

Abstract
Polo-like kinase 1 (Plk1) is a key regulator of mitotic progression and cell division in eukaryotes. It is highly expressed in tumor cells and considered a potential target for cancer therapy. Here, we report the discovery and application of a novel potent small-molecule inhibitor of mammalian Plk1, ZK-Thiazolidinone (TAL). We have extensively characterized TAL in vitro and addressed TAL specificity within cells by studying Plk1 functions in sister chromatid separation, centrosome maturation, and spindle assembly. Moreover, we have used TAL for a detailed analysis of Plk1 in relation to PICH and PRC1, two prominent interaction partners implicated in spindle assembly checkpoint function and cytokinesis, respectively. Specifically, we show that Plk1, when inactivated by TAL, spreads over the arms of chromosomes, resembling the localization of its binding partner PICH, and that both proteins are mutually dependent on each other for correct localization. Finally, we show that Plk1 activity is essential for cleavage furrow formation and ingression, leading to successful cytokinesis.
INTRODUCTION
The error-free segregation of chromosomes during cell division is necessary for the maintenance of correct ploidy and genomic integrity, and errors in cell division are presumed to lead to aneuploidy and cancer (Rajagopalan and Lengauer, 2004). To ensure that daughter cells receive the correct complement of chromosomes, two key events need to be coordinated. First, chromosomes must be equally segregated, a process that depends on the mitotic spindle. Second, cytokinesis, the process dividing the cell into two, must occur between the two sets of segregated chromosomes. Both of these processes require the activity of a key cell cycle regulator, the Polo-like kinase 1 (Plk1). Plks form a conserved subfamily of serine/threonine protein kinases. The first member to be identified was Polo in Drosophila melanogaster (Llamazares et al., 1991) and, subsequently, four Plk family members have been identified in mammals (Glover et al., 1998; Barr et al., 2004).
Plk1 contains an N-terminal kinase domain and a phosphopeptide-binding C-terminal regulatory polo-box domain (PBD; Leung et al., 2002; Elia et al., 2003b). In vertebrates Plk1 has been implicated in the activation of Cdk1-cyclin B upon entry into mitosis, centrosome maturation via the recruitment of the γ-tubulin ring complex (γ-TuRC), spindle formation, sister chromatid separation by cohesin removal from the chromosome arms, promotion of anaphase onset through direct phosphorylation of the APC/C complex as well as the inhibition of the APC/C inhibitor Emi1, and finally, mitotic exit and cytokinesis (reviewed in Barr et al., 2004). Fitting with these diverse functions, Plk1 localizes to the centrosomes, spindle poles, and kinetochores in prophase and metaphase, the central spindle in anaphase, and the midbody during cytokinesis. These localizations require the function of the PBD (Jang et al., 2002; Seong et al., 2002) and priming-kinases to generate phosphorylated docking sites that are subsequently recognized by the PBD (Elia et al., 2003b). The identification of these priming kinases is therefore important for understanding how Plk1 activity is controlled throughout mitosis as well as meiosis. In the early stages of mitosis, Cdk1 generates Plk1 docking sites (Elia et al., 2003a,b). In contrast, in anaphase Plk1 is able to self-prime its docking sites on proteins required for cytokinesis. Based on these findings, a model has been proposed to explain the temporal and spatial control of Plk1 activity (Neef et al., 2003, 2007).
Plk1 is overexpressed in a broad range of human tumors, and this is associated with poor prognosis in several types of cancer (Eckerdt et al., 2005; Takai et al., 2005). Its association with tumorigenesis indicates that Plk1 is an attractive kinase target for cancer drug development (Strebhardt and Ullrich, 2006). The targeted inactivation of essential kinases is often carried out by ATP-competitive small-molecule inhibitors that block their enzymatic activity. Small-molecule inhibitors have been successfully used for the inhibition of Aurora kinases (Keen and Taylor, 2004) and Cdks (Fischer et al., 2003). Most recently, efforts to identify new Plk inhibitors have led to the discovery of several potent inhibitors (McInnes et al., 2006; Peters et al., 2006; Strebhardt and Ullrich, 2006; Lansing et al., 2007; Lenart et al., 2007), and these new tools have been successfully used to study several aspects of Plk1 function.
Here we identify ZK-Thiazolidinone (TAL) as a novel ATP-competitive inhibitor of Plk1. Following an extensive characterization of this new inhibitor in vitro, we have addressed its specificity within intact cells by systematically analyzing its ability to counteract the role of Plk1 in previously established functions, notably sister chromatid separation, centrosome maturation, and bipolar spindle assembly. Our results confirm the requirement of Plk1 kinase activity for all these functions, indicating that TAL acts as a specific and potent Plk1 inhibitor in vivo. Having established TAL specificity, we have then used this novel inhibitor to gain further insights into the targeting and regulation of Plk1 at different times throughout mitosis and cytokinesis. Specifically, we have explored the relationship between the catalytic activity of Plk1 and the recently identified Plk1-interacting checkpoint helicase PICH (Baumman et al., 2007), and we have used TAL as a potent tool for studying Plk1 function at the central spindle during cytokinesis. Our results provide strong support for the recently proposed self-priming model of Plk1 targeting in anaphase cells (Neef et al., 2007) and help to explain the temporal regulation of Plk1 during mitotic exit and cytokinesis.
MATERIALS AND METHODS
Preparation of TAL Inhibitor
TAL was prepared as described (Schulze et al., 2006). For in vitro and in vivo experiments TAL was used from a stock solution (10 mM) in dimethyl sulfoxide (DMSO).
In Vitro Kinase Assays
Recombinant baculoviruses expressing full-length His-tagged Plk1 were produced using the BaculoGold kit according to manufacturer's instructions (BD Biosciences Pharmingen, San Diego, CA). Glutathione S-transferase (GST)-tagged Plk4 wild type (WT; residues 1–265) and kinase dead (KD; D154A) and His-tagged Aurora A (Kufer et al., 2002) were purified from E. coli. GST-tagged Aurora B (Neef et al., 2006) was purified from Sf9 cells. Cdk1 was obtained commercially (Upstate Biotechnology, Lake Placid, NY). In vitro phosphorylation reactions on different model substrates were carried out in BRB80 buffer (Stucke et al., 2004) in the case of Plk1, Plk4, and Cdk1 or a following described protocol for Aurora A (Kufer et al., 2002). Reactions were supplemented with 10 μM ATP and 2 μCi γ-32P-ATP (Amersham Pharmacia Biosciences, Piscataway, NJ). In vitro phosphorylation of Mklp2 was performed using 200 ng of substrate, 10 μM ATP and 2 μCi γ-32P -ATP, in a total volume of 20 μl BRB80 for 30 min at 30°C. Reactions were stopped by the addition of SDS sample buffer and heating to 95°C. Reactions products were visualized by SDS-PAGE followed by autoradiography.
To determine the half-maximal growth inhibition (IC50) of Plk1, activity assays were performed for 90 min at 22°C in presence of serial dilutions of inhibitor in a total volume of 31 μl using casein from bovine milk (Sigma, St. Louis, MO) as the substrate (0.66 μg/ml Plk1, 0.7 μM biotinylated casein, 50 mM HEPES, pH 7.5, 10 mM MgCl2, 3 mM MnCl2, 1 mM dithiothreitol, 0.01% Nonidet P40, 3% DMSO, 0.5 μM ATP, and 50 nCi γ-33P-ATP). Reactions were terminated by addition of 50 μl of SPA suspension (100 μM ATP, 10 mM EDTA, 0.2% Triton X-100, 2.5 mg/ml streptavidin-coated SPA beads [Amersham Pharmacia Biosciences] in phosphate-buffered saline). SPA beads were allowed to sediment over night at 22°C, and incorporated 33P was determined using a TopCount scintillation counter (Perkin Elmer-Cetus, Norwalk, CT). Dose–response curves were used to calculate IC50 values. The same procedure was followed to test a panel of 93 serine/threonine kinases.
