

Abstract
In a previous study, E47 HepG2 cells that overexpress human CYP2E1 were shown to be more sensitive to cisplatin than C34 cells that do not express CYP2E1. In this study, we found that this sensitivity was due to an earlier activation of ERK in the E47 cells than in C34 cells. Glutathione depletion by L-buthionine sulfoximine (BSO) enhanced cisplatin cytotoxicity via increasing production of reactive oxygen species (ROS) and activation of ERK. In contrast, elevation of glutathione by glutathione ethyl ester (GSHE) decreased cisplatin/BSO cytotoxicity by decreasing ROS production and ERK activation. Inhibition of ERK activation by U0126 protected against cisplatin/BSO cytotoxicity via inhibiting ROS production but not restoring intracellular glutathione content. Examination of the mode of cell death showed that U0126 inhibited cisplatin-induced necrosis but not apoptosis. Cisplatin induced apoptosis was caspases-dependent; BSO switched cisplatin-induced apoptosis to necrosis via decreasing activities of caspases, and GSHE switched cisplatin/BSO induced necrosis back to apoptosis through maintaining activities of caspases. Similar to GSHE, U0126 partially switched cisplatin/BSO induced necrosis to apoptosis via restoring activities of caspases. Cisplatin lowered levels of thioredoxin, especially in the presence of BSO. Although U0126 failed in restoring intracellular glutathione levels, it restored thioredoxin levels which maintain activities of the caspases. These results suggest that thioredoxin can replace glutathione to promote the active thiol redox state necessary for caspase activity, and thus glutathione and thioredoxin regulate the mode of cisplatin toxicity in E47 cells via redox regulation of caspase activities.
Keywords: cisplatin; extracellular signal-regulated kinases (ERK); CYP2E1; necrosis; apoptosis; glutathione; L-Buthionine-[R,S]-sulfoximine (BSO); thioredoxin
Introduction
Multiple mechanisms have been suggested in causing cisplatin-induced cytotoxicity, including intracellular accumulation of cisplatin, impaired DNA-repair processes, and decreased levels of cisplatin inactivating factors, such as glutathione and metallothioneins (1-2). Increased production of reactive oxygen species (ROS) appears to be closely associated with cisplatin-induced cytotoxicity (3-7). Glutathione depletion induces accumulation of ROS, leading to cell death (8). L-Buthionine-[R,S]-sulfoximine (BSO), which lowers cellular glutathione levels, enhances cisplatin-induced cytotoxicity to primary cultured rat hepatocytes and renal tubular epithelial cells, while L-cysteine, the precursor of GSH, protects against cisplatin cytotoxicity (9-10). Cytochrome P450 2E1 (CYP2E1), an active producer of ROS, enhances cisplatin-induced cytotoxicity (11). Administration of free radical scavengers or antioxidants inhibits the cisplatin-induced nephrotoxicity in animal models (3-5).
ROS contribute to cell death partly through effects on various cellular signaling pathways including the mitogen-activated protein kinase (MAPK) pathway (12-15). MAPK family members, including extracellular signal-regulated kinases (ERK1/2), c-JUN NH2-terminal protein kinase (JNK), and P38 MAPK, respond to several extracellular stimuli. These MAPK members participate in integrating extracellular signals to regulate cell proliferation, differentiation, cell survival and apoptosis (16). MAPK signaling pathways have been suggested to play a role in cisplatin-mediated cytotoxicity such as ERK in Hela cells (17) and human glioma cells (18), JNK/P38 MAPK in ovarian carcinoma cells (19), P38 MAPK in small cell lung carcinoma cells (20). Thus, depending on the conditions and cell type, different MAPK may play a role in cisplatin cytotoxicity.
E47 HepG2 cells that overexpress human CYP2E1 are more sensitive to cisplatin than C34 HepG2 cells that do not express CYP2E1 (11). CYP2E1 is an active producer of ROS (21), it can produce synergistic toxic effects with other hepatotoxins such as endotoxin (22, 23), and CYP2E1-mediated cytotoxicity includes both apoptotic and necrotic modes of cell death (24-26). In the present study, we investigated the role of MAPKs and of glutathione in cisplatin-induced cytotoxicity and in regulating the mode of cell death induced by cisplatin, including apoptosis and necrosis, in CYP2E1-overexpressing E47 cells.
