP stimulation in the presence and absence of inhibitors (Fig. 3C). The results on COX-2 expression were consistent with the data on PGE2 and TXB2 synthesis. Total inhibition of PGE2 and TXB2 synthesis by COX-2 specific inhibitors, celebrex and DuP697, indicates that it is the newly expressed COX-2 responsible for the synthesis. Interestingly, the synthesis of PGE2 was significantly increased and the production of TXB2 was totally blocked when the cells were treated with TXA2 synthase inhibitor UK37248, confirming that the synthesis of PGE2 and TXB2 is an interactive process which allows competition for the same substrate, PGH2, by the two synthases.
Activation of TPα by I-BOP Induced the Phosphorylation of Several Signaling Molecules in Related Signaling Pathways
To better define the pathways of I-BOP-induced COX-2 expression, we investigated the phosphorylation status and activation of several key signaling molecules in the signal transduction pathways. As shown in Fig. 4A, ERK was rapidly phosphorylated within 5 min and pERK level reached to the maximal amount at 20 min. Similarly, p38 MAPK was phosphorylated to a maximum at 20 min and pp38 MAPK returned to a basal level at 60 min. Comparable results were also obtained as shown in the same figure in relation to the phosphorylation of CREB, a transcription factor that binds cAMP response elements in DNA to increase transcription of concerned genes. Furthermore, high levels of phospho-p65 of NF-κB induced by I-BOP were observed at 10 – 40 min although levels began to decrease after 40 min (Fig. 4A). Since I-BOP-induced expression of COX-2 was inhibited by JAK inhibitor I, I-BOP-induced phosphorylation of Stat3 was also examined. The phosphorylation began to be observed at 5 min, stayed elevated and returned to a basal level at 60 min (Fig. 4B). JAK inhibitor I totally blocked the phosphorylation of Stat3 as expected. Consistent with the results presented in our recent publication [11], pGSK-3α/β induced by I-BOP was still observed at 40 min (Fig. 4A). The A549-pcDNA3 control cells exhibited little response to agonist stimulation (data not shown).
Figure 4. I-BOP-induced phosphorylation of ERK, CREB, NF-κB, p38 MAPK and GSK-3 in A549-TPα cells.
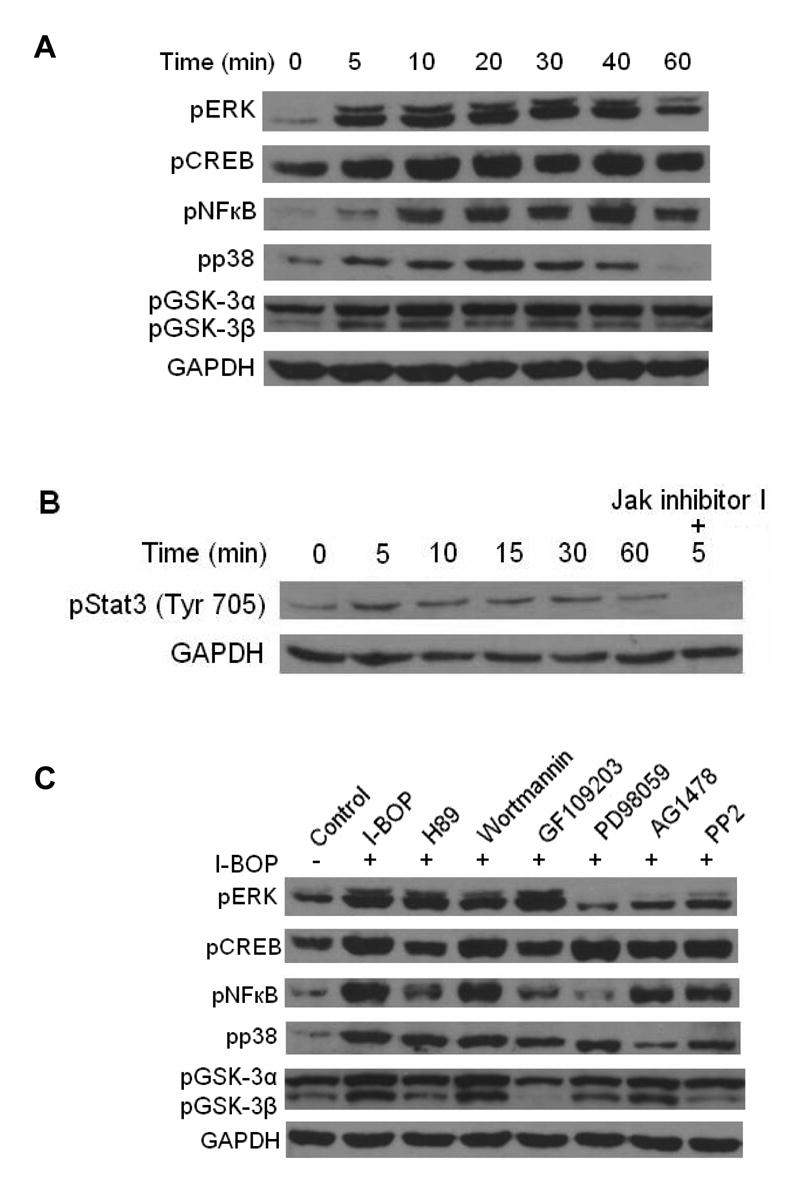
Cells were serum-starved for 16 h and then treated with 50 nM I-BOP or vehicle for varying length of time as indicated. The cell lysates were subjected to 12% SDS-PAGE and proteins were transferred to PVDF membranes for Western blotting analysis using antibodies against each phosphorylated signaling molecules as described under MATERIALS AND METHODS. The same membrane was erased after each probing and reprobed with a different antibody. GAPDH antibody was used to ensure equal protein loading. A. Time-dependent effects of I-BOP (50 nM, 5–60 min) on the phosphorylation of ERK, CREB, NF-κB, p38 MAPK and GSK-3 in A549-TPα cells. B. Time-dependent effects of I-BOP (50 nM, 5–60 min) on the phosphorylation of Stat3 in A549-TPα cells were carried out at the indicated time. Effect of JAK inhibitor on the phosphorylation of Stat3 was assessed by adding the inhibitor (10 μM) 30 min prior to stimulation by I-BOP for 5 min. C. Effect of various inhibitors on I-BOP-induced phosphorylation of ERK, CREB, NF-κB, p38 MAPK and GSK-3 in A549-TPα cells. Cells were serum-starved for 16 h and then treated with 10 μM H-89, 250 nM wortmannin, 250 nM GF109203X, 20 μM PD-98059, 2 μM AG1478 and 10 μM PP2 or vehicle (0.1% ethanol) for 30 min followed by stimulation with 50 nM I-BOP for 20 min. Cells were lysed and the lysates were subjected to SDS/PAGE and the phosphorylated signalling molecules were detected by Western blotting as described in A.
