

Abstract
We determined the effects of 10 daily exposures of intermittent hypoxia (IH; 1 h day−1; oxyhaemoglobin saturation = 80%) on muscle sympathetic nerve activity (MSNA, peroneal nerve) and the hypoxic ventilatory response (HVR) before, during and after an acute 20 min isocapnic hypoxic exposure. We also assessed the potential parallel modulation of the ventilatory and sympathetic systems following IH. Healthy young men (n = 11; 25 ± 1 years) served as subjects and pre- and post-IH measures of MSNA were obtained on six subjects. The IH intervention caused HVR to significantly increase (pre-IH = 0.30 ± 0.03; post-IH = 0.61 ± 0.12 l min−1 %SaO2−1). During the 20 min hypoxic exposure sympathetic activity was significantly greater than baseline and remained above baseline after withdrawal of the hypoxic stimulus, even though oxyhaemoglobin saturation had normalized and ventilation and blood pressure had returned to baseline levels. When compared to the pre-IH trial, burst frequency increased (P < 0.01), total MSNA trended towards higher values (P = 0.06), and there was no effect on burst amplitude (P = 0.82) during the post-IH trial. Following IH the rise in MSNA burst frequency was strongly related to the change in HVR (r = 0.79, P < 0.05) suggesting that these sympathetic and ventilatory responses may have common central control.
Acute exposure to hypoxia stimulates oxygen sensitive chemoreceptors located peripherally in the carotid body and centrally in the brainstem. The physiological consequences of stimulation of these excitatory regions are many and include abrupt increases in ventilation and circulatory adjustments including increased sympathetic vasomotor outflow. When a chemoreceptor stimulus (i.e. increased CO2 and/or decreased O2) is used to drive ventilation in humans, sympathetic activity is increased in a dose–response fashion (Somers et al. 1989; Morgan et al. 1995; Xie et al. 2000). Acute exposure to 20 min of combined hypoxia and hypercapnia causes increases in ventilation that are paralleled by increases in direct recordings of multiunit muscle sympathetic nerve activity (MSNA) (Morgan et al. 1995). After cessation of hypoxia and hypercapnia, ventilation and arterial oxyhaemoglobin saturation (SaO2) return to baseline values whereas the increases in sympathetic activity persist. Persistent sympathoexcitation has been reported by others and hypoxia, rather than hypercapnia, is the primary stimulus (Xie et al. 2001; Cutler et al. 2004; Tamisier et al. 2004a). With exposure to long-term hypoxia there are two reasons to suspect that tonic sympathetic activity is elevated. First, 4 weeks of sustained hypobaric hypoxia (5260 m) resulted in a 3-fold increase in normoxic MSNA in healthy humans which persisted for days to weeks upon return to sea level (Hansen & Sander, 2003). Second, in a pathological model of intermittent hypoxia (IH), patients with obstructive sleep apnoea show higher daytime MSNA compared to matched controls (Narkiewicz et al. 1999). Interpretation and comparison of these two models is difficult because the physiological changes in response to hypoxia will be dependent on the duration, severity and pattern of the exposure as well as the presence or absence of CO2. Nevertheless, it appears that repeated or sustained exposure to hypoxia results in sympathetic neural overactivity.
Most studies report human multiunit MSNA recordings as burst frequency (i.e. bursts min−1), which is generally believed to reflect the occurrence of sympathetic firings from the neurons in contact with or close proximity to the microelectrode. Some investigators report sympathetic activity as total MSNA or total activity which incorporates values of amplitude or area of the integrated bursts. Relatively few human studies have reported burst amplitude values, which are thought to be representative of the number or type of neuronal firings. Values of MSNA burst frequency and amplitude may provide unique information about the central regulation of sympathetic outflow in response to hypoxia. Indeed, there is some evidence from animal studies to suggest that hypoxia may affect each component of the sympathetic nerve activity signal independently (Malpas & Ninomiya, 1992a,b; Malpas et al. 1996). The manner by which these components are controlled is relevant to the understanding of the central generation of sympathetic ‘tone’ and cardiovascular regulation
The neurocirculatory effects of IH have not been studied extensively in humans and, to our knowledge, no study has examined the potential concurrent modulations within the ventilatory and sympathetic responses to hypoxia after long-term exposure to isocapnic intermittent hypoxia. With exposure to hypoxia, both the ventilatory and sympathetic drive are dependent on the carotid bodies and there are overlapping neural networks in the central nervous system; thus there is reason to consider the possibility that these two systems may undergo simultaneous augmentation. As such, the purposes of the present study were twofold. First, we tested the effects of 10 daily exposures to hypoxia on MSNA before, during and after an acute 20 min isocapnic hypoxic exposure. Second, we assessed the potential parallel modulation of the ventilatory and sympathetic systems post-IH. We hypothesized that our IH intervention would significantly increase sympathetic activity during the acute exposure and the persistent MSNA observed during normoxic recovery would be significantly elevated. We further hypothesized that the change in the MSNA response to hypoxia would be linked to the change in the ventilatory sensitivity to hypoxia.
Methods
Subjects
All experimental procedures and protocols were approved by the Clinical Screening Committee for Research of the University of British Columbia and conformed to the Declaration of Helsinki. Eleven men provided informed written consent prior to beginning the study. Subjects were young (25 ± 1 years), of normal weight (85 ± 4 kg) and height (180 ± 1 cm) and had a healthy range for body mass index (25 ± 9 kg m−2). All subjects were free from cardiovascular, pulmonary, and neurological disease. Subjects were excluded if they had a history of smoking, had ascended to altitude (> 3000 m) within 3 months prior to testing, were taking regular medication, or participated in competitive swimming, breath-hold diving, or any other form of apnoeic training. For screening purposes, all subjects performed pulmonary function testing using a calibrated spirometer (Spirolab II, Medical International Research, Rome, Italy). All subjects had a normal forced vital capacity (5.8 ± 0.2 l) and forced expired volume in 1 s (4.8 ± 0.1 l). Abstinence from caffeine, alcohol and exercise were required for 24 h prior to all testing procedures.