Materials and Methods
Cell cultures and cytotoxicity determination.
E47 cells are HepG2 cells that were transfected with pCI-CYP2E1 to overexpress human CYP2E1 followed by establishing stable cell lines via selection for G418 resistance, whereas C34 cells are HepG2 cells that were transfected with the pCi vector only and do not express CYP2E1 (27). Cells were cultured in minimal essential medium (MEM) supplemented with 10% fetal bovine serum plus 100 units/ml penicillin and 100 μg/ml streptomycin in 5% CO2 at 37°C. Before the experiment, cells were trypsinized and inoculated into 24-well or 6-well plates, at an amount of 1 × 105 (24-well) or 4 × 105 (6-well) cells/well. Cells were cultured in the plates for 2 days, and then varying concentrations of cisplatin (Sigma) were added. In some experiments, cisplatin was added after pretreatment of the E47 cells with 40 μM BSO (Sigma) to remove glutathione, or 5 mM glutathione ethyl ester (GSHE; Calbiochem, Temecula, CA), or 5 mM deferoxamine mesylate (DFO; Sigma) to increase glutathione, an iron chelator, or 0.1 mM Trolox (Calbiochem, Temecula, CA), an antioxidant, or 50μM MnTMP (Sigma), a mimic of superoxide distmase (SOD), or 50 μM Z-VAD-FMK, a pan caspase inhibitor, or 20 μM U0126, a specific inhibitor of ERK, or 20 μM SB203580, a specific inhibitor of P38 MAPK, or 20 μM SP600125, a specific inhibitor of JNK. Cisplatin-induced cytotoxicity was determined by reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as described previously (27).
Measurement of Intracellular ROS production
Fluorescence spectrophotometry was used to measure the levels of ROS, with 2′,7′-dichlorofluorescein diacetate (DCF-DA, Sigma) as the probe (28). DCF-DA readily diffuses through the cell membrane and is enzymatically hydrolyzed by intracellular esterases to the nonfluorescent DCFH, which can then be rapidly oxidized to highly fluorescent DCF in the presence of ROS. Cells incubated in medium alone or cells incubated under different treatment conditions were incubated with 5 μM DCF-DA in MEM for 30 min at 37°C in the dark. The cells were washed in PBS, trypsinized, and resuspended in 3 ml of PBS, and the intensity of fluorescence was immediately read in a fluorescence spectrophotometer (Perkin-Elmer 650−10S, Hitachi, Ltd.) at 503 nm for excitation and at 529 nm for emission. Results are expressed as arbitrary fluorescence units per mg protein.
Measurement of intracellular glutathione content
Cells were mixed with 5% trichloroacetic acid (TCA) and incubated at 4°C for 30 min to extract intracellular glutathione. The TCA supernatant was used to measure the glutathione content following the method of Tietze (29). Glutathione dissolved in 5% TCA solution was used as standard. Results are expressed as ng glutathione per mg protein.
Caspase-3, -8, and 9 activity
Caspase-3, -8 and -9 activity was determined by measuring enzymatic cleavage of the substrates Ac-DEVD-AMC, Z-IETD-AFC, and Ac-LEHD-AFC (Calbiochem, Temecula, CA), respectively. The substrates (final concentration 0.2 mM) were dissolved in assay buffer containing 20 mM HEPES (pH7.5), 10% glycerol, and 2 mM dithiothreitol. 10 μl of cell lysate was added to this assay buffer (0.2 ml) and incubated at 30°C overnight. The fluorescence associated with the released AMC (excitation at 380 nm, emission at 460 nm) or AFC (excitation at 400 nm, emission at 505 nm) was assayed in a PerkinElmer spectrofluorometer (Wellesley, MA). The data are expressed as arbitrary fluorescence units per milligram of protein.