PKA, PKC and EGFR Are Involved in I-BOP-stimulated Phosphorylation and Activation of Several Signaling Molecules
To elucidate molecular mechanisms of I-BOP-induced phosphorylation of signal molecules, we detected their phosphorylation following TPα activation in the presence of kinase inhibitors. I-BOP-induced ERK phosphorylation was diminished almost to a basal level by EGFR kinase inhibitor AG1478 and Src kinase inhibitor PP2, but was only slightly decreased by PKA inhibitor H89, PI 3-kinase inhibitor wortmannin and PKC inhibitor GF109203X (Fig. 4C) indicating that activation of EGFR but not PKA, PI 3-kinase and PKC is necessary to the phosphorylation of ERK.
In contrast, I-BOP-induced phosphorylation of p65 of NF-κB was significantly attenuated by MEK inhibitor PD98059, PKA inhibitor H89 and PKC inhibitor GF109203X, partially decreased by EGFR kinase inhibitor AG1478 and Src kinase inhibitor PP2, but not by PI-3kinase inhibitor wortmannin (Fig. 4C). These results indicate that ERK, PKA and PKC are significantly involved in NF-κB phosphorylation, but EGFR and Src participate only modestly in NF-κB phosphorylation. Furthermore, I-BOP-induced CREB phosphorylation was also attenuated by PKA inhibitor H89 and by PKC inhibitor GF109203X suggesting that PKA and PKC are important for CREB phosphorylation.
Finally, I-BOP-stimulated phosphorylation of p38 MAPK was attenuated by EGFR kinase inhibitor AG1478 and Src kinase inhibitor PP2 indicating that I-BOP- induced the activation of p38 MAPK through EGFR pathway (Fig. 4C). However, I-BOP-induced phosphorylation of GSK-3α/β was inhibited by PKA, PKC, ERK and Src inhibitors suggesting inactivation of GSK-3α/β could be achieved by several kinases except PKB and EGFR (Fig. 4C).
COX-2 Promoter Luciferase Reporter Assay
In order to further investigate the transcriptional regulation of human COX-2 gene by I-BOP, the luciferase reporter bearing various lengths of 5′-flanking regions of COX-2 gene promoter were employed [26]. Because of low transfection efficiency of plasmids in A549 cells, COX-2 promoter assay was carried out in AD293-TPα cells which exhibited high transfection efficiency and were characterized previously ( AD293-TPα cells were transfected with various promoter constructs and the effect of I-BOP on the luciferase activity of these constructs was studied. The transcriptional activities of luciferase bearing vectors from pXC 918 (−918 bp) to pXC44 (−44 bp) were determined following stimulation with 50 nM I-BOP. As shown in Fig. 5A, the stimulation ratio ranging from 8 to 12.6 fold was obtained by comparing the luciferase activity in I-BOP-treated cells with that of control. The stimulatory response of I-BOP was significantly decreased in pXC44 (−44 bp) indicating that a promoter region ranging from −80 to −44 bp was important for the I-BOP-stimulated response of COX-2 expression. Sequence analysis showed that a CRE consensus site (−57 to −53 bp) was present within this promoter region [24]. Another two significant decreases in promoter activity were found in pXC186 and pXC422 constructs indicating that promoter regions ranging from −250 to −186 bp and −487 to −422 bp were also important for the I-BOP-stimulated response of COX-2 expression. Sequence analysis of these promoter regions reveals that a distal NF-κB (−446 to −437 bp) site and a proximal NF-κB (−223 to −214 bp) site exist in the two promoter regions respectively [24] indicating that NF-κB motifs also played an important role in the regulation of I-BOP-induced COX-2 expression.