Cell Culture and Synchronization
Human MCF7, NCI-H460, DU145, HeLa S3, mouse B16-F10, and Caco-2 cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were grown at 37°C under 5% CO2, either in DMEM (HeLa S3) or in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum and penicillin and streptomycin (100 U/ml and 100 mg/ml, respectively). Thymidine arrest was performed for 14 h at a concentration of 4 mM. Nocodazole and taxol arrests were performed for 16 h at concentrations of 0.2 μg/ml and 1 μg/mL, respectively. Cells arrested in metaphase were obtained by treating cells with 25 μM noscapine or 20 μM VS-83 for 10 h and subsequently 2 h with 20 μM MG132.
In Vitro Cell Proliferation Assays
Cells were seeded in 96-well plates at 1500 (Caco-2), 3000 (NCI-H460, HeLa), or 5000 (MCF7, DU 145) cells/well. Cells were allowed to adhere for 24 h and then fresh growth medium plus serial dilutions of inhibitor compounds were added. The final concentration of the solvent DMSO was 0.5%. After 4 d of continuous incubation, the cells were fixed with glutaraldehyde and stained with crystal violet, and the absorbance was recorded at 595 nm. All measurements were done in quadruplicates. The values were normalized to the absorbance of solvent-treated cells (= 100%), and the absorbance of a reference plate, which was fixed at the time point of compound application (= 0%). IC50 was determined as compound concentration that was required to achieve 50% inhibition of cellular growth.
Fluorescence-activated Cell Sorting Analysis
HeLaS3 (Figure 1C) or HeLaS3 and MCF-7 cells (Supplementary Figure 2) were incubated with 0.5% DMSO or 1 μM TAL at different time points (Figure 1C) or various concentrations of TAL for 24 h (Supplementary Figure 2). Cell suspensions were fixed with 80% ethanol, permeabilized by treatment for 5 min with 0.25% Triton X-100 in PBS, and incubated with 0.1% RNase and 10 μg/ml propidium iodide. Cellular DNA content was determined by flow cytometry using FACSCalibur (BD Biosciences Clontech, San Jose, CA) system and CellQuest software (Becton-Dickinson, Lincoln Park, NJ).
Figure 1.

TAL inhibits specifically Plk1 and cause a prometaphase-like mitotic arrest. (A) Chemical structure of the ATP-competitive kinase inhibitor, ZK-Thiazolidinone (TAL). (B) Mklp2 was treated with buffer alone (−Plk1) or Plk1 with increasing amounts of TAL and radioactive ATP for 30 min and then analyzed by SDS-PAGE and autoradiography ([32P]). Comassie blue staining showed equal amounts of Mklp2 in all samples. (C) FACS analysis was performed in HeLa S3 cells treated for 24 h with DMSO as a control (1) or for 12 h, 12 h + 12 h release, 24 h, and 48 h with 1 μM TAL (2, 3, 4, and 5, respectively). The percentage of cells with less than 2N (pre-G1), 2N (G1 and S), or 4N (G2/M) content are shown. (D) Mad2-positive staining in a monopolar cell treated with 1 μM TAL. Cells were stained for Mad2 (red), α-tubulin (green), and DNA was stained with DAPI (blue). Scale bar, 10 μm. (E) Mitotic index from cells after transfection with GL2, BubR1, and Mad2 siRNA-oligos for 48 h, treated with either DMSO or 1 μM TAL for the last 12 h. (300 mitotic cells for each combination, n = 2). (F) Selected live-cell images of HeLa cells expressing GFP-tagged Histon-H2B treated with DMSO or 1 μM TAL. Time = 0 min indicates the onset of chromosome condensation.
Transient Transfection and Small Interfering RNA
Plasmid transfections were performed using FuGENE 6 reagent (Roche Diagnostics, Indianapolis, IN) according to manufacturer's instructions. Small interfering RNA (siRNA) duplexes were transfected using Oligofectamine (Invitrogen). In siRNA experiments, BubR1 siRNA duplexes (5′-GGAGATCCTCTACAAAGGG) were purchased from Qiagen (Hilden, Germany). Plk1, Mad2, BubR1, Eg5, and PICH (oligo 1) were depleted using previously published duplexes (Stucke et al., 2004; Hanisch et al., 2006; Baumman et al., 2007), and the GL2 duplex (Elbashir et al., 2001) was used for control. For rescue experiments, the Plk1-RNAi plasmid was transfected simultaneously with myc-Plk1 WT or KD constructs, following established protocols (Hanisch et al., 2006).
Mitotic Chromosome Spreads
HeLa S3 cells were either treated with VS-83 or TAL overnight. Mitotic cells were collected by shake-off. Chromosome spreads were obtained following established protocols (Hanisch et al., 2006).
RESULTS
TAL Specifically Inhibits Plk1 In Vitro and In Vivo
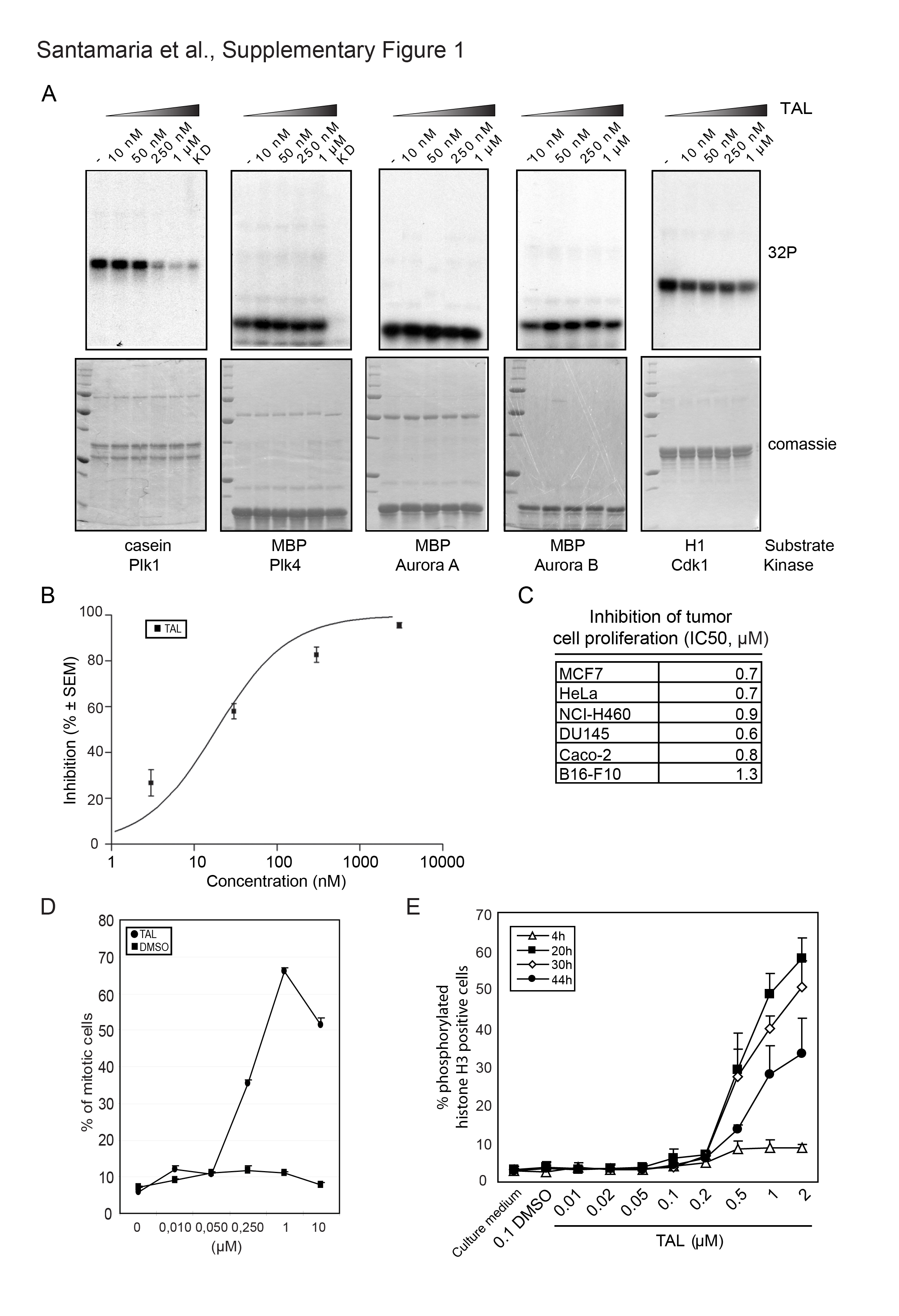
Screening of the Schering compound collection for inhibition of Plk1 kinase activity resulted in the identification of the Thiazolidinone lead series. The Thiazolidinones represent a novel chemical class of kinase inhibitors. They do not contain either of three preferred structural motifs of ATP-competitive kinase inhibitors (Perola, 2006) and, in particular, they are distinct from previously described Plk1 kinase inhibitors (Andrews et al., 2004; Davis-Ward et al., 2004; Bearss et al., 2006; Perola, 2006; Steegmaier et al., 2007). The compound characterized here, ZK-Thiazolidinone (Figure 1A) is hereafter referred to as TAL. As assayed under specific in vitro conditions TAL inhibited human Plk1 with an IC50 of 19 ± 12 nM (Supplementary Figure 1B; for assay conditions see Materials and Methods). When tested against a panel of 93 serine/threonine and tyrosine kinases, the compound showed high selectivity for Plk1 (Supplementary Table 1 and Supplementary Figure 1A). The only other kinases showing significant sensitivity to TAL were the two closest relatives of Plk1: Plk2 and Plk3, which were inhibited at IC50 of lower than 100 nM (data not shown). In contrast, no significant inhibition could be seen for Plk4 (Supplementary Figure 1A). We also tested the ability of TAL to inhibit Plk1 activity using the physiological Plk1 target Mklp2 as a substrate (Neef et al., 2003) and found strong inhibition of Mklp2 phosphorylation by 250 nM TAL (Figure 1B). Taking together, these results show that TAL potently and selectively inhibits Plk1 in vitro.