Western blots
Cell lysate proteins (40 μg) were resolved by electrophoresis using SDS–PAGE and then transferred to nitrocellulose membranes for blotting with antibodies specific for pERK, ERK, pP38 MAPK, P38 MAPK, pJNK, JNK, TRX (Santa Cruz Biotechnology, Santa Cruz, CA), CYP2E1 (a gift from Dr. Jerome Lasker, Hackensack Biomedical Research Institute, Hackensack, NJ), or β-actin (Sigma) antibody, followed by horseradish peroxidase-conjugated secondary antibody. Proteins were visualized by radiography using the ECL Western detection reagent (Amersham Bioscience, Piscataway, NJ). Blots were quantified using the UN-SCAN-IT automated digitizing system version 5.1 (Silk Scientific, Inc.), and the results expressed as the ratio of p-ERK/ERK.
Fluorescence microscopy
Nuclear morphology of the cells was examined to identify the mode of cell death. Nuclear morphology with characteristic features of apoptosis or necrosis is considered a very reliable determinant for distinguishing the type of cell death in cell culture (30, 31). The adherent cells were trypsinized and suspended in PBS along with the floating cells, and the collected cells were divided into two tubes. One tube was stained with the cell membrane-impermeant DNA-specific dye propidium iodide (PI; Sigma) for 5 min at 37°C (final concentration 0.5 μM), and then the cells were observed under the fluorescence microscope using the appropriate filter to examine necrotic cells. Necrosis was determined on the basis of positive (red color) PI staining of the nucleus, which is indicative of loss of membrane integrity (32). The other tube was fixed with cold methanol, the cells were washed in PBS, and the above procedures with PI repeated to examine apoptotic cells (33). Condensed or fragmented nuclei were counted as apoptotic cells. Apoptotic or necrotic cells were quantitated in randomly selected 5 high-power fields. In some experiments, PI was added directly into culture plate wells to stain the adherent cells.
In order to further confirm apoptosis, Annexin V-FITC Apoptosis Detection Kit (BioVision, Mountain View, CA) was used to stain the cells followed by observation under the fluorescence microscope using the appropriate filter to examine apoptotic cells. Cells that have bound Annexin V-FITC show green staining in the plasma membrane.
LDH leakage
Lactate dehydrogenase (LDH) leakage was assayed using the cytotoxicity detection kit (Roche Diagnostics GmbH, Penzberg, Germany). The cytotoxicity was expressed as percentage LDH release: 100% × LDHout/(LDHout + LDHin).
SiRNA study
Thioredoxin siRNA, scrambled siRNA and transfection reagent were purchased from Santa Cruz Biotechnology, Santa Cruz, CA. MEK1 and MEK2 siRNA kits were purchased from Cell Signaling Technology, Beverly, MA. Transfections were performed according to the protocols provided by the respective commercial company.
Statistics
Statistical evaluation was carried out by analysis of variance (ANOVA) followed by Student-Newman-Keuls post hoc test. Results were considered statistically different if P was less than 0.05.