Figure 5. Analysis of I-BOP-responsive elements in the 5′-flanking region of COX-2 promoter.



Luciferase vectors bearing various lengths of COX-2 promoter were constructed as described previously (27). Plasmid transient transfection was performed as described in the MATERIALS AND METHODS. A The potential responsive elements in the 5′-flanking regions are indicated. AD293-TPα cells transfected with 1.0 μg of the indicated reporter containing different regions of COX-2 promoter were treated with 50 nM I-BOP or vehicle for 24 h. Cell lysates were then prepared, and luciferase activity was assayed. Each group was performed in triplicates. The expression ratio (Fold/Basal) of I-BOP-treated cells to control was shown. Values were means ±S.D. of four independent experiments. B The mutated sequences of each responsive element are shown in the lower case. C and D The C/EBP, CRE or NF-κB motifs (both proximal (p) and distal (d)) are important for COX-2 gene activation. AD293-TPα cells transfected with 1.0 μg of the indicated reporter containing respective mutation of COX-2 promoter were treated with 50 nM I-BOP (black column) or vehicle (white column) for 24 h. Cell lysates were then prepared, and luciferase activity was assayed. Each group was performed in triplicates. In the left panel of each figure, three independent assays were performed, and the relative fold of I-BOP inducibility of wild and mutant promoters was compared. Statistical analysis was performed by Student’s t-test. * and ** are P values less than 0.05 and 0.01 respectively when compared with I-BOP stimulated level of wild promoters. E. Effects of various inhibitors on the promoter activity induced by I-BOP. A promoter (pXC918)-luciferase construct was transfected into AD293-TPα cells which were later treated with 50 nM I-BOP as indicated in A. The expression ratio (Fold/Basal) of I-BOP-treated cells to control in the absence and the presence of inhibitors was shown. Values were means ±S.D. of four independent experiments. * and ** are P values less than 0.05 and 0.01 respectively when compared with I-BOP stimulated level of COX-2 promotor-luciferase activity.
To further define the role of CCAAT/enhancer-binding protein (C/EBP) and CRE motifs in the basal and I-BOP-induced COX-2 gene regulation, the reporter expression controlled by the wild type or mutant −207/+49 regions (Fig. 5B & C) of human COX-2 gene promoter was assessed in transient transfection experiments. As shown in Fig. 5B & C, a mutation at the C/EBP motif caused 60~70% loss of basal promoter activity, but only a 20~30% decrease in relative fold of I-BOP-induced increase of the promoter activity by comparing a wild type and a mutant C/EBP. In contrast, a mutation at CRE motif lost 20~30% of basal promoter activity and 40~50% of relative fold of I-BOP-induced activity by comparing a wild type and a mutant CRE. The stimulatory effect of I-BOP was significantly attenuated when both C/EBP and CRE sites were mutated (−207/+49 mCE/C) indicating that both C/EBP and CRE motifs are necessary for COX-2 promoter activation. These results demonstrated that C/EBP is more important in regulating the basal COX-2 expression, while CRE motif is more important in I-BOP response, analogous to that of EGF-induced COX-2 promoter activity in A431 cells [26].
To further study the role of NF-κB motifs in the basal and I-BOP-induced COX-2 gene regulation, the reporter expression controlled by the wild type or mutant −531/+49 regions (Fig. 6D) of human COX-2 gene promoter was assessed in transient transfection experiments. The mutation of two basic pairs at distal NF-κB motif lost 40~50% of basal promoter activity and 20~30% of relative fold of I-BOP-induced activity by comparing a distal wild type and a mutant NF-κB. In contrast, the mutation of two basic pairs at the proximal NF-κB motif of COX-2 promoter resulted in a 30~40% loss of relative fold of I-BOP-induced promoter activity, but little effect on basal promoter activity by comparing a proximal wild type and a mutant NF-κB. The stimulatory effect of I-BOP was decreased by 40~50% when both NF-κB motif sites were mutated (−531/+49 mNF-κB (d+p)) indicating that both NF-κB motifs are necessary for COX-2 promoter activation. These results demonstrated that the distal NF-κB motif is more important in regulating the basal COX-2 expression, while the proximal NF-κB motif is more important in regulating the I-BOP-induced COX-2 expression. This is consistent with hypoxia-induced COX-2 expression in human vascular endothelial cell where the proximal NF-κB motif is also more important in regulating the hypoxia-induced COX-2 expression [28]. These results are in line with the findings on the expression of COX-2 protein and the phosphorylation of NF-κB in the presence of NF-κB inhibitors described above. Taken together, both NF-κB and CRE motifs play a critical role in the regulation of I-BOP-induced transcription of COX-2 gene in AD293-TPα cells.
Figure 6. Signaling pathways involved in TPα-mediated COX-2 Expression.