Experimental protocol
The experimental protocol is shown in Fig. 1. All subjects reported to the lab for a familiarization trial (Day 1) where they became acquainted with masked breathing and HVR procedures. Subjects underwent two experimental days (Days 2 and 14) where continuous ventilatory, cardiovascular and MSNA data were obtained. The experimental days consisted of a four-phased protocol: (i) 10 min of normoxic eupnoea that served as the baseline period; (ii) the HVR procedure; (iii) 20 min of isocapnic hypoxia exposure; (iv) 20 min of normoxic breathing, which was considered the recovery period. During the 20 min hypoxic exposure, 100% N2 was added to the inspiratory circuit as needed to maintain SaO2 at 80%. Subjects were exposed to IH on Days 3–13 where isocapnic hypoxia was administered for 1 h such that SaO2 was maintained at 80%.
Figure 1. Experimental protocol.

Subjects completed each of the 14 days consecutively. Familiarization (Day 1) consisted of pulmonary function testing, anthropometric measurements, and a practice HVR. On the pre-and post-intermittent hypoxia (IH) trials (Days 2 and 14) subjects were instrumented for collection of muscle sympathetic nerve activity, and cardiovascular and ventilatory parameters. During the hypoxic intervention (Days 3–13) subjects breathed a hypoxic inspirate such that oxyhaemoglobin saturation was maintained at 80% and end-tidal carbon dioxide was kept at baseline levels.
General procedures
Subjects were studied in the semi-recumbent position. Inspiratory flow was determined using a calibrated pneumotachograph (Hans-Rudolph 3813, Kansas City, MO, USA) connected to the inspired side of a face mask. From the inspiratory flow signal, tidal volume (VT) and breathing frequency (fb) values were determined and minute ventilation 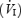 was calculated as the product of fb and VT. Calibrated gas analysers were used to monitor end-tidal oxygen (PET,O2) and carbon dioxide (PET,CO2) (S-3A/I and CD-3A, Applied Electrochemistry, Pittsburg, PA, USA). Heart rate was determined from a standard bipolar limb lead electrocardiogram (ADInstruments, ML110, 5303, Colorado Springs, CO, USA). Arterial oxyhaemoglobin saturation was measured at the finger using a pulse oximeter (Ohmeda 3740, Louisville, CO, USA). Beat-by-beat blood pressure was obtained using noninvasive photoplethysmography at the finger and calibrated according to manufacturer specifications (Finometer, Finapres Medical System, Arnhem, the Netherlands). Automated cuff measurements of blood pressure (BPM-100, VSM Medtech Ltd, Vancouver, Canada) were taken from the arm every 2 min and used to normalize the beat-by-beat values. Mean arterial blood pressure (MAP) was calculated from systolic blood pressure (SBP) and diastolic blood pressure (DBP) values. All data was acquired using an analog-to-digital converter (Powerlab/16SP ML 795, ADInstruments) and sampled at 1 kHz. Data were stored on a computer for offline analysis (Chart v5.02, ADInstruments).
was calculated as the product of fb and VT. Calibrated gas analysers were used to monitor end-tidal oxygen (PET,O2) and carbon dioxide (PET,CO2) (S-3A/I and CD-3A, Applied Electrochemistry, Pittsburg, PA, USA). Heart rate was determined from a standard bipolar limb lead electrocardiogram (ADInstruments, ML110, 5303, Colorado Springs, CO, USA). Arterial oxyhaemoglobin saturation was measured at the finger using a pulse oximeter (Ohmeda 3740, Louisville, CO, USA). Beat-by-beat blood pressure was obtained using noninvasive photoplethysmography at the finger and calibrated according to manufacturer specifications (Finometer, Finapres Medical System, Arnhem, the Netherlands). Automated cuff measurements of blood pressure (BPM-100, VSM Medtech Ltd, Vancouver, Canada) were taken from the arm every 2 min and used to normalize the beat-by-beat values. Mean arterial blood pressure (MAP) was calculated from systolic blood pressure (SBP) and diastolic blood pressure (DBP) values. All data was acquired using an analog-to-digital converter (Powerlab/16SP ML 795, ADInstruments) and sampled at 1 kHz. Data were stored on a computer for offline analysis (Chart v5.02, ADInstruments).
Hypoxic ventilatory response
We assessed the hypoxic ventilatory response (HVR) using previously described methods (Guenette et al. 2004; Koehle et al. 2005; Foster et al. 2006). Subjects breathed room air from a mixing chamber and 100% N2 was added to the inspiratory circuit to evoke a gradual drop in SaO2 to 75% over an approximate 5 min period. Isocapnia was maintained during the test by the manual addition of CO2 to the inspiratory circuit. The FI,O2 was determined by analysing gas sampled from the proximal side of the inspiratory valve. Minute ventilation was plotted as a function of SaO2 and the slope of the linear regression was taken to represent the HVR. The relationship between 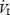 and SaO2 was considered acceptable when linearity was demonstrated.
and SaO2 was considered acceptable when linearity was demonstrated.
Sympathetic nerve activity
Direct intraneural recordings of multiunit postganglionic MSNA were acquired from the peroneal nerve using microneurography (Vallbo et al. 1979). Briefly, the technique involved inserting a recording needle into the common peroneal nerve to record electrical nerve activity. After location by surface and subcutaneous stimulation, a tungsten microelectrode was inserted for recording purposes (tip diameter 10 μm, 35 mm long, Fredrick Haer, Bowdoinham, ME, USA). Nerve signals were rectified, amplified (total gain 50 000; custom-built microneurography preamplifier and amplifier, Yale University, New Haven, CT, USA), band pass filtered (300–5000 Hz), and integrated (time constant 100 ms; Integrator model B937C, Bioengineering, University of Iowa, Iowa City, IA, USA). Signals were monitored via audio representation of burst activity (Audio Monitor, model AM8, Grass Product Group, Warwick, RI, USA) and visual inspection of the mean voltage neurogram. The accuracy of the recording site was confirmed (pre and post) using the following criteria: (i) demonstration of pulse synchronous bursts occurring 1.2–1.4 s after a QRS complex; (ii) activation with breath hold or Valsalva manoeuvre; (iii) no activation upon pinch, stroking of the skin, or startle stimuli; and (iv) a signal to noise ratio of > 3: 1. Offline, the integrated neurogram was time shifted to align each burst with its corresponding R-wave and high pass filtered to set the noise threshold to zero. For the purposes of quantification, MSNA was expressed as burst frequency (bursts min−1) and total activity (product of burst frequency and peak amplitude; arbitrary units).