Results
Cell death induced by cisplatin is ERK-dependent
In order to examine the role of MAPKs in cisplatin cytotoxicity, E47 and C34 cells were pretreated for 1 h with specific inhibitors of P38 MAPK, JNK, and ERK (SB203580, SP600125, and U0126, respectively), and then varying concentrations of cisplatin were added. After incubation for 24 h, MTT assays were performed to examine cytotoxicity. In E47 cells, concentration-dependent cisplatin cytotoxicity was observed as 0.5 mM of cisplatin led to more than 80% loss of cell viability, while 0.25 mM of cisplatin led to 50% loss of cell viability (Fig. 1 A). In C34 cells, 0.5 mM of cisplatin caused 50% loss of cell viability, but 0.25 mM of cisplatinn did not cause loss of cell viability (Fig. 1 A). Among the three specific inhibitors of MAPK, only the ERK inhibitor U0126 decreased the loss of cell viability induced by cisplatin in both E47 and C34 cells (Fig. 1 A), suggesting that ERK, but not P38 MAPK and JNK, played an important role in the cisplatin cytotoxicity. The inhibition of toxicity by U0126 also suggests that cisplatin may activate ERK under these experimental conditions. Phosphorylation of ERK (ERK activation) was assayed by western blotting analysis. Because 0.25 mM cisplatin led to cell death in E47 cells but not in C34 cells, 0.25 mM of cisplatin was selected for all the following experiments. As shown in Fig. 2 A, cisplatin induced a sustained ERK activation in both C34 and E47 cells, but in E47 cells ERK activation occurred earlier. For example, after 1 or 4 h of cisplatin treatment, ERK was already activated (pERK/ERK ratios of 0.08 and 0.15, respectively) and with further activation at 8, 16 and 24 h (pERK/ERK ratios of 0.3, 0.5 and 0.85, respectively) (Fig. 2 A). In C34 cells, no activated ERK was observed at the earlier times (1 and 4 h); ERK activation was observed at 8 h (pERK/ERK ratios of 0.07) but this was weaker than that in E47 cells. At later times (16 and 24 h), ERK activation was comparable to that found in E47 cells (Fig. 2 A). On the contrary, no activation of P38 MAPK or of JNK was observed through 24 h of treatment with cisplatin (Fig 2 A), which probably explains the lack of effect of SB203580 and SP600125 on cisplatin toxicity (Fig 1A). Cisplatin did not affect CYP2E1 expression in the E47 cells throughout the treatment time (Fig 2 A). U0126 inhibited this ERK activation in both C34 and E47 cells after 24 h incubation (Fig. 2 B). This suggests that U0126 inhibited cisplatin cytotoxicity via inhibiting ERK activation. In order to test if earlier activation of ERK contributes to the sensitivity of E47 cells to cisplatin, U0126 was added at different times after cisplatin, and then cell viability was measured. As shown in Fig. 1 B, addition of U0126 at 1−8 h a fter cisplatin prevented cisplatin cytotoxicity in C34 and E47 cells. However, addition of U0126 at 16 h after cisplatin prevented cisplatin cytotoxicity in C34 cells but not in E47 cells. These results are consistent with the notion that sustained ERK activation contributed to cisplatin cytotoxicity both in E47 and C34 cells, and an earlier ERK activation may have caused the E47 cells to be more sensitive to cisplatin.
Fig.1.

(A) Effects of U0126, SB203580, and SP600125 on cisplatin-induced loss of cell viability. Cells were seeded into 24-well plates at 1×105 cells/well and after 2 days' preincubation, 20 μM of SB203580, 20 μM of SP600125, or 20 μM of U0126, or DMSO control were added for 1 h, following by addition of varying concentrations of cisplatin, and readdition of SB203580, SP600125, and U0126. After incubation for 24 h at 37°C, MTT assays were performed to evaluate cell viability. Data are shown as means±SD. * P<0.05, compared with CP group. CP, cisplatin; SB, SB203580; SP, SP600125; U0, U0126.
(B) Effect of the timed addition of U0126 on cisplatin-induced loss of cell viability. Cells were seeded into 24-well plates at 1×105 cells/well and after 2 days' preincubation, 20 μM of U0126 or DMSO control was added 1 h before (CP U-1), concurrent with (CP U+0), or 1, 4, 8, or 16 h (CP U+1, +4, +8, +16, respectively) after cisplatin treatment, MTT assays were performed to evaluate cell viability after cisplatin treatment for 24 h at 37ºC . Data are shown as means±SD. ** P<0.01, compared with Control; ## P<0.01, compared with CP group. CP, cisplatin.
Fig. 2.

A. Effect of cisplatin on activation of MAPKs. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 0.25 mM of cisplatin was added. After incubation for different times at 37°C, cells were collected, washed, and then sonicated. Cell lysates were used for Western blotting analysis of phosphorylated and total levels of MAPK.
B. Effect of cisplatin concentration on the activation of ERK in the absence or presence of U0126. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 20 μM of U0126 or DMSO control was added for 1 h, following by addition of varying concentrations of cisplatin and readdition of U0126. After incubation for 24 h at 37°C, cells were collected, washed, and then sonicated. Cell lysates were used for Western blotting analysis to detect phosphorylated ERK (activated ERK) and total ERK. CP, cisplatin; U0, U0126.