Finally, I-BOP-induced promoter activity was determined in the absence and presence of various inhibitors tested above to support the findings by Western blot and PG analysis. Fig. 5E shows that the results of relative promoter activity in the absence and presence of inhibitors are consistent with those of relative protein expression and PG levels.
Discussion
In this study, A549 cells stably transfected with TPα cDNA were used to investigate the induction of COX-2 expression and the associated mechanisms of TPα-mediated COX-2 expression and increased synthesis of PGE2 and TXB2. We found that TP agonist, I-BOP, elicited a dose and time-dependent increase in COX-2 expression and in PGE2 and TXB2 synthesis through TPα. Induction of COX-2 expression by a TP agonist would naturally lead to increased PGE2 and TXB2 synthesis although we are not sure if the coupling enzymes, PGE synthase and thromboxane synthase, are also induced. However, both microsomal PGE synthase and thromboxane synthase are known to over-express in tumors [29, 30], whereas the key PG catabolic enzyme, 15-hydroxyprostaglandin dehydrogenase, is demonstrated to under-express in tumors [31]. In fact, the expressions of COX-2 and 15-hydroxyprostaglandin dehydrogenase were shown to be regulated reciprocally [32]. The consequence is that amplified tumor tissue levels of PGE2 and TXB2 are expected. Previously, it was shown that TXB2 level was much higher in human lung tumor tissues than in non-tumor tissues [33]. Increase in tissue level of TXB2 is known to be the result of an enhancement of TXAS activity leading to increased synthesis of TXA2 which may induce the expression of COX-2. The expression of COX-2 and the elevation of its metabolite levels have been broadly studied in both experimental animals and human tumors [34, 35]. COX-2 up-regulation has been observed in lung cancer as well as in cancers of colon, stomach, liver, pancreas, breast, and skin and thought to play a critical role in cancer progression. Although COX-2 up-regulation can be achieved by cytokines, growth factors and tumor promoters, many of these agents may act indirectly by inducing synthesis of PGs [36]. Among the PGs that have been reported to induce the expression of COX-2 are COX-derived products, PGE2, PGF2α and TXA2, which appear to constitute a positive autoamplyfying pathway that may stimulate cell growth and proliferation by enhancing the production of PGs. Induction of COX-2 expression by PGE2 has been reported in several cell lines [20, 21, 37]. However, induction of COX-2 expression by a TXA2 mimetic has only been shown in human endothelial cells while studying platelet-vascular endothelial cell interactions [38]. Our report represents the first demonstration that a TP agonist may induce COX-2 expression in a cancer cell line.
To determine the signaling pathways that mediate the induction of COX-2 and the synthesis of PGE2 and TXB2 by I-BOP in A549-TPα cells, we used pharmacological inhibitors of various signaling pathways to determine if blockade of the activity of specific signaling molecules would block the induction of COX-2 and the synthesis of PGE2 and TXB2 by I-BOP. These inhibitors were used at concentrations proven to be optimal for each signaling molecule. We found that inhibitors of PKA, PKC, p38 MAPK, JAK, Src, EGFR and ERK except inhibitors of JNK and GSK-3 led to a decrease of I-BOP-induced expression of COX-2 and synthesis of PGE2 and TXB2 indicating that multiple signaling molecules and pathways were involved in TP-mediated COX-2 expression and PG synthesis. The role of each signaling molecule and the interplay of these molecules in each pathway for TP-mediated induction of COX-2 remain to be determined.
Signaling pathways that are critical for cell growth and proliferation are members of the mitogen-activated protein kinases (MAPKs). Three principal members are: ERK1/2, also known as p42/44 MAPK, p38 MAPK and JNK/SAPK [39]. ERK1/2 signaling pathway has been implicated as a key regulator of cell proliferation. ERK1/2 is activated through ras/raf/MEK cascade and is important in mediating signals induced by cytokines, growth factors and tumor promoters [40]. ERK1/2 can be also activated by transactivation of EGFR and subsequent mediation by ras/raf/MEK pathway [41]. Activated ERK1/2 is then translocated to the nucleus to activate key transcriptional factors involved in the expression of growth- and proliferation- related genes including COX-2 [42]. Protein kinases A, B and C are kinases that activate intermediary signaling molecules such as ras and raf. Similarly, p38 MAPK and JNK/SAPK can be activated through their specific signaling molecules and are then translocated to the nucleus to activate responsive transcriptional factors involved in cell growth and proliferation [43, 44]. Each of these three MAPKs has been shown to be involved in the expression of COX-2 [45]. Our studies also indicate that both ERK and p38 MAPK pathways are involved in TP-mediated COX-2 expression. These two pathways were previously found to be responsible for PGE2-mediated COX-2 expression in human non-pigmented ciliary epithelial cells [37]. The fact that inhibitors of PKA, PKC, Src and EGFR are able to block I-BOP-induced COX-2 expression is due to these signaling molecules being upstream of ERK1/2 activation. However, JNK pathway did not appear to be involved in TP-mediated COX-2 expression since JNK inhibitor did not exhibit any significant inhibitory effect although this pathway was found to be operative in porcine aortic smooth muscle cells [46]. The discrepancy may be due to the difference between two different cell types. Another independent pathway, JAK signaling which activates Stat3, has been shown to be involved in COX-2 expression [47]. Our studies also indicate that JAK signaling may participate in TP-mediated COX-2 expression.