Data and statistical analyses
Ventilatory, cardiovascular and MSNA values were averaged over 5 min sections during baseline, hypoxic exposure and recovery. Values for MSNA are reported as percentage change from baseline. Repeated measures analysis of variance procedures (Statistica v.6.1, Statsoft Inc., Tulsa, OK, USA) were performed to determine: (i) the difference between periods of baseline, the hypoxic exposure and recovery; and (ii) the differences between pre-and post-10 days of hypoxic exposure. When significant F ratios were detected, Tukey's post hoc analysis was applied to determine where the differences resided. Linear correlation analysis was used to examine the relationship between the change in HVR and the change in maximum burst frequency obtained in the last 5 min of the hypoxic exposure for both pre- and post-IH trials. Student's paired t test was preformed to analyse the difference between pre- and post-IH values of HVR. The level of significance was set at P < 0.05 for all statistical comparisons. Data are expressed as means ± s.e.m.
Results
Effects of IH on MSNA
Pre- and post-IH measures of MSNA were successfully acquired from six subjects. Figure 2 is a representative trace of MSNA and the corresponding variables from one subject (post-IH trial). Although ventilation, MAP and SaO2 all returned to baseline values during normoxic recovery, there was a long-lasting elevation in MSNA. This was a consistent observation in all subjects for all trials. Figure 3 displays the percentage increases from baseline for each 5 min period for MSNA during the pre- and post-IH trials. For each trial, burst frequency and amplitude increased significantly within the first 5 min of the hypoxic exposure and remained elevated throughout the hypoxic exposure and during the entire period of normoxic recovery (P < 0.001). The increase in burst frequency was higher post-IH compared to pre-IH (P < 0.001) whereas no significant difference between pre- and post-IH was observed for burst amplitude (P > 0.05). Total MSNA increased significantly during the pre- and post-IH hypoxic exposures (P < 0.001). There was a trend for total MSNA to be higher during the post-IH trial (P = 0.06).
Figure 2. Effects of isocapnic hypoxia on muscle sympathetic nerve activity.

Representative trace for one subject during the post-experimental trial. Traces represent 30 s of data obtained from baseline, hypoxic exposure, and normoxic recovery. Although respiratory (tidal volume, VT), cardiovascular (mean arterial pressure, MAP), and oxyhaemoglobin saturation (SaO2) values all returned to baseline values during recovery, muscle sympathetic nerve activity remained elevated. The persistent sympathoexcitation was observed in all subjects for all trials. Note that the partial pressure of carbon dioxide (PCO2) was maintained at baseline levels throughout experimentation.
Figure 3. The effect of intermittent hypoxia (IH) on the percent change in muscle sympathetic nervous activity (MSNA) (n = 6).

Values represent 5 min segments for pre-IH (open bars) and post-IH (filled bars). A, burst frequency increased significantly over time and remained elevated after the hypoxic exposure, and was significantly higher post-IH. B, amplitude increased significantly over time and remained elevated after the 20 min hypoxic exposure; however, no significant difference between pre- and post-IH was observed. C, total MSNA increased significantly over time and remained elevated after the hypoxic exposure. There was a trend for total MSNA to be higher post-IH (P = 0.06). *Significantly different from baseline (P < 0.05). †Significantly different from pre-IH (P < 0.05).
Effects of IH on cardiovascular and ventilatory measurements
Cardiovascular and ventilatory measurements were acquired from all subjects (n = 11) and are presented in Table 1. Compared to baseline, the 20 min hypoxic exposure resulted in significant increases in HR, SBP and MAP (P < 0.001) and there was no change in DBP (P > 0.05). These variables quickly returned to baseline values upon termination of the hypoxic exposure. There was no effect of IH on cardiovascular variables (P > 0.05). For both pre- and post-IH, significant increases in 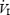 and VT (P < 0.001) were observed during the first 5 min of the hypoxic exposure and returned to baseline values during recovery. During the hypoxic exposure, there was no difference between pre- and post-IH values for
and VT (P < 0.001) were observed during the first 5 min of the hypoxic exposure and returned to baseline values during recovery. During the hypoxic exposure, there was no difference between pre- and post-IH values for 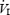 or VT (P > 0.05). During the pre-IH trial, fb increased from baseline within the first 5 min of the hypoxic exposure (P < 0.001) and remained elevated for 15 min into recovery (P < 0.05), eventually returning to baseline values during the last 5 min. Post-IH, increases in fb were observed (P < 0.01) in the same time course as seen in the pre-IH trial but returned to baseline values 15 min into the hypoxic exposure.
or VT (P > 0.05). During the pre-IH trial, fb increased from baseline within the first 5 min of the hypoxic exposure (P < 0.001) and remained elevated for 15 min into recovery (P < 0.05), eventually returning to baseline values during the last 5 min. Post-IH, increases in fb were observed (P < 0.01) in the same time course as seen in the pre-IH trial but returned to baseline values 15 min into the hypoxic exposure.
Table 1.