TAL Treatment Results in an Increased Mitotic Index Due to Checkpoint Activation
Consistent with Plk1 inhibition, TAL inhibited the proliferation of various human and mouse tumor cell lines with an IC50 of 0.2–1.3 μM (Supplementary Figure 1C). Because Plk1 activity is essential for mitotic progression, we analyzed the effect of TAL on the cell cycle of HeLa S3 and MCF7 cells by flow cytometry. Within 24 h of treatment with increasing concentrations of the compound, TAL induced an accumulation of HeLa S3 and MCF7 cells with a 4N DNA content, indicative of a G2/M arrest (Supplementary Figure 2). Concomitantly, a striking increase in the mitotic index (Supplementary Figure 1D) and an increase in phospho-histone H3 staining (Supplementary Figure 1E) could be observed. At concentrations above 10 μM, increasing cell death was observed, dependent on cell line and duration of exposure (data not shown), in agreement with the reported effect of Plk1 depletion in tumor cells (Liu and Erikson, 2003; Liu et al., 2006). We also tested the reversibility of 1 μM TAL treatment of HeLa S3 cells. After 12 h of treatment, a release for 12 h into fresh medium readily reversed the G2/M block (Figure 1C). A mitotic arrest was maintained during a 24 h treatment, but after 48 h an elevated proportion of cells with sub-2N DNA content was observed, indicative of cell death (Figure 1C). Because of the clear accumulation of mitotic cells in response to 12 h treatment with 1 μM TAL (Figure 1C), the absence of cell death and the reversibility of the block, we used 1 μM for 12 h as a standard condition for most of the following experiments.
The predominant phenotype seen upon depletion of Plk1 is a cell cycle arrest in prometaphase (Sumara et al., 2004; Van Vugt et al., 2004). In line with these observations, treatment of asynchronously growing HeLa S3 cells with 1 μM TAL resulted in a prometaphase-like arrest characterized by frequent monopolar spindles and kinetochores that were positive for Mad2 (Figure 1D), indicating that kinetochores were either not attached to the spindle or not fully occupied by microtubules. Depletion of either BubR1 or Mad2, two key regulators of the spindle assembly checkpoint (SAC), suppressed the mitotic arrest caused by TAL treatment (Figure 1E), demonstrating its checkpoint dependence. Live cell microscopy also confirmed that TAL-treated cells failed to form a normal metaphase plate and instead arrested with monopolar spindles, whereas control cells entered anaphase ∼30 min after nuclear envelope breakdown (Figure 1F and Supplementary Movies S1 and S2).
TAL Treatment Produces Mitotic Defects Expected for Plk1 Inhibition
To further validate the specificity of TAL for Plk1 within living cells, we examined the ability of this compound to interfere with selected processes that are known to require Plk1 activity, notably centrosome maturation, sister chromatid cohesion, and spindle formation. These processes have previously been studied extensively, and they all concern early stages of mitosis (Barr et al., 2004). Most recently, they have also been used to validate the specificity of other, structurally distinct Plk1 inhibitors (McInnes et al., 2006; Peters et al., 2006; Lansing et al., 2007; Lenart et al., 2007).
The first process we examined is centrosome maturation at the G2/M transition. At this stage of the cell cycle, additional γ-TuRCs are recruited to the centrosome to enhance microtubule nucleation at the onset of mitosis (Khodjakov and Rieder, 1999; Palazzo et al., 2000). A role for Plk1 in centrosome maturation was originally established through antibody injection (Lane and Nigg, 1996) and more recently confirmed by siRNA-mediated Plk1 depletion (Sumara et al., 2004; Hanisch et al., 2006). After treatment of cells with TAL, γ-tubulin recruitment to centrosomes (identified by pericentrin staining) was clearly impaired (Figure 2A), very similar to the phenotype seen after depletion of Plk1 (Sumara et al., 2004; Hanisch et al., 2006). For control, untreated cells or cells displaying a similar cell cycle arrest phenotype due to inhibition of the kinesin-related motor Eg5 (Blangy et al., 1995) were analyzed in parallel and found to show normal γ-tubulin recruitment (Supplementary Figure 3 and Figure 2A). Interestingly, in TAL-treated cells, Plk1 itself failed to localize to centrosomes (see Figure 5 and Supplementary Figure 4), in agreement with a recent study using the Plk1 inhibitor BI 2536 (Lenart et al., 2007). In human cells, Plk1 depletion also impairs the recruitment of Aurora A kinase to the centrosome (De Luca et al., 2006; Hanisch et al., 2006), which might provide a plausible explanation for the Aurora A requirement in centrosome maturation observed in invertebrates (Hannak et al., 2001; Berdnik and Knoblich, 2002). The availability of TAL afforded a unique opportunity to address the question of whether Plk1 activity is required for Aurora A recruitment. As shown in Figure 2B, Aurora A failed to localize to centrosomes in TAL-treated cells, similar to the phenotype seen in Plk1-depleted cells, but not in untreated cells or Eg5-depleted cells, analyzed for control (Supplementary Figure 3 and Figure 2B). Instead, in both TAL-treated cells and Plk1-depleted cells, Aurora A associated predominantly with spindle microtubules (Figure 2B). Eg5 localization was not affected upon TAL treatment (Figure 2C) nor were the levels of Plk1, Eg5, γ-tubulin, and Aurora A proteins (Figure 2D). These results not only confirm that Plk1 acts upstream of Aurora A (De Luca et al., 2006; Hanisch et al., 2006), but further demonstrate that Plk1 activity is required for Aurora A recruitment to centrosomes.
Figure 2.

Centrosome maturation is impaired in TAL-treated cells. HeLa S3 cells were transfected with Eg5 or Plk1 siRNA-oligos for 36 h or treated with 1 μM TAL for 12 h and then stained for γ-tubulin, Aurora A, or Eg5 (red; A, B, and C, respectively), pericentrin (A and B), or α-tubulin (C; green), and DNA was stained with DAPI (blue). Scale bar, 10 μm. (D) Lysates from nocodazole- (0.2 μg/mL) or taxol- (1 μg/ml) arrested cells, treated with DMSO or 1 μM TAL were used for Western blot analysis. Membranes were probed for Plk1, Eg5, γ-tubulin, Aurora A, and α-tubulin as loading control.
Figure 5.