ERK phosphorylation mediated by TP activation was extensively studied by several groups [8–10]. We have demonstrated that I-BOP induced rapid activation of ERK primarily through EGFR pathway since EGFR inhibitor blocked phosphorylation almost totally. TP-mediated transactivation of EGFR in eventual activation of ERK has also been reported in several cell lines [8–10]. In addition to EGFR, other signaling pathway such as activation of PKC was shown to involve in ERK activation in these cell lines although we did not see PKC being involved in a significant way in A549 cells. Krysan et al. [48] reported that PGE2 activates ERK signaling and cell proliferation in an EGFR-independent manner and that PKC but not PKA mediates ERK activation in a subset of lung cancer cell lines not including A549 cells. It appears that different receptors in different cell lines utilize separate signaling pathways in activating ERK. On the other hand, TP-mediated activation of NF-κB involves PKC since its inhibitor blocked phosphorylation of p65 of NF-κB. PKC may directly target the IκB leading to its degradation by the proteasome degradation pathway [49]. Our results indicate that TP-mediated activation of NF-κB involved PKC as well as PKA and ERK. Similar results were observed in NF-κB activation by hypoxia, IL-1β and lipoteichoic acid (LTA) in different cell lines [31, 50, 51].
In addition to the involvement of ERK, evidences were provided that p38 MAPK but not JNK/SAPK signaling mediated I-BOP-induced COX-2 expression and that the activation of p38MAPK involved Src and EGFR as assessed by inhibitor studies. Similarly, JAK/Stat3 pathway was also found to be involved in I-BOP induced COX-2 expression since the activation of Stat3 and the COX-2 expression were blocked by JAK inhibitor. Previous evidences showed that EGF activated Stat3 through Src, EGFR and JAK in A549 cells [52]. Therefore, I-BOP may activate Stat3 through Src, EGFR and JAK pathway since I-BOP may transactivate EGFR. Recently, we showed that I-BOP induced phosphorylation of GSK-3 and cyclin D1 expression in a PKA dependent manner in HEK293-TPα cells [11]. We also observed that I-BOP induced phosphorylation and inactivation of GSK-3 and I-BOP-induced COX-2 expression was slightly increased in the presence of GSK-3 inhibitor in A549-TPα cells as demonstrated in this study. It is known that GSK-3 phosphorylation and inactivation result in the accumulation and transport of β-catenin into the nucleus and the activation of β-catenin/Tcf/Lef transcription pathway which induces the expression of oncogenes such as cyclin D1, COX-2 and CD44 [53, 54]. We believe that GSK-3 phosphorylation is mediated by I-BOP induced activation of PKA and PKC and that β-catenin/Tcf/Lef pathway is a viable pathway for I-BOP-induced expression of COX-2. In summary, at least four signaling pathways, ERK, p38MAPK, JAK and GSK-3α/β-catenin/Tcf/Lef, are involved in I-BOP-induced COX-2 expression.
The activation of key molecules, ERK, p38 MAPK, CREB, NF-κB and GSK involved in COX-2 transcription was further examined by studying the phosphorylation of these molecules. I-BOP induced the phosphorylation of each of these five molecules within 5–10 min. EGFR inhibitor AG1478 and Src inhibitor PP2 significantly blocked I-BOP-induced phosphorylation of ERK and p38, but not that of CREB and NF-κB indicating that these two transcription factors are not the primary targets of ERK and p38. In contrast, PKA inhibitor H89 and PKC inhibitor GF 109203X significantly inhibited I-BOP-induced phosphorylation of CREB and NF-κB, but modestly block that of ERK and p38 indicating that the sites of action of PKA and PKC are CREB and NF-κB. ERK inhibitor PD98059 significantly inhibited the phosphorylation of ERK as expected and also of NF-κB indicating that ERK may participate in the activation of NF-κB. PI 3-kinase/PKB inhibitor wortmannin caused little decrease in the I-BOP-induced phosphorylation of these key signaling molecules and modest reduction of the induction of COX-2 as well as the synthesis of PGE2 and TXB2 indicating that PKB/Akt may not be involved significantly in the TP-mediated signaling pathway. In summary, both CREB and NF-κB appear to be critical transcriptional factors responsible for TP-mediated expression of COX-2. CREB activation depended primarily on PKA although PKC could be also involved, whereas NF-κB activation relied on PKA, PKC and ERK.
Both CREB and NF-κB have been shown to control the induced transcription of the COX-2 gene by IL-1β, TNF-α and others [36]. The COX-2 promoter region does have one CRE, one C/EBP, and two NF-κB motifs which are responsible for the induced transcription of the COX-2 gene [26]. We investigated the role of CREB, C/EBP and NF-κB in the regulation of the transcription of COX-2 gene by I-BOP using luciferase reporter assay with different promoter deletion regions and mutation sites of COX-2 promoter. Our results demonstrated that CRE, C/EBP and NF-κB motifs play an important role in the regulation of TPα-mediated COX-2 gene transcription. CREB and proximal NF-κB were required for regulating I-BOP-induced COX-2 transcription, whereas C/EBP and distal NF-κB were important for regulating basal COX-2 expression in A549-TPα cells. These results are similar to those found in EGF-induced COX-2 expression in A431 cells [23, 24]. Furthermore, the results obtained above using Western blot for COX-2 expression are in consistent with the data obtained by the luciferase reporter assay with the COX-2 promoter indicating that the above-mentioned signaling molecules and pathways play important roles in TP-mediated regulation of COX-2 expression.