Effect of glutathione de pletion on cisplatin toxicity and activation of ERK
E47 cells have elevated levels of glutathione, which appears to reflect a metabolic adaptation to the CYP2E1-generated oxidative stress (34). Depletion of glutathione can induce cell toxicity and death (8). The relationship between glutathione and ERK activation in the actions of cisplatin in E47 cells was therefore examined. Cells were pretreated with 40 μM of BSO (an inhibitor of glutamate cysteine ligase) overnight to deplete intracellular glutathione, and then 0.25 mM of cisplatin was added. As shown in Fig. 3 A, cisplatin alone induced about a 40% loss of cell viability in E47 cells, but BSO plus cisplatin induced a 95% loss of cell viability. BSO alone under these conditions had no effect on cell viability (data not shown). U0126 partially protected against this BSO plus cisplatin enhanced cell death. BSO enhanced cisplatin-induced ERK activation, and U0126 prevented this BSO-enhanced ERK activation (Fig. 3 B). Pretreatment with 5 mM of GSHE for 1 h blunted the E47 cell death induced by cisplatin alone or the enhanced cell death of cisplatin in combination with BSO (Fig. 3 C). ERK activation was also inhibited by GSHE (Fig. 3 D). Thus glutathione loss potentiated ERK activation by cisplatin in E47 cells. We next examined the glutathione level in order to validate the actions of BSO. As shown in Fig. 4 A, after treatment with 0.25 mM of cisplatin, E47 intracellular glutathione content decreased about 25% after 4 h of cisplatin treatment, 40% at 8 h, and 60% at 16 h. However, to our surprise, U0126 had no effect on the glutathione level (Fig 4A) even though it protected against cisplatin toxicity (Fig. 1A). This inidicates that the protection by U0126 is not related to elevating glutathione. An 85% loss of glutathione was caused by BSO alone or cisplatin plus BSO, but U0126 did not increase glutathione levels (Fig. 4 A), even though the U0126 partially protected against the potentiated toxicity produced by cisplatin plus BSO (Fig 3 A).
Fig. 3.

A. Effects of U0126 on BSO-enhanced cisplatin cytotoxicity. E47 cells were seeded into 24-well plates at 1×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin and BSO re-addition. 20 μM of U0126 or DMSO control was added 1 h prior to cisplatin. MTT assays were performed to evaluate cytotoxicity. * P<0.05, compared with Control group; # P<0.05, compared with CP group; $ P<0.05, compared with BSO+CP group.
B. Effects of U0126 on BSO-enhanced ERK activation. E47 cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin with BSO re-addition. 20 μM of U0126 or DMSO control was added 1 h prior to cisplatin. After incubation for 16 h at 37°C, cells were collected, washed, and then sonicated. Cell lysates were applied for Western blotting analysis to detect phosphorylated ERK and total ERK.
C. Effects of GSHE on BSO-enhanced cisplatin cytotoxicity. The procedures for cell and drug treatment were similar to (A) except for addition of 5mM GSHE instead of U0126. $ P<0.05, compared with Control; * P<0.05, compared with CP group; && P<0.01, compared with CP group; ## P<0.01, compared with CP+BSO group.
D. Effects of GSHE on BSO-enhanced ERK activation. The procedures for cell and drug treatment were similar to (B) except for addition of 5mM GSHE instead of U0126. CP, cisplatin; U, U0126; GSHE, glutathione ethyl ester.
Fig.4.

Effects of U0126 on glutathione levels and ROS production induced by cisplatin alone or cisplatin plus BSO.
A. Effect of U0126 on glutathione levels. E47 cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin with BSO re-addition. 20 μM of U0126 or DMSO control was added 1 h prior to cisplatin. After incubation at 37°C for 4, 8, and 16 h, respectively, glutathione levels were measured as described in MATERIALS AND METHODS. ** P<0.01, compared with CP group.