Plk1 and PICH spread over chromatid arms upon TAL addition. (A and B) HeLa S3 cells were transfected with Eg5 or Plk1 siRNA-oligos for 36 h or treated with 1 μM TAL for 12 h. Cells were fixed and stained for PICH and Plk1 (A and B, respectively; green), CREST (red), and DNA (blue). Scale bar, 10 μm. (C) Lysates from nocodazole (0.2 μg/mL) or taxol (1 μg/ml) arrested cells, treated with DMSO or 1 μM TAL were used for Western blot analysis. Membranes were probed for PICH, phospho-H3, cyclin-B, and α-tubulin as loading control. (D) HeLaS3 cells were treated under different conditions: first column, DMSO for 12 h; second column, TAL for 12 h; third column, PICH-siRNA oligo for 36 h; and fourth column, PICH-siRNA oligo for 36 h together with TAL during the last 12 h. Cells were fixed and stained for PICH (red) and Plk1 (green), and DNA was stained with DAPI (blue). Scale bar, 10 μm.
Most recently, evidence has emerged for additional roles of Plk1 in spindle formation and maintenance (Sumara et al., 2004; Peters et al., 2006) as well as in kinetochore-microtubule attachment and chromosome congression (Hanisch et al., 2006; Lenart et al., 2007). To determine whether Plk1 activity is required for the maintenance of spindle bipolarity, HeLa S3 cells synchronized to display bipolar spindles (see Materials and Methods) were treated with TAL and followed by live cell videomicroscopy over time (Supplementary Movie S3 and Figure 3A). In parallel, cells were fixed and stained with anti-α-tubulin antibodies (Figure 3B). Although control cells (treated with DMSO solvent only) readily maintained bipolar spindles and chromosomes in a metaphase plate (Figure 3A and data not shown), in TAL-treated cells bipolar spindles progressively collapsed to yield monopolar microtubule arrays (Figure 3, B and C), and metaphase plates became disorganized (Figure 3, A and B). Most likely, this phenotype results from a shortening of kinetochore fibers (K-fibers) for the benefit of microtubule bundles that extend from the spindle pole to the cell periphery (Figure 3B, 60 and 120 min, arrows). These results confirm and extend those recently reported by Kapoor and colleagues (Peters et al., 2006) using a chemically distinct Plk1 inhibitor.
Figure 3.

Plk1 activity is required for bipolar spindle maintenance. (A) Cells were arrested with 20 μM VS-83 for 12 h and released into 20 μM MG132 for 2 h to enrich in metaphase cells. Then 1 μM TAL was added and cells were followed by live-cell imaging. Representative stills from live-cell images of HeLa cells expressing GFP-tagged Histone-H2B are shown. Time = 0 represents the time of addition of the inhibitor. (B) HeLa S3 cells were arrested with 25 μM noscapine during 10 h and release into 20 μM MG132. After 2-h MG132 arrest, 1 μM TAL was added, and cells were fixed at indicated times and stained for α-tubulin (green), and DNA was stained with DAPI (blue). Representative images of phenotypes observed at each time point are shown. Scale bar, 10 μm. (C) Quantification of mono- or bipolar spindles in TAL-treated cells; 300 mitotic cells for each data point, n = 2.
To explore the suspected role of Plk1 in K-fiber stabilization, K-fibers were stained with anti-HURP antibodies (Sillje et al., 2006) before and after cold treatment (Rieder and Borisy, 1981). For comparison, monoastral spindles were produced in control cells by treatment with VS-83, a small-molecule inhibitor of Eg5 (Sarli et al., 2005). Although HURP-positive K-fibers could readily be seen in both VS-83 and TAL-treated cells at time zero (Figure 4A, left), cold treatment for 20 min virtually abolished HURP staining in TAL-treated cells but not in VS-83 treated cells (Figure 4A, right). Furthermore, unlike the situation in VS-83 cells, the thin and long microtubules observed in TAL-treated cells did not end at kinetochores, as shown by costaining with CREST kinetochore serum (Figure 4A, bottom panels). These data lend strong support to the view that Plk1 activity is required to maintain stable kinetochore-microtubule interactions.
Figure 4.

(A) Plk1 activity is required for K-fiber stabilization and chromatid arm separation. HeLa S3 cells were treated for 12 h with 20 μM VS-83 or 1 μM TAL and before fixation and permeabilization with PTEMF; cells were incubated for 20 min in ice-cold growth medium. Cells were stained for HURP (far red), CREST (red), and α-tubulin (green), and DNA was stained with DAPI (blue). (B) DAPI-stained chromosome spread prepared from HeLa S3 treated for 10 h with 20 μM VS-83 or 1 μM TAL.
As a final validation of TAL specificity, we examined a third well-established function of vertebrate Plk1, namely the removal of cohesins from the arms of sister chromatids during prophase (Losada et al., 2002; Sumara et al., 2002; Losada, 2007) As shown by the analysis of chromosomes spreads, sister chromatid arms remained paired in cells treated with TAL, whereas the expected X-shaped chromosomes, indicative of arm separation, were seen in control cells treated with VS-83 (Figure 4B). Collectively, our analysis of the effect of TAL on centrosome maturation, bipolar spindle formation, and sister chromatid cohesion demonstrate that this Plk1 inhibitor produces phenotypes that are entirely consistent with our current understanding of Plk1 function, lending strong support for its in vivo specificity.
Having established confidence in the reliability of TAL as a new research tool to study Plk1 function, we next proceeded to use TAL for a more detailed analysis of Plk1 in relation to two recently identified interaction partners, notably PICH implicated in SAC function (Baumman et al., 2007) and PCR1, a putative protein scaffold playing a key role in cytokinesis (Neef et al., 2007).
Spreading of Plk1 and PICH over Chromatid Arms upon Plk1 Inactivation
Having identified PICH as a prominent Plk1-interaction partner and substrate in prometaphase, we took advantage of TAL to explore the consequences of Plk1 inhibition on the localization of PICH and Plk1 (Figure 5). To provide controls with monoastral spindles, Eg5-depleted cells were analyzed in parallel. On inhibition of Plk1 by TAL, PICH was found to spread over chromatid arms, similar to the phenotype seen in Plk1-depleted cells (Figure 5A). This confirms that Plk1 is required to remove PICH from chromatid arms (Baumman et al., 2007) and further demonstrates a requirement for Plk1 activity. Interestingly, when compared with (Eg5 depleted) control cells, Plk1 itself was also found to spread over chromatid arms in response to TAL treatment (Figure 5B), suggesting that Plk1 activity is required to concentrate this kinase at the kinetochore. To mimic the situation seen in TAL-treated cells, we overexpressed myc-tagged kinase dead (Plk1 KD) in Plk1-depleted cells. Cells moderately overexpressing myc-tagged Plk1 KD showed staining of chromatid arms very similar to that seen for endogenous Plk1 in TAL-treated cells, whereas Plk WT analyzed for control remained concentrated at kinetochores (Supplementary Figure 4B). These results indicate that Plk1 interacts with a docking partner on chromatid arms, whose localization is sensitive to Plk1 activity.
Because the chromosomal localization of PICH is clearly controlled by Plk1 (Figure 5A and Baumman et al., 2007), we considered this protein a likely candidate for the recruitment of Plk1 over chromatid arms. To test this hypothesis, we first used TAL to confirm that the electrophoretic mobility shift seen in PICH during mitotic arrest is due to Plk1 activity (Figure 5C). Then, we analyzed the influence of PICH depletion on Plk1 localization with or without TAL treatment (Figure 5D). Compared with DMSO-treated control cells, where Plk1 was concentrated at kinetochores, TAL induced the spread of Plk1 over chromatid arms. In contrast, upon depletion of PICH, TAL treatment no longer induced this redistribution, demonstrating that PICH is required for relocalization of TAL-inactivated Plk1 to chromatid arms (Figure 5D). Collectively, the above data strongly suggest that PICH is the major Plk1 interaction partner on chromosome arms.