In conclusion, our study demonstrated that at least four signaling pathways, ERK, p38MAPK, JAK and β-catenin/TCF/LEF, are involved in I-BOP-induced COX-2 expression. Transcription factors NF-κB, CREB, C/EBP and STAT3 are downstream signaling molecules that interact with the COX-2 promoter and play important roles in TPα-mediated expression of COX-2 and synthesis of PGE2 and TXB2. A summary of signaling pathways leading to TPα-mediated expression of COX-2 is shown in Figure 6.
Acknowledgments
This work was supported in part by grants from the NIH (HL-46296) and the Kentucky Lung Cancer Research Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Needleman P, Moncada S, Bunting S, Vane JR, Hamberg M, Samuelsson B. Identification of an enzyme in platelet microsomes which generates thromboxane A2 from prostaglandin endoperoxides. Nature. 1976;261(5561):558–560. doi: 10.1038/261558a0. [DOI] [PubMed] [Google Scholar]
- 2.Needleman P, Minkes M, Raz A. Thromboxanes: Selective biosynthesis and distinct biological activities. Science. 1976;193(4248):163–165. doi: 10.1126/science.945611. [DOI] [PubMed] [Google Scholar]
- 3.Hamberg M, Svensson J, Samuelsson B. Thromboxanes: A new group of biologically active compounds from prostaglandin endoperoxides. Proc Natl Acad Sci USA. 1975;72(8):2994–2998. doi: 10.1073/pnas.72.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirata M, Hayashi Y, Ushikubi F, Yokota Y, Kageyama R, Nakanishi S, Narumiya S. Cloning and expression of cDNA for a human thromboxane A2 receptor. Nature. 1991;49(6310):617–620. doi: 10.1038/349617a0. [DOI] [PubMed] [Google Scholar]
- 5.Raychowdhury MK, Yukawa M, Collins LJ, McGrail SH, Kent KC, Ware JA. Alternative splicing produces a divergent cytoplasmic tail in the human endothelial thromboxane A2 receptor. J Biol Chem. 1994;269(30):19256–19261. [PubMed] [Google Scholar]
- 6.Shenker A, Goldsmith P, Unson CG, Spiegel AM. The G-protein coupled to the thromboxane A2 receptor in human platelets is a member of the novel Gq family. J Biol Chem. 1991;266(14):9309–9313. [PubMed] [Google Scholar]
- 7.Hirata T, Ushibuki F, Kakizuka A, Okuma M, Nurumiya S. Two thromboxane A2 receptor isoforms in human platelets – Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest. 1996;97(4):949–956. doi: 10.1172/JCI118518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallet C, Blaie S, Levy-Toledano S, Habib A. Epidermal-growth-factor receptor and metalloproteinases mediate thromboxane A2-dependent extracellular-signal-regulated kinase activation. Biochem J. 2003;371(pt3):733–742. doi: 10.1042/BJ20021030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Y, Tang S, Zhou S, Ware JA. The thromboxane A2 receptor activates mitogen-activated protein kinase via protein kinase C-dependent Gi coupling and Src-dependent phosphorylation of the epidermal growth factor receptor. J Pharmacol Exp Ther. 2001;296(2):426–433. [PubMed] [Google Scholar]
- 10.Miggin SM, Kinsella BT. Regulation of extracellular signal-regulated kinase cascades by alpha- and beta-isoforms of the human thromboxane A2 receptor. Mol Pharmacol. 2002;61(4):817–831. doi: 10.1124/mol.61.4.817. [DOI] [PubMed] [Google Scholar]
- 11.Yan W, Tai HH. Glycogen synthase kinase-3 phosphorylation, T-cell factor signaling activation and cell morphology change following stimulation of thromboxane receptor α. J Pharmacol Exp Ther. 2006;371(1):267–274. doi: 10.1124/jpet.105.096826. [DOI] [PubMed] [Google Scholar]
- 12.Morinelli TA, Zhang LM, Newman WH, Meier KE. Thromboxane A2/prostaglandin H2-stimulated mitogenesis of coronary artery smooth muscle cells involves activation of mitogen-activated protein kinase and S6 kinase. J Biol Chem. 1994;269(8):5693–5698. [PubMed] [Google Scholar]
- 13.Lin X, Ramamurthy SK, Le Breton GC. Thromboxane A receptor-mediated cell proliferation, survival and gene expression in oligodendrocytes. J Neurochem. 2005;93(2):257–268. doi: 10.1111/j.1471-4159.2004.02969.x. [DOI] [PubMed] [Google Scholar]
- 14.Daniel TO, Liu H, Morrow JD, Crews BC, Marnett LJ. Thromboxane A2 is a mediator of cyclooxygenase-2 endothelial migration migration and angiogenesis. Cancer Res. 1999;59(18):4574–4577. [PubMed] [Google Scholar]
- 15.Nie D, Lamberti M, Zacharek A, Li L, Szekeres K, Tang K, Chen Y, Honn KV. Thromboxane A2 regulation of endothelial migration, angiogenesis and tumor metastasis. Biochem Biophys Res Commun. 2000;267(1):245–251. doi: 10.1006/bbrc.1999.1840. [DOI] [PubMed] [Google Scholar]