B. Effect of U0126 on ROS accumulation. The procedures for cell and drug treatment were the same as (A). ROS production was measured as described in MATERIALS AND METHODS. * P<0.05 and ** P<0.01, compared with CP group; ## P<0.01, compared with CP+BSO group.
C. Effect of GSHE on ROS accumulation. The procedures for cell and drug treatment were similar to (A) except that 5 mM GSHE instead of U0126 was used. * P<0.05, compared with Control; $ P<0.05, compared with CP group; ## P<0.01, compared with CP+BSO group. CP, cisplatin; U, U0126.
Besides glutathione, ROS play an important role in cisplatin cytotoxicity, therefore we examined the relationship between ROS accumulation and ERK activation. As shown in Fig. 4 B, cisplatin did not increase ROS accumulation at 4 h, but it increased ROS 2-fold at 8 h and 5-fold at 16 h. The increase in ROS production may play a role in the cisplatin-induced toxicity and ERK activation. ROS accumulation caused by cisplatin in C34 cells did not increase at 8 h (data not shown), indicating that, like ERK activation, cisplatin elevation of ROS accumulation occurred earlier in E47 cells than C34 cells. U0126 blocked ROS accumulation caused by cisplatin (Fig. 4 B), even though glutathione levels were not restored (Fig 4 A). These results suggest that ERK activation and ROS accumulation are associated with each other. BSO alone did not lead to ROS accumulation (data not shown), but it enhanced cisplatin-induced ROS accumulation at 8 h and 16 h (Fig. 4 B). The enhanced ROS accumulation found with cisplatin plus BSO may play a role in the enhanced toxicity and elevated activation of ERK found with cisplatin plus BSO. U0126 blocked this enhanced ROS production (Fig. 4 B) even though U0126 did not restore the low glutathione levels (Fig. 4 A). Increasing glutathione by pretreatment with GSHE blocked the ROS accumulation caused by cisplatin alone or in combination with BSO (Fig 4 C), which probably is important for the protection by GSHE against cisplatin toxicity (Fig. 3 C) and activation of ERK (Fig. 3 D).
To further support a connection between ERK activation and ROS production, MEK1/2 siRNA instead of U0126 was used to inhibit ERK activation. In the presence of a scrambled siRNA, cisplatin activated ERK (Fig 5A). This ERK activation was inhibited partially by MEK1 siRNA (siRNA1) or MEK2 siRNA (siRNA2) (Fig 5A). Cisplatin-induced accumulation of ROS was decreased partially (Fig 5B) and cell viability was increased partially (Fig 5C) by siRNA1 and siRNA2. These results, along with the U0126 results, are consistent with the notion that ERK activation promoted ROS production and cytotoxicity.
Fig.5.
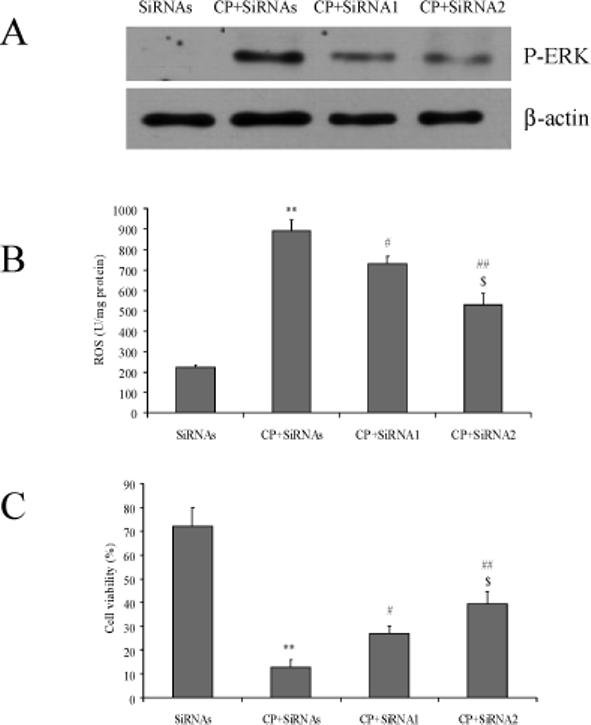
A. Effects of MEK1/2 SiRNA on ERK activation. 100 nM SiRNA was added and E47 cells were incubated for 24 h, followed by addition of 0.5 mM of CP for 16 h. Cells were collected, washed, and then sonicated. Cell lysates were used for Western blotting analysis of phosphorylated and total levels of ERK. SiRNAs, scramble SiRNA; SiRNA1, MEK1 SiRNA; SiRNA2, MEK2 SiRNA.