Cardiovascular and ventilatory measures pre and post exposure to intermittent hypoxia (IH)
| Hypoxia | Recovery | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline eupnoea | 5 min | 10 min | 15 min | 20 min | 25 min | 30 min | 35 min | 40 min | |
| Pre-IH | |||||||||
| SBP (mmHg) | 111 ± 3 | 118 ± 3* | 117 ± 3* | 120 ± 3* | 120 ± 3* | 112 ± 3 | 114 ± 3 | 114 ± 3 | 113 ± 3 |
| DBP (mmHg) | 67 ± 3 | 67 ± 3 | 66 ± 4 | 66 ± 2 | 65 ± 3 | 69 ± 2 | 67 ± 3 | 69 ± 4 | 67 ± 3 |
| MAP (mmHg) | 89 ± 2 | 98 ± 4* | 97 ± 4* | 101 ± 4* | 99 ± 4* | 90 ± 3 | 91 ± 2 | 91 ± 2 | 91 ± 2 |
| HR (beats min−1) | 57 ± 3 | 68 ± 4* | 68 ± 4* | 67 ± 4* | 67 ± 4* | 56 ± 3 | 57 ± 3 | 57 ± 3 | 60 ± 3 |
| 7.3 ± 1.0 | 10.9 ± 1.4* | 10.6 ± 1.5* | 10.3 ± 1.4* | 9.8 ± 1.4* | 7.4 ± 1.1 | 7.4 ± 1.1 | 7.5 ± 1.2 | 7.5 ± 1.2 | |
| fb (breaths min−1) | 12.6 ± 1.2 | 14.6 ± 1.1* | 14.9 ± 1.1* | 14.7 ± 0.9* | 15.2 ± 1.0* | 14.6 ± 1.1* | 14.8 ± 1.18 | 14.4 ± 1.1* | 13.5 ± 1.2 |
| VT (l) | 0.81 ± 0.12 | 1.10 ± 0.08* | 0.99 ± 0.11* | 0.92 ± 0.90* | 0.92 ± 0.13* | 0.79 ± 0.12* | 0.75 ± 0.13* | 0.74 ± 0.09 | 0.80 ± 0.15 |
| PET,CO2 (mmHg) | 41.4 ± 0.8 | 42.0 ± 1.1 | 41.2 ± 1.2 | 41.1 ± 1.4 | 41.3 ± 1.2 | 41.5 ± 0.8 | 41.2 ± 1.0 | 41.3 ± 1.1 | 41.6 ± 1.3 |
| SaO2 (%) | 97.3 ± 0.3 | 80.9 ± 1.0* | 80.8 ± 1.0* | 80.5 ± 0.8* | 80.4 ± 1.0* | 94.4 ± 0.4 | 97.4 ± 0.2 | 97.4 ± 0.2 | 97.4 ± 0.2 |
| Post-IH | |||||||||
| SBP (mmHg) | 113 ± 4 | 119 ± 4* | 121 ± 4* | 117 ± 3* | 120 ± 4* | 116 ± 3 | 114 ± 4 | 115 ± 3 | 116 ± 3 |
| DBP (mmHg) | 71 ± 2 | 71 ± 3 | 70 ± 3 | 70 ± 3 | 69 ± 2 | 68 ± 3 | 69 ± 2 | 69 ± 2 | 70 ± 3 |
| MAP (mmHg) | 90 ± 3 | 96 ± 3* | 98 ± 3* | 94 ± 2* | 97 ± 4* | 94 ± 3* | 92 ± 3 | 92 ± 3 | 92 ± 2 |
| HR (beats min−1) | 59 ± 4 | 69 ± 5* | 67 ± 4* | 68 ± 4* | 67 ± 4* | 57 ± 3 | 58 ± 4 | 58 ± 3 | 58 ± 3 |
| 6.4 ± 1.2 | 9.8 ± 1.7* | 9.7 ± 1.8* | 9.3 ± 1.7* | 8.9 ± 1.6* | 6.3 ± 1.1 | 6.4 ± 1.2 | 6.5 ± 1.2 | 6.6 ± 1.2 | |
| fb (breaths min−1) | 12.9 ± 1.0 | 14.2 ± 1.1* | 14.0 ± 1.2* | 13.8 ± 1.1 | 13.2 ± 1.1 | 12.7 ± 1.0 | 12.7 ± 1.1 | 12.6 ± 0.9 | 12.2 ± 0.9 |
| VT (l) | 0.75 ± 0.07 | 1.00 ± 0.06* | 1.00 ± 0.07* | 1.00 ± 0.07* | 0.96 ± 0.07* | 0.71 ± 0.04 | 0.72 ± 0.03 | 0.75 ± 0.03 | 0.75 ± 0.05 |
| PET,CO2 (mmHg) | 41.2 ± 0.9 | 41.7 ± 1.2 | 41.6 ± 1.4 | 41.2 ± 1.5 | 41.3 ± 1.7 | 41.7 ± 1.2 | 40.9 ± 1.2 | 41.0 ± 1.3 | 41.4 ± 1.2 |
| SaO2 (%) | 97.4 ± 0.1 | 79.7 ± 0.5 | 80.5 ± 0.5 | 80.2 ± 0.7 | 79.9 ± 0.3 | 94.6 ± 0.3 | 97.6 ± 0.4 | 97.7 ± 0.3 | 97.7 ± 0.1 |
SBP = systolic blood pressure; DBP = diastolic blood pressure; MAP = mean arterial pressure; HR = heart rate;  ; fb = breathing frequency; VT = tidal volume; PET,CO2 = end-tidal partial pressure of carbon dioxide; SaO2 = oxyhaemoglobin saturation.
; fb = breathing frequency; VT = tidal volume; PET,CO2 = end-tidal partial pressure of carbon dioxide; SaO2 = oxyhaemoglobin saturation.
Significantly different from baseline (P < 0.05).
Association of HVR with MSNA
Exposure to IH caused mean values for HVR to increase from 0.30 ± 0.07 to 0.61 ± 0.29 l min−1 %S−1aO2 (P < 0.01). All subjects showed an increase in HVR. Figure 4A displays individual and mean HVR data for all subjects (n = 11) and Fig. 4B those for subjects who completed MSNA trials (n = 6). Figure 5 shows the relationship between the change in HVR and MSNA burst frequency. The burst frequency value was taken from the last 5 min of the hypoxic exposure from each trial (pre- and post-IH) and is expressed as the percentage change from baseline. There was a significant positive correlation between post-IH values for MSNA and the HVR (r = 0.79; P < 0.05).
Figure 4. The effect of IH on the hypoxic ventilatory response (HVR).

Data are given as individual (○) and the group mean (•) values. Top, HVR data for all subjects (n = 11). Bottom, HVR data for subjects from whom MSNA data was collected (n = 6). *Significantly different from pre-IH.
Figure 5. Relationship between the change (post-IH – pre-IH) in hypoxic ventilatory response (ΔHVR) and muscle sympathetic nerve activity (Δ burst frequency).

Burst frequency values were obtained from the final 5 min of the hypoxic exposures.