Inhibition of Plk1 Activity by TAL Leads to Cytokinesis Failure
In the past, it has been difficult to pinpoint a specific role for Plk1 in postanaphase events, because of multiple requirements for this kinase at earlier stages of mitosis. Thus, in the final series of experiments, we used TAL to explore the purported role of Plk1 in cytokinesis (Mundt et al., 1997; Carmena et al., 1998; Nigg, 1998; van Vugt and Medema, 2005). TAL treatment of cells released from an MG132 induced metaphase phase arrest caused extensive multinucleation, confirming the requirement for Plk1 for cell division (Supplementary Figure 5, A and B). To examine this phenotype in more detail, we performed live cell imaging on asynchronous cultures of HeLa S3 cells treated with TAL. In cells that were in prophase or prometaphase at the time of TAL addition, bipolar spindle collapsed into a monopole (Figure 6A), arguing that TAL exerts its Plk1-inhibitory effect rapidly after addition. Of the cells showing already aligned chromosomes when the drug was added, spindle bipolarity was similarly lost in 30% of cells, whereas 70% progressed into anaphase and segregated their chromosomes normally but then failed to complete cytokinesis, resulting in binucleation (Figure 6A). This argues that events early in anaphase are regulated by Plk1 and crucial to ensure proper cytokinesis. Finally, cells that were already in telophase or cytokinesis at the time of TAL addition completed normal division (Figure 6A). Careful examination of cells that had aligned chromosomes at the time of TAL addition revealed that most of these cells (>70%; Figure 6B) failed to display cleavage furrow ingression, again indicating an early requirement for Plk1 activity in this process (Figure 6C, bottom, upper two rows, and Supplementary Movie S4). In contrast, cells already in anaphase at the time of TAL addition mostly showed transient furrow ingression (>90%; Figure 6B), and yet cytokinesis ultimately failed (Figure 6C, bottom, lower two rows, and Supplementary Movie S5).
Figure 6.

Plk1 activity is required for successful cytokinesis. (A) Quantification of the percentage of cells in different stages of mitosis that upon TAL treatment collapsed into monopolar spindles or went through mitosis completing normal division or failing in cytokinesis. Approximately 100 cells were counted in two independent experiments performed by live-cell imaging. (B) The failure in cytokinesis with or without furrow ingression was analyzed and plotted for the cells in metaphase or anaphase from the previous experiment. (C) Representative stills from these movies are shown. Time = 0 represents the stage in mitosis at the time of addition of the inhibitor. Top panels, a control cell that progress normally through mitosis (DMSO); bottom panels, cells that fail in cytokinesis with or without furrow formation and ingression (TAL panels, upper two rows and lower two rows, respectively).
The furrowing defects in Plk1-inhibited cells suggested that some aspects of cleavage furrow formation were impaired. We therefore looked at the localization of the RhoA GTPase, the essential regulator of actomyosin dynamics during cytokinesis, and its upstream guanine nucleotide exchange factor (GEF) ECT2 to the equatorial cell cortex and central spindle, respectively. Strikingly, TAL suppressed RhoA recruitment to the equatorial cell cortex, implying that Plk1 acts upstream of RhoA (Figure 7A). We then investigated the effects of TAL on ECT2, because the localized activation of RhoA at the cleavage furrow is dependent on this GEF (Yuce et al., 2005). This showed that ECT2 was lost from the central spindle in TAL-treated cells (Figure 7B), explaining why RhoA is lost from the equatorial cell cortex and cleavage furrow ingression fails upon Plk1 inhibition.
Figure 7.

Plk1 activity is required for proper furrow ingression. (A and B) Control HeLa cells (left, DMSO) and cells treated for 30 min with 1 μM TAL (right) were fixed and stained for RhoA or ECT2 (A and B, respectively; green), actin (B; red), and DNA (blue). Cells in early and late anaphase stages are shown in the control.
Plk1 targeting during anaphase has been suggested to control its localization during anaphase and cytokinesis through a self-priming feedback mechanism (Neef et al., 2003, 2007). To further test this hypothesis, we investigated two Plk1 docking partners at the central spindle required for cytokinesis, notably the kinesin-6 family motor Mklp2 and the central spindle microtubule-associated protein PRC1. Control cells treated with DMSO showed normal staining for Plk1, Mklp2, and PRC1 (Figure 8A, left). In contrast, after Plk1 inhibition with TAL the central spindle was highly disorganized and Plk1 staining was lost during anaphase and telophase (Supplementary Figure 5C; Figure 8A, right). This suggests that Plk1 must generate a docking site on its major central spindle-binding partners. Phosphorylation of Mklp2 by Plk1 is necessary for Mklp2 function and Plk1 localization to the central spindle in late anaphase and telophase (Neef et al., 2003), but so far the consequences of this phosphorylation for the Mklp2 localization could not be addressed. After TAL treatment the central spindle localization of Mklp2 was strongly reduced and a more cortical staining appeared (Figure 8A, right). Because Mklp2 is required for the relocalization of Aurora B from the kinetochores to the central spindle (Gruneberg et al., 2006), we asked whether the localization of Aurora B was altered in TAL-treated cells. As shown in Supplementary Figure 5C, Aurora B also showed a cortical localization after inhibitor treatment, most likely representing the pool of Aurora B bound to the displaced Mklp2, whereas the central spindle localization of Aurora B was diminished. PRC1 localization at the central spindle is also affected in Mklp2-depleted cells (Neef et al., 2007). Therefore, we analyzed PRC1 localization after treatment with TAL. After inhibition of Plk1 activity, PRC1 localized to the disorganized central spindle in early anaphase (Figure 8A). However, in late anaphase/telophase, PRC1 failed to form a focused band and spread out along the entire length of the microtubule bundle (Figure 8A, right), resembling the localization in Mklp2-depleted cells (Neef et al., 2007). In addition, very strong microtubule bundles were observed, resembling the phenotype generated upon depletion of endogenous PRC1 and concomitant rescue with a docking site mutant of PRC1 (S602A mutant), which is unable to bind and localize Plk1 (Neef et al., 2007). Phosphospecific antibodies to this site (pT602) specifically stained the central spindle in control cells, and this staining was lost upon Plk1 inhibition (Figure 8A, right), confirming the specific phosphorylation by Plk1 on this site. In contrast, staining for Mklp1 and for phospho-S911 Mklp1, an Aurora B phosphosite (Neef et al., 2006), were comparable with or without TAL treatment (Figure 8A).
Figure 8.

Plk1 activity is required for its interaction with the anaphase spindle. (A) Control HeLa cells treated for 30 min with DMSO (left) or 1 μM TAL (right) were fixed and stained for the indicated antibodies. PRC1, PRC1-pT602, Mklp2, Mklp1, and Mklp1-pS911 are shown in green, Plk1 in red, and DNA in blue. (B) HeLa cells were arrested in nocodazole (50 ng/ml) for 14 h, then washed, and released in fresh medium for 80 min. After 20-min release, the inhibitor was added to sample three to a final concentration of 1 μM and incubated for additional 60 min. PRC1 was precipitated from the lysates with a specific antibody, and the precipitates were analyzed by Western blot with antibodies to PRC1pT481, PRC1pT602, and Plk1. The same blot was reprobed for total PRC1. The asterisk shows a cross-reactive band of the PRC1pT602 antibody.
Finally, we used the TAL inhibitor to confirm that the PRC1-T602 phosphorylation by Plk1 correlates with Plk1 binding in vivo. As shown previously, Plk1 and PRC1 did not interact in metaphase (Figure 8B, 0 min nocodazole release), when PRC1 is phosphorylated at T481 by Cdk1 (Neef et al., 2007). After 80-min release from nocodazole, cells entered anaphase, as shown by the dephosphorylation of PRC1 at T481and the phosphorylation at PRC1 T602 by Plk1. In these anaphase cells, Plk1 precipitated with PRC1. However, when the release was performed in the presence of TAL, T602 was not phosphorylated and the amount of Plk1 precipitated together with PRC1 was significantly reduced. Together with the previously reported data this shows that active Plk1 is required for the generation of its own binding site on PRC1 in anaphase. Thus, the use of TAL in the above experiments provides strong support for the previously proposed self-priming model (Neef et al., 2003, 2007).