- 16.Giovannucci E, Rimm EB, Stampfer MJ, Colditz GA, Ascherio A, Willett WC. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann Int Med. 1994;121(4):241–246. doi: 10.7326/0003-4819-121-4-199408150-00001. [DOI] [PubMed] [Google Scholar]
- 17.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T, Su LK, Levin B. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyopsis. N Eng J Med. 2000;342(26):1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 18.Bakhle YS. COX-2 and cancer: a new approach to an old problem. Br J Pharmacol. 2001;134(6):1137–1150. doi: 10.1038/sj.bjp.0704365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276(21):18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 20.Hinz B, Brune K, Pahl A. Prostaglandin E2 upregulates cyclooxygenase-2 expression in lipoplysaccharide-stimulated RAW 264.7 macrophages. Biochem Biophys Res Commun. 2000;272(3):744–748. doi: 10.1006/bbrc.2000.2859. [DOI] [PubMed] [Google Scholar]
- 21.Minghett L, Polazz A, Nicolini C, Creminon C, Levi G. Upregulation of cyclooxygenase-2 expression in cultured microglia by prostaglandin E2, cyclic AMP and non-steroidal anti-inflammatory drugs. Eur J Neurosci. 1997;9(5):934–940. doi: 10.1111/j.1460-9568.1997.tb01444.x. [DOI] [PubMed] [Google Scholar]
- 22.Hida T, Yatabe Y, Achiwa H, et al. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, especially in adenocarcinomas. Cancer Res. 1998;58:3761–3764. [PubMed] [Google Scholar]
- 23.Wolff H, Saukkonen K, Anttila S, et al. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 24.Zhou H, Yan F, Tai HH. Phosphorylation and desensitization of the human thromboxane receptor-α by G-protein-coupled receptor kinases. J Pharmacol Exp Ther. 2001;298(3):1243–1251. [PubMed] [Google Scholar]
- 25.Wang JM, Ko CY, Chen LC, Wang WL, Chang WC. Functional role of NF-IL6β and its sumoylation and acetylation modifications in promoter activation of cyclooxygenase- 2 gene. Nucleic Acids Res. 2006;34(1):217–231. doi: 10.1093/nar/gkj422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LC, Chen BK, Chang JM, Chang WC. Essential role of c-Jun induction and coactivator p300 in epidermal growth factor-induced gene expression of cyclooxygenase-2 in human epidermoid carcinoma A431 cells. Biochim Biophys Acta. 2004;1683(1–3):38–48. doi: 10.1016/j.bbalip.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 27.Harding L, Wang Z, Tai HH. Stimulation of prostaglandin E2 synthesis by interleukin-1β is amplified by interferon but inhibited by interleukin-4 in human aminion-derived WISH cells. Biochim Biophys Acta. 1996;1310(1):48–52. doi: 10.1016/0167-4889(95)00144-1. [DOI] [PubMed] [Google Scholar]
- 28.Schmedtje JF, Jr, Ji YS, Liu WL, DuBois RN, Runge MS. Hypoxia induces cyclooxgenase-2 via the NF-κB p65 transcription factor in human vascular endothelial cells. J Biol Chem. 1997;272(1):601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 29.Moussa O, Yordy JS, Abol-Enein H, Sinha D, Bissada NK, Halushka PV, Ghoneim MA, Watson DK. Prognostic and functional significance of thromboxane synthase gene overexpression in invasive bladder cancer. Cancer Res. 2005;65(24):11581–11587. doi: 10.1158/0008-5472.CAN-05-1622. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is over-expressed in non-small cell lung cancer. Clin Cancer Res. 2001;7(9):2669–2674. [PubMed] [Google Scholar]
- 31.Ding Y, Tong M, Liu S, Moscow J, Tai HH. NAD+-linked 15-hydroxyprostaglandin dehydrogenase behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005;26(1):65–72. doi: 10.1093/carcin/bgh277. [DOI] [PubMed] [Google Scholar]
- 32.Tong M, Ding Y, Tai HH. Reciprocal regulation of cyclooxygenase-2 and 15-hydroxyprostaglandin dehydrogenase expression in A549 human lung adenocarcinoma cells. Carcinogenesis. 2006;27(11):2170–2179. doi: 10.1093/carcin/bgl053. [DOI] [PubMed] [Google Scholar]
- 33.Chen GG, Lee TW, Yip JH, Xu H, Lee IK, Mok TS, Warner TD, Yim AP. Increased thromboxane B2 levels are associated with lipid peroxidation and Bcl-2 expression in human lung carcinoma. Cancer Lett. 2006;234(2):193–198. doi: 10.1016/j.canlet.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 34.Kutchera W, Jones DA, Matsunami N, Groden J, McIntyre TM, Zimmerman GA, White RL, Prescott SM. Prostaglandin H synthase 2 is expressed abnormally in human colon cancer: evidence for a transcriptional effect. Proc Natl Acad Sci USA. 1996;93(10):4816–4820. doi: 10.1073/pnas.93.10.4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxgenase-2 in human lung carcinoma. Cancer Res. 1998;58(22):4997–5001. [PubMed] [Google Scholar]