Discussion
The present study provides a novel human model of long-term sympathetic overactivity in response to intermittent hypoxia. It also provides an opportunity to study the mechanisms involved in long-term neural control of the circulation. The major findings are twofold. First, 10 days of intermittent hypoxia resulted in significant increases in sympathetic vasomotor outflow to skeletal muscle during isocapnic hypoxia that was mediated by increases in burst frequency rather than burst amplitude. We interpret this observation to indicate that both components of MSNA are regulated independently in response to intermittent hypoxia. Second, following intermittent hypoxia the rise in MSNA burst frequency was strongly related to the change in hypoxic ventilatory response. This unique observation can be explained by two potential points of modulation within the central nervous system: (i) concurrent changes of separate regions controlling the sympathetic and ventilatory systems, or (ii) an alteration of one region that has common control over both the sympathetic and ventilatory systems.
Effect of IH on MSNA
Pre-IH, 20 min of isocapnic hypoxia significantly increased burst frequency, amplitude and total MSNA which was observable within the first 5 min and remained elevated for the entirety of the exposure. Post-IH, MSNA responses to the 20 min of isocapnic hypoxia followed an identical pattern over time, but significant increases in burst frequency (P < 0.01), a trend towards higher total MSNA (P = 0.06), and no effect on burst amplitude (P = 0.82) were demonstrated (Fig. 3). Hypoxia mediated sympathoexcitation is commonly expressed as burst frequency and our results are in good agreement with the findings of others (Morgan et al. 1995; Xie et al. 2001). Mechanisms for hypoxia-mediated sympathetic overactivity are complex and not fully understood. Patients with obstructive sleep apnoea show higher MSNA burst frequency values compared to matched controls during daytime periods of normoxia (Narkiewicz et al. 1998). After 4 weeks of sustained high altitude, sea level burst frequency remains increased by 300% of baseline in healthy subjects (Hansen & Sander, 2003). Collectively, it appears that sympathetic overactivation occurs with long-term exposure to hypoxia and our data support these observations. We did not demonstrate significant changes in MSNA during baseline normoxic conditions; however, our intervention did show an increase during acute hypoxia and the ensuing normoxic recovery period. The most obvious explanation for the discrepancy between our study and the above-mentioned investigations is the hypoxic protocols employed have used different total exposure time and severity of hypoxia. The larger increases in MSNA burst frequency in obstructive sleep apnoea or at high altitude could be attributable to the additional sympathoexcitatory effects related to coexisting complications associated with pathology, changes in the partial pressure of arterial CO2, hypobaria and anxiety associated with travel to altitude.
There are relatively few studies that have examined sympathoexcitatory stimulation on burst amplitude values obtained from multiunit recordings in humans. Controversy exists with regards to what burst amplitude represents physiologically. It has been suggested that changes in burst amplitude reflect either an alteration in the total number of neurons recruited or an alteration in the population of neurons recruited (Ninomiya et al. 1993; Malpas, 1998). In addition, there have been conflicting reports of alterations to burst amplitude in the literature. Xie et al. (2001) cited no change in burst amplitude after 20 min of isocapnic hypoxia (mean SaO2 = 80 %) whereas others have reported a 210% increase in total amplitude after 25–30 min of hypoxia (mean SaO2 = 75 %) (Leuenberger et al. 1991). In our study we found that burst amplitude was increased acutely with 20 min of hypoxia but unlike burst frequency, burst amplitude was not additionally augmented with IH. Our data suggest that sympathetic outflow is enhanced by an increased neuronal firing rate with no effect on the type or number of neurons recruited. To date, no other study has examined the long-term effects of hypoxia on burst amplitude in humans but results obtained from experimental animals provide some insight into the selective responsiveness of each component of the MSNA signal in the current study. In anaesthetized cats hypoxia increased burst amplitude, but not burst frequency of renal sympathetic nerve activity (Malpas & Ninomiya, 1992a). This agrees with the present study and suggests that burst amplitude and frequency are independently activated in response to afferent stimulation.
Mechanisms controlling the sympathoexcitation
In humans exposed to acute hypoxia, sympathoexcitation is initiated at the carotid bodies (Ryan et al. 1995) and the peripheral chemoreflex is likely to be the primary pathway activated. In rats, direct activation of central chemoreceptors can independently generate increases in sympathetic outflow (Guyenet, 2000); however, this is only achieved with severe levels of hypoxia (FI,O2 = 0.10) (Sun & Reis, 1994). In our study, to maintain SaO2 at 80% the FI,O2 ranged between 0.12 and 0.15. Consequently, it is unlikely that direct central chemoreception made a major contribution to the observed sympathoexcitation in our study. We are confident that the observed sympathoexcitation in our study is the result of hypoxia alone as baseline values for cardiorespiratory variables were identical between experimental days, and PET,CO2 was kept constant throughout all trials (see Table 1).
The mechanisms underlying the sustained elevation in MSNA after acute hypoxia are unknown. We considered the possibility that sympathetic outflow remained elevated because of persistent chemical stimuli. It is possible that that there were persistent changes in the intracellular milieu of the carotid body and/or central chemoreceptors. However, PET,O2, SaO2 and ventilation all quickly returned to baseline values upon cessation of breathing the hypoxic inspirate, which argues that there was normalization at the receptor sites.
Increases in ventilation and sympathoexcitation are both mediated by input from carotid body afferents. Therefore it is not surprising that they respond in parallel with exposure to hypoxia, but it is curious that the return to normoxia does not yield a similar pattern in ventilation and MSNA; three possible mechanisms exist. First, neurotransmitter release and/or neural synapses in areas of the central nervous system that control sympathetic outflow may take a considerable length of time to normalize (Neubauer & Sunderram, 2004). Second, there may be retention of endogenous peripheral vasodilatory substances maintaining baroreceptor engagement and consequently sympathetic outflow (Tamisier et al. 2004a,b). Production of endogenous vasodilatatory substances, such as prostaglandins, adenosine and nitric oxide (Marshall, 2000) have been reported during acute hypoxia and have been implicated in initiating a cascade of events leading to a reduction in blood pressure, unloading the baroreflex and resulting in enhanced sympathetic vasomotor outflow in an attempt to restore blood pressure (Halliwill & Minson, 2002). Indeed, several studies have examined chemoreceptor and baroreceptor reflexes under acute hypoxic conditions but the results of these investigations have been contradictory with some showing reduced baroreflex sensitivity (Bhattacharva et al. 1973; Halliwill & Minson, 2002; Halliwill et al. 2003) and others reporting no change in sensitivity (Cooper et al. 2005; Spicuzza et al. 2005). It has also been reported that rather than a change in baroreflex sensitivity there is a resetting of baroreflex control of both heart rate and sympathetic activity to higher levels (Halliwill & Minson, 2002; Halliwill et al. 2003). Germane to the present study is the observation that upon return to sea level following prolonged high altitude exposure (4 weeks; 5250 m) persistent sympathoexcitation is not related to baroreflex activity (Hansen & Sander, 2003). The relative contribution of baroreflex activity was not measured in our study and we cannot completely rule out its potential influence, but based on the above we would predict that it would likely play a minor role. Third, it may simply be that the gain in the chemoreflex control of sympathetic activity is greater than for ventilatory activity in humans (Morgan et al. 1995).