DISCUSSION
Specific Inhibition of Plk1 by TAL In Vitro and In Vivo
Here we have characterized TAL, a new potent and specific inhibitor of Plk1. Importantly, TAL is built on a novel chemical scaffold and structurally distinct from other Plk1 inhibitors recently described (McInnes et al., 2006; Peters et al., 2006; Lansing et al., 2007; Lenart et al., 2007). Because specificity is a general concern when using small molecule inhibitors to study biological processes, results can be viewed with increased confidence whenever chemically distinct compounds produce consistent phenotypes. Using TAL, we have been able to confirm functions previously attributed to Plk1, attesting to the selectivity of this compound. In addition, the use of TAL has allowed us to enhance our understanding of Plk1 function in mitotic spindle formation, chromosome arm separation, and the temporal and spatial control of Plk1 during mitosis and cytokinesis. These findings are discussed in more detail in the following sections.
Plk1 Activity in Mitotic Spindle Formation and Chromosome Congression
Bipolar spindle formation and maintenance involves the remodeling of the microtubule nucleating and organizing capacities of the spindle poles, as well as proper kinetochore-microtubule attachment. Plk1 is involved in both processes and may be important to help coordinate these events. It has previously been shown that Plk1 is needed to recruit γ-tubulin to the centrosomes in order to enhance microtubule nucleation activity in preparation of bipolar spindle formation (Khodjakov and Rieder, 1999; Palazzo et al., 2000). Our present study not only confirms that this function depends on Plk1 activity, but also demonstrates that Plk1 activity is needed for Aurora A localization to the centrosomes. This is significant because Aurora A, together with its partner TPX2, is known to be required for normal spindle formation (Kufer et al., 2002; Tsai et al., 2003; Eyer and Maller, 2004). Once mitotic spindles have formed, Plk1 is also required for the maintenance of spindle bipolarity, as shown by the use of two chemically distinct Plk1 inhibitors (Peters et al., 2006 and our present study). This intriguing observation suggests that Plk1 substrates involved initially in spindle formation must remain phosphorylated and/or that additional substrates need to be phosphorylated in order to maintain the bipolarity of already formed spindles. With regard to chromosome congression, our present data strengthen the emerging view that Plk1 contributes to stabilize kinetochore-microtubule attachments. This may reflect a role for Plk1 in the generation of spindle forces that stabilize kinetochore-microtubule attachments (Sumara et al., 2004; Hanisch et al., 2006; Lenart et al., 2007), potentially through direct phosphorylation of Plk1 substrates at the kinetochore. With the aid of Plk1 inhibitors such as TAL it should be possible to further address these issues.
Mutual Dependency of Plk1 and PICH in Chromosome Localization
Two mitotic kinases, Polo-like kinase and Aurora B, have been implicated in cohesin release during mitotic prophase (Losada et al., 2002; Sumara et al., 2002). Plk1 can directly phosphorylate cohesin subunits, but a direct interaction between Plk1 and cohesin has not been reported and it is not know how phosphorylation leads to the dissociation of cohesin from chromosomes. Also, other factors such as Wapl are clearly involved in regulating cohesin dissociation (Gandhi et al., 2006; Kueng et al., 2006). Here, we show that Plk1 spread over chromatid arms when its activity is inhibited with TAL. A similar redistribution after Plk1 depletion was previously observed for PICH, one of the binding partners of Plk1 during mitosis (Baumman et al., 2007). Our results with the TAL inhibitor directly link the localization of PICH over chromatid arms to Plk1 activity. Moreover, our data clearly demonstrate interdependency between these two proteins in that Plk1 remains at kinetochores upon TAL treatment of PICH-depleted cells. Under normal circumstances, we envision that Plk1 gets recruited to PICH (through a Cdk1-dependent priming event) and that a PICH-Plk1 complex has the ability to spread over chromatid arms. Our present results clearly indicate that PICH is a major interaction partner of Plk1 on chromatid arms and it will be interesting to explore whether this complex is mechanistically related to the removal of cohesins by Plk1. In any case, in response to phosphorylation by Plk1, PICH is released from chromatid arms, resulting in its concentration at the centromere/kinetochore (Baumman et al., 2007).
Temporal Regulation of Plk1 Localization and Activity
Plk1 regulates different processes in a spatially and temporally controlled manner through its specific recruitment to different substrates and subcellular structures that have been phosphorylated by appropriate priming kinases (Elia et al., 2003a). The prevailing evidence indicates that Cdk1 is a major priming kinase, albeit not the only one, able to create Plk1 docking sites (Rauh et al., 2005; Yamaguchi et al., 2005; Oshimori et al., 2006; Qi et al., 2006; Baumman et al., 2007), but there is also evidence that Plk1 is itself able to generate docking sites, particularly during late stages of mitotic progression when Cdk1 levels have fallen (Neef et al., 2003, 2007). In a recent study, Plk1 levels at both kinetochores and centrosomes were reported to be strongly reduced in response to inhibition of Plk1 activity (Lenart et al., 2007), and Plk1 has been reported to create a docking site on the kinetochore protein PBIP-1 (Kang et al., 2006). However, after exposure of cells to TAL we did not observe a strong reduction in Plk1 levels at kinetochores, although this was difficult to quantify in view of the spreading of Plk1 over chromatid arms (see above). In agreement with Peters and coworkers (Lenart et al., 2007), we also found that the centrosome localization of Plk1 during early mitosis was sensitive to Plk1 inhibition by TAL. Potential interaction partners of Plk1 at the centrosome have previously been identified (Casenghi et al., 2003; Oshimori et al., 2006), and it will be interesting to explore through what mechanism(s) Plk1 activity is required for centrosomal localization of this kinase.
Plk1 Function in Cytokinesis
Before the availability of small-molecule inhibitors, defining the precise roles of Plk1 in cytokinesis was difficult because of the early prometaphase arrest produced by Plk1 depletion. Using the advantage of chemical inhibitors to block Plk1 activity in a temporally controlled manner, we show that the addition of TAL to asynchronously growing cell populations produces widely different phenotypes, depending on the exact time of addition. In particular, when cells were already about to enter anaphase at the time of TAL addition, the inhibition of Plk1 abolished both cleavage furrow formation and ingression. In contrast, when cells had already progressed to late anaphase at the time of TAL addition, furrow formation and partial ingression could be observed, before furrows regressed and cells failed to undergo cytokinesis. Collectively, these data demonstrate that the exact cell cycle position is critical for the phenotypic consequences of Plk1 inhibition.
During anaphase, antiparallel microtubules are bundled by kinesin motors and microtubules-associated proteins, mainly PRC1 (reviewed in Glotzer, 2005). Here, we have shown that TAL treatment causes the disorganization of the central spindle and concomitant Plk1 displacement, suggesting that this phenotype may result from the lack of phosphorylation on the prominent Plk1-docking partners PRC1 and Mklp2, both of which have previously been shown to be required for Plk1 recruitment to the central spindle. We further show that Plk1 is a critical upstream activator of the ECT2-RhoA cascade that is required to initiate cytokinesis. This conclusion has been confirmed independently with other small-molecule inhibitors (Brennan et al., 2007; Petronckzi et al., 2007) and also through a chemical genetics approach using an allele-specific Plk1 inhibitor (Burkard et al., 2007). Because ECT2 can activate RhoA globally, additional mechanisms are required to restrict the equatorial localization of RhoA. Here, we show that TAL treatment inhibits both the localization of ECT2 at the central spindle and the recruitment of RhoA to the equatorial cell cortex in anaphase. Conceivably, Plk1 may control the localization and activation of RhoA by directly affecting RhoA itself or its upstream regulators, such as CYK-4 and ECT2. Clearly, TAL represents a useful tool to further explore the role of Plk1 in cytokinesis and to search for the direct substrates that are relevant to this process.
The Future of Plk1 Inhibitors
Clearly, Plk1 inhibitors such as the compound TAL described here represent powerful research tools. They hold great promise for the identification and characterization of new Plk1 substrates at the centrosomes, kinetochores, and central spindle, and this in turn will help to better understand the spatial and temporal regulation of mitotic progression and cell division. Whether Plk1 inhibitors will also prove valuable in a therapeutic context remains to be seen. Recently, the crystal structure of a Plk1 kinase domain mutant (T210V) in a complex with the nonhydrolyzable ATP analogue adenylylimidodiphosphate (AMPPNP) has been reported (Kothe et al., 2007). This and additional structural information, notably of the kinase domain in either the active or inhibitor-bound state, will be very helpful for the optimization of inhibitors, such as to achieve anti-proliferative effects in tumor cells at submicromolar drug concentrations. Another crucial step toward clinical success will be to understand how the mitotic arrest caused by the inhibition of Plk1 is linked to the induction of cell death. Finally, it will be attractive to monitor potential synergistic effect of Plk1 inhibitors with other compounds that target, for example, the mitotic spindle apparatus.