- 36.Herschman HR. Prostaglandin synthase 2. Biochim Biophys Acta. 1996;1299(1):125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- 37.Rosch S, Ramer R, Brune K, Hinz B. Prostaglandin E2 induces cylcooxygenase-2 expression in human non-pigmented ciliary epithelial cells through activation of p38 and p42/44 mitogen-actiavted protein kinases. Biochem Biophys Res Commun. 2005;338(2):1171–1178. doi: 10.1016/j.bbrc.2005.10.051. [DOI] [PubMed] [Google Scholar]
- 38.Caughey GE, Cleland LG, Gambler JR, James MJ. Upregulation of endothelial cyclooxygenase-2 and prostanoid synthesis by platelets – role of thromboxane A2. J Biol Chem. 2001;276(41):37839–37845. doi: 10.1074/jbc.M010606200. [DOI] [PubMed] [Google Scholar]
- 39.Bokemeyer D, Sorokin A, Dunn MJ. Multiple intracellular MAP kinase signaling cascades. Kidney Int. 1996;49(5):1187–1198. doi: 10.1038/ki.1996.172. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101(8):2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 41.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signaling by G-protein-coupled receptors. Nature. 1996;379(6565):557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 42.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17(11 Reviews):1395–413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 43.Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J. The stress-activated protein kinase subfamily of c-jun kinase. Nature. 1994;369(6476):156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 44.Frasch SC, Nick JA, Fadok VA, Bratton DL, Worthen GS, Henson PM. p38 mitogen-activated protein kinase-dependent and –independent intracellular signal transduction pathways leading to apoptosis in human neutrophils. J Biol Chem. 1998;273(14):8389–8397. doi: 10.1074/jbc.273.14.8389. [DOI] [PubMed] [Google Scholar]
- 45.McGinty A, Foschi M, Chang YW, Han J, Dunn MJ, Sorokin A. Induction of prostaglandin endoperoxide synthase 2 by mitogen-activated protein kinase cascades. Biochem J. 2000;352 (pt 2):419–424. [PMC free article] [PubMed] [Google Scholar]
- 46.Miggin SM, Kinsella BT. Thromboxane A2 receptor mediated activation of the mitogen activated protein kinase cascades in human uterine smooth muscle cells. Biochim Biophys Acta. 2001;1539(1–2):147–162. doi: 10.1016/s0167-4889(01)00103-3. [DOI] [PubMed] [Google Scholar]
- 47.Koon HW, Zhao D, Zhan Y, Simeonidis S, Moyer MP, Pothoulakis C. Substance P stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J Immunol. 2006;176(8):5050–5059. doi: 10.4049/jimmunol.176.8.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krysan K, Reckamp KL, Dalwadi H, Sharma S, Rozengurt E, Dohadwala M, Dubinett SM. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65(14):6275–6281. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- 49.Chen J. Ubiquitin signaling in the NF-κB pathway. Nature Cell Biol. 2005;7(8):758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang B, Xu S, Hou X, Pimentel DR, Brecher P, Cohen RA. Temporal control of NF-κB activation by ERK differentially regulates interleukin-1β-induced gene expression. J Bio Chem. 2004;279(2):1323–1329. doi: 10.1074/jbc.M307521200. [DOI] [PubMed] [Google Scholar]
- 51.Chang YC, Li PC, Chen BC, Chang MS, Wang JL, Chiu WT, Lin CH. Lipoteichoic acid-induced nitric oxide synthase expression in RAW 264.7 macrophages is mediated by cyclooxygenase-2, prostaglandin E2, protein kinase A, p38 MAPK, and nuclear factor-κB pathways. Cellular signaling. 2006;18(8):1235–1243. doi: 10.1016/j.cellsig.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 52.Song L, Turkson J, Carras JG, Karras JG, Jove R, Haura EB. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene. 2002;22(27):4150–4165. doi: 10.1038/sj.onc.1206479. [DOI] [PubMed] [Google Scholar]
- 53.Wong NA, Pignatelli M. Beta-catenin--a linchpin in colorectal carcinogenesis. Am J Pathol. 2002;160(2):389–401. doi: 10.1016/s0002-9440(10)64856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thiel A, Heinonen M, Rintahaka J, Hallikainen T, Hemmes A, Dixon DA, Haglund C, Ristimaki A. Expression of cyclooxygenase-2 is regulated by glycogen synthase kinase-3β in gastric cancer cells. J Biol Chem. 2006;281(8):4564–4569. doi: 10.1074/jbc.M512722200. [DOI] [PubMed] [Google Scholar]