Effects of IH on ventilation
Increase in HVR following IH is a well documented phenomenon (Garcia et al. 2000; Foster et al. 2005; Katayama et al. 2005). Three hours of daily hypoxia (FI,O2 = 12 %) has been shown to cause HVR to increase 38–44% (Katayama et al. 2005). In the present study, HVR increased by 49%. We interpret this to mean that our IH protocol elicited a significant alteration in the hypoxic ventilatory control system. The increases in HVR can be attributed to enhanced carotid sinus nerve afferent activity (Nielsen et al. 1988) and may be the result of changes in cellular mechanisms in the carotid body, such as oxygen free radicals, haeme proteins and ion channels (Prabhakar & Overholt, 2000). It should be noted that carotid body sensitivity in rats can be affected differently depending on the hypoxic protocol used and is dependent on duration, pattern and severity (Peng & Prabhakar, 2004). Using a human model, we previously found no difference in HVR after short duration IH (12% O2 separated by 5 min of normoxia for 1 h) compared to long duration IH (30 min of 12% O2) (Foster et al. 2005). The disparity between these two studies may be due to differences between species and the total time exposed to hypoxia. Within the central nervous system, dopamine, an inhibitory transmitter, has been shown to respond to carotid sinus afferents and with chronic hypoxia, time-dependent reductions in the dopamine-2 receptor are observed in ventilatory control centres in the brain which could lead to increases in excitatory activity in these regions (Dwinell et al. 2000). It has also been proposed that central nervous system plasticity following IH is related to brain-derived neurotrophic factor synthesis and serotonin-dependent mechanisms (Mitchell et al. 2001). However, these putative possibilities have not been established in humans and a greater extrapolation to our findings is beyond the scope of our data.
Effects of IH on concurrent sympathetic and ventilatory activity
After the IH intervention we showed a significant relationship between the maximum change in burst frequency and HVR (Fig. 5). Coupled alterations in the ventilatory and sympathetic system have been previously shown after long-term exposure to hypoxia where changes in noradrenaline and tidal volume are correlated (Asano et al. 1997). Furthermore, in patients with sleep apnoea, hypoxia causes a greater increase in ventilation and MSNA (after the sympathetic-inhibitory influence of breathing was eliminated) compared to controls (Narkiewicz et al. 1999). Plasticity, or a persistent change in the neural control system, is a fundamental property of neural systems. In our study we have shown that the respiratory system demonstrates plasticity as does the sysmpathetic nervous system in response to IH. The plasticity can be attributed to enhanced central neural integration of chemoafferent inputs and/or carotid body plasticity. We favour the concept that enhanced integration occurred centrally where two specific explanations can account for the observed linkage between the sympathetic and ventilatory systems. First, a common central control region may undergo modulation with IH, such as the nucleus tractus solitarii (Guyenet, 2000). Second, overlapping neural structures or separate control centres for each system may be modulated to a similar degree with IH, such as cell groups in the medulla oblongata. While we believe our interpretation is justified we acknowledge the correlative nature of these findings and further studies are required to answer questions of cause and effect.
Effects of IH on cardiovascular measurements
During the 20 min exposure to hypoxia MAP, SBP and heart rate increased and returned to baseline values within 5 min of normoxic recovery. DBP was not significantly altered during the hypoxic exposure or recovery. There was no effect of IH on blood pressure at any stage of the experimental protocol. We have previously shown that 12 days of short duration, but not long duration, IH increase SBP and DBP sensitivity to progressive hypoxia with no effect on heart rate sensitivity (Foster et al. 2005). We postulated that a unique engagement of the carotid body with the short duration IH leads to a greater degree of sympathetic stimulation and downstream cardiovascular consequences. This is supported by other studies where rats subjected to intermittent hypoxia (FI,O2≈ 3 – 5 %) every 30 s, 7 h per day for 35 days show increases in resting blood pressure that are dependent on the carotid body afferents and sympathetic innervation of the kidney (Fletcher et al. 1992a,b). The fact that we did not see significant changes in cardiovascular measures may either be the result of an IH intervention that was too mild or brief to cause cardiovascular alterations, or measures of cardiovascular activity during sustained hypoxia not being effective in detecting changes in cardiovascular sensitivity. We did not show a parallel change in heart and MSNA in the present study. It does not appear that IH changes heart rate sensitivity as we (Foster et al. 2005) and others (Katayama et al. 2001) have shown. Our study was not designed to examine this relationship but our finding is suggestive of differential sympathetic and vagal responses to IH.
Methodological considerations
Data were obtained on two separate days and are therefore subject to day-to-day variability. We have previously shown that HVR values show a day-to-day coefficient of variation of 27% (Koehle et al. 2005). In the present study, our IH intervention exceeded this (Fig. 4) and we believe it to be unlikely that the increase in HVR we observed was due to day-to-day variation. Multiunit recordings of MSNA on separate days will be comprised of a different subset of neurons during each trial as electrode placement is never identical. However, MSNA burst frequency shows ∼15–20% variability between days (Sundlof & Wallin, 1977; Vallbo et al. 1979; Hansen & Sander, 2003). This is consistent with the present study were the average within-subject coefficient of variation for MSNA burst frequency was 16.2 ± 5.6%. In response to IH we observed significant increases in MSNA burst frequency in each subject without exception. This argues that it is unlikely that the effect we observed was simply due to normal day-to-day variability. Nonetheless, to minimize the influence of day-to-day variability on our findings we employed four specific strategies. First, our primary analysis of muscle sympathetic activity was based on burst frequency values. Second, we normalized the exposure and recovery data as a percentage of each individual's baseline (Xie et al. 2001). Third, baseline shifts in the mean voltage neurogram were excluded from analysis. Fourth, we ensured that baseline conditions were identical between days for ventilatory and cardiovascular variables (Table 1). Interpretation of our burst amplitude data must be made with caution as there is relatively little information regarding the predictability of burst amplitude (Malpas, 1998).