Supplementary Material
ACKNOWLEDGMENTS
We thank the project team at Bayer Schering Pharma for their contribution and Anja Wehner for technical assistance. We are grateful to the “checkpoint-lab” for insightful discussions, Jorge Martinalbo for initial advice, Sabine Elowe for critical reading of the manuscript, and Thomas Gaitanos for help with data analysis. We also thank Vasiliki Sarli (University of Leipzig, Institute for Organic Chemistry, Leipzig, Germany), Athanassios Giannis (University of Leipzig, Institute for Organic Chemistry, Leipzig, Germany), Stefan Hümmer (Max-Plank Institute for Biochemistry, Chemical Genetics, Independent Research Group, Martinsried, Germany), and Thomas Mayer (Max-Plank Institute for Biochemistry, Chemical Genetics, Independent Research Group, Martinsried, Germany) for providing VS-83 compound, Robert Habedanck (Max-Plank Institute for Biochemistry, Department of Cell Biology, Martinsried, Germany) for providing Plk4 kinase, Jens Westendorf for help with the flow cytometer, and all members of the Nigg department for kindly sharing reagents and discussion. This work was supported by the Max-Planck Society.
Abbreviations used:
- GEF
guanine nucleotide exchange factor
- K-fiber
kinetochore fiber
- γ-TuRC
γ-tubulin ring complex
- PBD
Polo-box domain
- Plk1
Polo-like kinase
- Plk1 KD
Plk1 kinase dead
- Plk1 WT
Plk1 wild type
- SAC
spindle assembly checkpoint
- TAL
ZK-Thiazolidinone.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E07-05-0517) on August 1, 2007.

 The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
REFERENCES
- Andrews C., III, et al. Thiophene compounds. WO2004/014899. International patent. 2004
- Barr F. A., Sillje H.H.W., Nigg E. A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004;5:429–441. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- Baumman C., Körner R., Hofmann K., Nigg E. A. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. doi: 10.1016/j.cell.2006.11.041. [DOI] [PubMed] [Google Scholar]
- Bearss D., Vankayalapati H., Grand C. Inhibitors of polo-like kinase-1. WO2006/124996. International patent. 2006
- Berdnik D., Knoblich J. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr. Biol. 2002;12:640–647. doi: 10.1016/s0960-9822(02)00766-2. [DOI] [PubMed] [Google Scholar]
- Blangy A., Lane H., d'Herin P., Harper M., Kress M., Nigg E. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- Brennan I., Peters U., Kapoor T., Straight A. Polo-like kinase controls vertebrate spindle elongation and cytokinesis. PLoS ONE. 2007;2:e409. doi: 10.1371/journal.pone.0000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard M., Randall C., Larochelle S., Zhang C., Shokat K., Fisher R., Jallepalli P. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. USA. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmena M., Riparbelli M., Minestrini G., Tavares A., Adams R., Callaini G., Glover D. Drosophila polo kinase is required for cytokinesis. J. Cell Biol. 1998;143:659–671. doi: 10.1083/jcb.143.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casenghi M., Meraldi P., Weinhart U., Duncan P. I., Korner R., Nigg E. A. Polo-like kinase 1 regulates Nlp, a centrosome protein involved in microtubule nucleation. Dev. Cell. 2003;5:113–125. doi: 10.1016/s1534-5807(03)00193-x. [DOI] [PubMed] [Google Scholar]
- Davis-Ward R., Mook R., Neeb M., Salovich J. Pyrimidine compounds. WO2004/074244. International patent. 2004
- De Luca M., Lavia P., Guarguaglini G. A functional interplay between Aurora-A, Plk1 and TPX2 at spindle poles: Plk1 controls centrosomal localization of Aurora-A and TPX2 spindle association. Cell Cycle. 2006;5:296–303. doi: 10.4161/cc.5.3.2392. [DOI] [PubMed] [Google Scholar]
- Eckerdt F., Yuan J., Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005;24:267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Elia A., Cantley L., Yaffe M. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003a;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- Elia A., Rellos P., Haire L., Chao J., Ivins F., Hoepker K., Mohammad D., Cantley L., Smerdon S., Yaffe M. B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003b;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- Evan G., Lewis G., Ramsay G., Bishop J. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyer P., Maller J. Regulation of Xenopus Aurora A activation by TPX2. J. Biol. Chem. 2004;279:9008–9015. doi: 10.1074/jbc.M312424200. [DOI] [PubMed] [Google Scholar]
- Fischer P., Endicott J., Meijer L. Cyclin-dependent kinase inhibitors. Prog. Cell Cycle Res. 2003;5:235–248. [PubMed] [Google Scholar]
- Gandhi R., Gillespie P. J., Hirano T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr. Biol. 2006;16:2406–2417. doi: 10.1016/j.cub.2006.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M. The molecular requirements for cytokinesis. Science. 2005;307:1735–1739. doi: 10.1126/science.1096896. [DOI] [PubMed] [Google Scholar]
- Glover D. M., Hagan I. M., Tavares A.A.M. Polo-like kinases: a team that plays throughout mitosis. Genes Dev. 1998;12:3777–3787. doi: 10.1101/gad.12.24.3777. [DOI] [PubMed] [Google Scholar]
- Gruneberg U., Neef R., Li X., Chan E., Chalamalasetty R., Nigg E., Barr F. KIF14 and citron kinase act together to promote efficient cytokinesis. J. Cell Biol. 2006;172:363–372. doi: 10.1083/jcb.200511061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch A., Wehner A., Nigg E. A., Sillje H.H.W. Different Plk1 functions show distinct dependencies on Polo-Box domain-mediated targeting. Mol. Biol. Cell. 2006;17:448–459. doi: 10.1091/mbc.E05-08-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannak E., Kirkham M., Hyman A. A., Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J. Cell Biol. 2001;155:1109–1116. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill E., Clarke M., Barr F. The Rab6-binding kinesis, Rab6-KIFL, is required for cytokinesis. EMBO J. 2000;19:5711–5719. doi: 10.1093/emboj/19.21.5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y., Lin C., Ma S., Erikson R. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Natl. Acad. Sci. USA. 2002;99:1984–1989. doi: 10.1073/pnas.042689299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y., et al. Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol. Cell. 2006;24:409–422. doi: 10.1016/j.molcel.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Keen N., Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat. Rev. Cancer. 2004;4:927–936. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- Khodjakov A., Rieder C. L. The sudden recruitment of γ-tubulin to the centrosome at the onset of mitosis and its dynamic exchange throughout the cell cycle, do not require microtubules. J. Cell Biol. 1999;146:585–596. doi: 10.1083/jcb.146.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothe M., et al. Structure of the catalytic domain of human Polo-like kinase 1. 2007;46:5960–5971. doi: 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- Kueng S., Hegemann B., Peters B. H., Lipp J. J., Schleiffer A., Mechtler K., Peters J.-M. Wapl controls the dynamic association of cohesin with chromatin. Cell. 2006;127:955–967. doi: 10.1016/j.cell.2006.09.040. [DOI] [PubMed] [Google Scholar]
- Kufer T., Sillje H., Korner R., Gruss O., Meraldi P., Nigg E. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol. 2002;158:617–623. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane H., Nigg E. Antibody microinjection reveals an essential role for human polo-like kinase 1 (Plk1) in the functional maturation of mitotic centrosomes. J. Cell Biol. 1996;135:1701–1713. doi: 10.1083/jcb.135.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansing T. J., et al. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol. Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- Lenart P., Petronczki M., Steegmaier M., Di Fiore B., Lipp J., Hoffmann M., Rettig W., Kraut N., Peters J. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of Polo-like kinase 1. Curr. Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- Leung G. C., Hudson J. W., Kozarova A., Davidson A., Dennis J. W., Sicheri F. The Sak polo-box comprises a structural domain sufficient for mitotic subcellular localization. Nat. Struct. Mol. Biol. 2002;9:719–724. doi: 10.1038/nsb848. [DOI] [PubMed] [Google Scholar]
- Liu X., Erikson R. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA. 2003;100:5789–5794. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Lei M., Erikson R. Normal cells, but not cancer cells, survive severe Plk1 depletion. Mol. Cell. Biol. 2006;26:2093–2108. doi: 10.1128/MCB.26.6.2093-2108.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamazares S., Moreira A., Tavares A., Girdham C., Spruce B., Gonzalez C., Karess R., Glover D., Sunkel C. Polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991;5:2153–2165. doi: 10.1101/gad.5.12a.2153. [DOI] [PubMed] [Google Scholar]
- Losada A. Cohesin regulation: fashionable ways to wear a ring. Chromosoma. 2007;116:321–329. doi: 10.1007/s00412-007-0104-x. [DOI] [PubMed] [Google Scholar]
- Losada A., Hirano M., Hirano T. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 2002;16:3004–3016. doi: 10.1101/gad.249202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes C., et al. Inhibitors of Polo-like kinase reveal roles in spindle-pole maintenance. Nat. Chem. Biol. 2006;2:608–617. doi: 10.1038/nchembio825. [DOI] [PubMed] [Google Scholar]
- Mundt K. E., Golsteyn R. M., Lane H. A., Nigg E. A. On the regulation and function of human Polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem. Biophys. Res. Commun. 1997;239:377–385. doi: 10.1006/bbrc.1997.7378. [DOI] [PubMed] [Google Scholar]
- Neef R., Gruneberg U., Kopajtich R., Li X., Nigg E. A., Sillje H., Barr F. A. Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat. Cell Biol. 2007;9:436–444. doi: 10.1038/ncb1557. [DOI] [PubMed] [Google Scholar]
- Neef R., Klein U. R., Kopajtich R., Barr F. A. Cooperation between mitotic kinesins controls the late stages of cytokinesis. Curr. Biol. 2006;16:301–307. doi: 10.1016/j.cub.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Neef R., Preisinger C., Sutcliffe J., Kopajtich R., Nigg E. A., Mayer T. U., Barr F. A. Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J. Cell Biol. 2003;162:863–876. doi: 10.1083/jcb.200306009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg E. A. Polo-like kinases: positive regulators of cell division from start to finish. Curr. Opin. Cell Biol. 1998;10:776–783. doi: 10.1016/s0955-0674(98)80121-x. [DOI] [PubMed] [Google Scholar]
- Oshimori N., Ohsugi M., Yamamoto T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006;8:1095–1101. doi: 10.1038/ncb1474. [DOI] [PubMed] [Google Scholar]
- Palazzo R., Vogel J., Schaneckenberg B., Hull D., Wu X. Centrosome maturation. Curr. Top. Dev. Biol. 2000;49:449–470. doi: 10.1016/s0070-2153(99)49021-0. [DOI] [PubMed] [Google Scholar]
- Perola E. Minimizing false positives in kinase virtual screens. Proteins: Struct. Funct. Bioinf. 2006;64:422–435. doi: 10.1002/prot.21002. [DOI] [PubMed] [Google Scholar]
- Peters U., Cherian J., Kim J., Kwok B., Kapoor T. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat. Chem. Biol. 2006;2:618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- Petronckzi M., Glotzer M., Kraut N., Peters J. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev. Cell. 2007;12:713–725. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Qi W., Tang Z., Yu H. Phosphorylation- and polo-box-dependent binding of Plk1 to Bub1 is required for the kinetochore localization of Plk1. Mol. Biol. Cell. 2006;17:3705–3716. doi: 10.1091/mbc.E06-03-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan H., Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- Rauh N. R., Schmidt A., Bormann J., Nigg E. A., Mayer T. U. Calcium triggers exit from meiosis II by targeting the APC/C inhibitor XErp1 for degradation. Nature. 2005;437:1048–1052. doi: 10.1038/nature04093. [DOI] [PubMed] [Google Scholar]
- Rieder C., Borisy G. The attachment of kinetochores to the pro-metaphase spindle in PtK1 cells. Recovery from low temperature treatment. Chromosoma. 1981;82:693–716. doi: 10.1007/BF00285776. [DOI] [PubMed] [Google Scholar]
- Sarli V., Huemmer S., Sunder-Plassmann N., Mayer T. U., Giannis A. Synthesis and biological evaluation of novel Eg5 Inhibitors. Chem. Biol. Chem. 2005;6:2005–2013. doi: 10.1002/cbic.200500168. [DOI] [PubMed] [Google Scholar]
- Schulze V., Eis L., Wortmann O., Kosemund G., Prien G., Siemeister G., Hess-Stumpp H., Eberspächer U., Brittain D., Islam I. Meta-substituted thiazolidinones, the production thereof and their use as a medicaments. WO2006/063806. International patent. 2006
- Seong Y., Kamijo K., Lee J., Fernandez E., Kuriyama R., Miki T., Lee K. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem. 2002;277(35):32282–32293. doi: 10.1074/jbc.M202602200. [DOI] [PubMed] [Google Scholar]
- Sillje H.H.W., Nagel S., Korner R., Nigg E. A. HURP Is a Ran-Importin β-regulated protein that stabilizes kinetochore microtubules in the vicinity of chromosomes. Curr. Biol. 2006;16:731–742. doi: 10.1016/j.cub.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Steegmaier M., et al. BI 2536, a potent and selective inhibitor of Polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Strebhardt K., Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- Stucke V., Baumann C., Nigg E. Kinetochore localization and microtubule interaction of the human spindle checkpoint kinase Mps1. Chromosoma. 2004;113:1–15. doi: 10.1007/s00412-004-0288-2. [DOI] [PubMed] [Google Scholar]
- Sumara I., Gimenez-Abian J., Gerlich D., Hirota T., Kraft C., de la Torre C., Ellenberg J., Peters J. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 2004;14:1712–1722. doi: 10.1016/j.cub.2004.09.049. [DOI] [PubMed] [Google Scholar]
- Sumara I., Vorlaufer E., Stukenberg P. T., Kelm O., Redemann N., Nigg E., Peters J. The dissociation of cohesin from chromosomes in prophase is regulated by Polo-like kinase. Mol. Cell. 2002;9:515–525. doi: 10.1016/s1097-2765(02)00473-2. [DOI] [PubMed] [Google Scholar]
- Takai N., Hamanaka R., Yoshimatsu J., Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- Tsai M.-Y., Wiese C., Cao K., Martin O., Donovan P., Ruderman J., Prigent C., Zheng Y. A Ran signalling pathway mediated by the mitotic kinase Aurora A in spindle assembly. Nat. Cell Biol. 2003;5:242–248. doi: 10.1038/ncb936. [DOI] [PubMed] [Google Scholar]
- Van Vugt M., van de Weerdt B., Vader G., Janssen H., Calafat J., Klompmaker R., Wolthuis R., Medema R. Polo-like kinase-1 is required for bipolar spindle formation but is dispensable for anaphase promoting complex/Cdc20 activation and initiation of cytokinesis. J. Biol. Chem. 2004;279:36841–36854. doi: 10.1074/jbc.M313681200. [DOI] [PubMed] [Google Scholar]
- van Vugt M.A.T.M., Medema R. H. Getting in and out of mitosis with Polo-like kinase-1. Oncogene. 2005;24:2844–2859. doi: 10.1038/sj.onc.1208617. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Goto H., Yokoyama T., Sillje H., Hanisch A., Uldschmid A., Takai Y., Oguri T., Nigg E., Inagaki M. Phosphorylation by Cdk1 induces Plk1-mediated vimentin phosphorylation during mitosis. J. Cell Biol. 2005;171:431–436. doi: 10.1083/jcb.200504091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuce O., Piekny A., Glotzer M. An ECT2-centraspindlin complex regulates the localization and function of RhoA. J. Cell Biol. 2005;170:571–582. doi: 10.1083/jcb.200501097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}