Summary
This is the first study to show that 10 daily exposures to isocapnic hypoxia in humans can increase sympathetic outflow during acute hypoxia and recovery. The primary new findings are that (i) 10 days of IH caused a significant increase in MSNA burst frequency during acute hypoxia, and (ii) the increases in burst frequency showed a strong relationship with the increase in HVR. Since burst frequency and HVR were modulated concurrently after our intervention this suggests that these physiological responses may have common central control. We also observed an increase in burst amplitude with exposure to acute hypoxia which is maintained during normoxic recovery. However, the increase in burst amplitude was not augmented with our intervention whereas burst frequency was. This implies that IH may differentially affect each component of the MSNA signal or that each component is under separate central control.
Acknowledgments
We thank our subjects for their enthusiastic participation
This study was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Foundation for Innovation. P.M.K. was supported by a postdoctoral fellowship from NSERC. N.T.A. was supported by a scholar award from the Michael Smith Foundation for Health (MSFHR). A.W.S. was supported by a scholar award from the MSFHR and a new investigator award from the Canadian Institutes of Health Research.
References
- Asano K, Mazzeo RS, McCullough RE, Wolfel EE, Reeves JT. Relation of sympathetic activation to ventilation in man at 4300 m altitude. Aviat Space Environ Med. 1997;68:104–110. [PubMed] [Google Scholar]
- Bhattacharva J, Cunningham DJ, Howson MG, Lee MJ, Sleight P. The effects of mild hypoxia with constant PA,CO2 and ventilation on the setting and sensitivity of the baroreceptor-cardiac-depressor reflex in man. J Physiol. 1973;234:112P–114P. [PubMed] [Google Scholar]
- Cooper VL, Pearson SB, Bowker CM, Elliott MW, Hainsworth R. Interaction of chemoreceptor and baroreceptor reflexes by hypoxia and hypercapnia – a mechanism for promoting hypertension in obstructive sleep apnoea. J Physiol. 2005;568:677–687. doi: 10.1113/jphysiol.2005.094151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler MJ, Swift NM, Keller DM, Wasmund WL, Smith ML. Hypoxia-mediated prolonged elevation of sympathetic nerve activity after periods of intermittent hypoxic apnea. J Appl Physiol. 2004;96:754–761. doi: 10.1152/japplphysiol.00506.2003. [DOI] [PubMed] [Google Scholar]
- Dwinell MR, Huey KA, Powell FL. Chronic hypoxia induces changes in the central nervous system processing of arterial chemoreceptor input. Adv Exp Med Biol. 2000;475:477–484. doi: 10.1007/0-306-46825-5_46. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992a;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Qian W, Miller CC, 3rd, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension. 1992b;19:555–561. doi: 10.1161/01.hyp.19.6.555. [DOI] [PubMed] [Google Scholar]
- Foster GE, McKenzie DC, Milsom WK, Sheel AW. Effects of two protocols of intermittent hypoxia on human ventilatory, cardiovascular and cerebral responses to hypoxia. J Physiol. 2005;567:689–699. doi: 10.1113/jphysiol.2005.091462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster GE, McKenzie DC, Sheel AW. Effects of enhanced human chemosensitivity on ventilatory responses to exercise. Exp Physiol. 2006;91:221–228. doi: 10.1113/expphysiol.2005.032276. [DOI] [PubMed] [Google Scholar]
- Garcia N, Hopkins SR, Powell FL. Effects of intermittent hypoxia on the isocapnic hypoxic ventilatory response and erythropoiesis in humans. Respir Physiol. 2000;123:39–49. doi: 10.1016/s0034-5687(00)00145-6. [DOI] [PubMed] [Google Scholar]
- Guenette JA, Diep TT, Koehle MS, Foster GE, Richards JC, Sheel AW. Acute hypoxic ventilatory response and exercise-induced arterial hypoxemia in men and women. Respir Physiol Neurbiol. 2004;143:37–48. doi: 10.1016/j.resp.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Neural structures that mediate sympathoexcitation during hypoxia. Respiration Physiol. 2000;121:147–162. doi: 10.1016/s0034-5687(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Minson CT. Effect of hypoxia on arterial baroreflex control of heart rate and muscle sympathetic nerve activity in humans. J Appl Physiol. 2002;93:857–864. doi: 10.1152/japplphysiol.01103.2001. [DOI] [PubMed] [Google Scholar]
- Halliwill JR, Morgan BJ, Charkoudian N. Peripheral chemoreflex and baroreflex interactions in cardiovascular regulation in humans. J Physiol. 2003;552:295–302. doi: 10.1113/jphysiol.2003.050708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J, Sander M. Sympathetic neural overactivity in healthy humans after prolonged exposure to hypobaric hypoxia. J Physiol. 2003;546:921–929. doi: 10.1113/jphysiol.2002.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama K, Sato K, Matsuo H, Hotta N, Sun Z, Ishida K, Iwasaki K, Miyamura M. Changes in ventilatory responses to hypercapnia and hypoxia after intermittent hypoxia in humans. Respir Physiol Neurobiol. 2005;146:55–65. doi: 10.1016/j.resp.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Katayama K, Shima N, Sato Y, Qiu JC, Ishida K, Mori S, Miyamura M. Effect of intermittent hypoxia on cardiovascular adaptations and response to progressive hypoxia in humans. High Alt Med Biol. 2001;2:501–508. doi: 10.1089/152702901753397063. [DOI] [PubMed] [Google Scholar]
- Koehle MS, Foster GE, McKenzie DC, Sheel AW. Repeated measurement of hypoxic ventilatory response as an intermittent hypoxic stimulus. Respir Physiol Neurobiol. 2005;145:33–39. doi: 10.1016/j.resp.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Leuenberger U, Gleeson K, Wroblewski K, Prophet S, Zelis R, Zwillich C, Sinoway L. Norepinephrine clearance is increased during acute hypoxemia in humans. Am J Physiol. 1991;261:H1659–H1664. doi: 10.1152/ajpheart.1991.261.5.H1659. [DOI] [PubMed] [Google Scholar]
- Malpas SC. The rhythmicity of sympathetic nerve activity. Prog Neurobiol. 1998;56:65–96. doi: 10.1016/s0301-0082(98)00030-6. [DOI] [PubMed] [Google Scholar]
- Malpas SC, Bendle RD, Head GA, Ricketts JH. Frequency and amplitude of sympathetic discharges by baroreflexes during hypoxia in conscious rabbits. Am J Physiol. 1996;271:H2563–H2574. doi: 10.1152/ajpheart.1996.271.6.H2563. [DOI] [PubMed] [Google Scholar]
- Malpas SC, Ninomiya I. Effect of chemoreceptor stimulation on the periodicity of renal sympathetic nerve activity in anesthetized cats. J Auton Nerv Syst. 1992a;37:19–28. doi: 10.1016/0165-1838(92)90141-3. [DOI] [PubMed] [Google Scholar]
- Malpas SC, Ninomiya I. A new approach to analysis of synchronized sympathetic nerve activity. Am J Physiol. 1992b;263:H1311–H1317. doi: 10.1152/ajpheart.1992.263.4.H1311. [DOI] [PubMed] [Google Scholar]
- Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiol Scand. 2000;168:561–573. doi: 10.1046/j.1365-201x.2000.00709.x. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Baker TL, Nanda SA, Fuller DD, Zabka AG, Hodgeman BA, Bavis RW, Mack KJ, Olson EB., Jr Invited review: Intermittent hypoxia and respiratory plasticity. J Appl Physiol. 2001;90:2466–2475. doi: 10.1152/jappl.2001.90.6.2466. [DOI] [PubMed] [Google Scholar]
- Morgan BJ, Crabtree DC, Palta M, Skatrud JB. Combined hypoxia and hypercapnia evokes long-lasting sympathetic activation in humans. J Appl Physiol. 1995;79:205–213. doi: 10.1152/jappl.1995.79.1.205. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Cooley RL, Dyken ME, Somers VK. Sympathetic activity in obese subjects with and without obstructive sleep apnea. Circ. 1998;98:772–776. doi: 10.1161/01.cir.98.8.772. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circ. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Neubauer JA, Sunderram J. Oxygen-sensing neurons in the central nervous system. J Appl Physiol. 2004;96:367–374. doi: 10.1152/japplphysiol.00831.2003. [DOI] [PubMed] [Google Scholar]
- Nielsen AM, Bisgard GE, Vidruk EH. Carotid chemoreceptor activity during acute and sustained hypoxia in goats. J Appl Physiol. 1988;65:1796–1802. doi: 10.1152/jappl.1988.65.4.1796. [DOI] [PubMed] [Google Scholar]
- Ninomiya I, Malpas SC, Matsukawa K, Shindo T, Akiyama T. The amplitude of synchronized cardiac sympathetic nerve activity reflects the number of activated pre- and postganglionic fibers in anesthetized cats. J Auton Nerv Syst. 1993;45:139–147. doi: 10.1016/0165-1838(93)90125-e. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Overholt JL. Cellular mechanisms of oxygen sensing at the carotid body: heme proteins and ion channels. Respir Physiol. 2000;122:209–221. doi: 10.1016/s0034-5687(00)00160-2. [DOI] [PubMed] [Google Scholar]
- Ryan ML, Hedrick MS, Pizarro J, Bisgard GE. Effects of carotid body sympathetic denervation on ventilatory acclimatization to hypoxia in the goat. Respir Physiol. 1995;99:215–224. doi: 10.1016/0034-5687(94)00096-i. [DOI] [PubMed] [Google Scholar]
- Somers VK, Mark AL, Zavala DC, Abboud FM. Contrasting effects of hypoxia and hypercapnia on ventilation and sympathetic activity in humans. J Appl Physiol. 1989;67:2101–2106. doi: 10.1152/jappl.1989.67.5.2101. [DOI] [PubMed] [Google Scholar]
- Spicuzza L, Porta C, Bramanti A, Maffeis M, Casucci G, Casiraghi N, Bernardi L. Interaction between central-peripheral chemoreflexes and cerebro-cardiovascular control. Clin Auton Res. 2005;15:373–381. doi: 10.1007/s10286-005-0284-5. [DOI] [PubMed] [Google Scholar]
- Sun MK, Reis DJ. Hypoxia selectively excites vasomotor neurons of rostral ventrolateral medulla in rats. Am J Physiol. 1994;266:R245–R256. doi: 10.1152/ajpregu.1994.266.1.R245. [DOI] [PubMed] [Google Scholar]
- Sundlof G, Wallin BG. The variability of muscle nerve sympathetic activity in resting recumbent man. J Physiol. 1977;272:383–397. doi: 10.1113/jphysiol.1977.sp012050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamisier R, Nieto L, Anand A, Cunnington D, Weiss JW. Sustained muscle sympathetic activity after hypercapnic but not hypocapnic hypoxia in normal humans. Respir Physiol Neurobiol. 2004a;141:145–155. doi: 10.1016/j.resp.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Tamisier R, Norman D, Anand A, Choi Y, Weiss JW. Evidence of sustained forearm vasodilatation after brief isocapnic hypoxia. J Appl Physiol. 2004b;96:1782–1787. doi: 10.1152/japplphysiol.01241.2003. [DOI] [PubMed] [Google Scholar]
- Vallbo AB, Hagbarth KE, Torebjork HE, Wallin BG. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol Rev. 1979;59:919–957. doi: 10.1152/physrev.1979.59.4.919. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Crabtree DC, Puleo DS, Goodman BM, Morgan BJ. Neurocirculatory consequences of intermittent asphyxia in humans. J Appl Physiol. 2000;89:1333–1339. doi: 10.1152/jappl.2000.89.4.1333. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Morgan BJ. Exposure to hypoxia produces long-lasting sympathetic activation in humans. J Appl Physiol. 2001;91:1555–1562. doi: 10.1152/jappl.2001.91.4.1555. [DOI] [PubMed] [Google Scholar]