B. Effects of MEK1/2 SiRNA on ROS production. The procedures for cell and SiRNA treatment were the same as (A). ROS production was measured as described in MATERIALS AND METHODS. ** P<0.01, compared with the SiRNAs group; # P<0.05 and ## P<0.01, compared with CP+SiRNAs group; $ P<0.05, compared with CP+SiRNA1.
C. Effects of MEK1/2 SiRNA on cell viability. The procedures for cell and SiRNA treatment were the same as (A). MTT assays were performed to measure cell viability. ** P<0.01, compared with SiRNAs group; # P<0.05 and ## P<0.01, compared with the CP+SiRNAs group; $ P<0.05, compared with CP+SiRNA1.
Role of ERK activation in cisplatin-induced cell necrosis and apoptosis
Cisplatin induces both necrosis and apoptosis (35, 36). Since the MTT assay cannot distinguish the mode of cell death, experiments with PI staining were carried out to evaluate the mode of cell death caused by cisplatin. Necrosis was determined on the basis of positive PI staining (red color) of the nucleus in non-permeabilized cells, indicative of the loss of membrane integrity (32). As shown in Fig. 6 A, after exposure to 0.25mM of cisplatin alone, no necrotic cell death was detected at 4 and 8 h. About 15% necrotic cells were detected after exposure for 16 h to cisplatin. Treatment with BSO plus cisplatin caused 20% necrosis at 8 h and 95% necrosis at 16 h. These results were confirmed by a LDH leakage assay; thus, LDH leakage, reflective of necrosis, was detected at 16 h but not at 4 or 8 h after cisplatin treatment, and treatment with BSO plus cisplatin elevated LDH leakage at 16 h (Fig 6 B). Thus, cisplatin toxicity is largely apoptotic but switches to necrosis when glutathione is depleted. U0126 decreased the cell necrosis induced by cisplatin alone or especially in combination with BSO (Fig. 6A, 6B).
Fig.6.
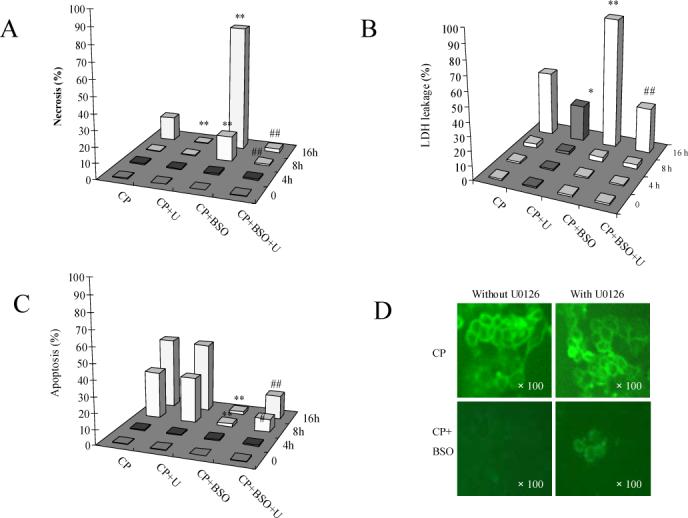
A. Effects of U0126 on cisplatin-induced necrosis. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin with BSO re-addition. 20 μM of U0126 or DMSO control was added 1 h prior to cisplatin. After incubation at 37°C for 16 h, cell necrosis was measured by PI staining as described in MATERIALS AND METHODS. CP, cisplatin; U, U0126. ** P<0.01, compared with CP group; ## P<0.01, compared with CP+BSO group.
B. LDH leakage. The procedures for cell and drug treatment were similar to (A) except that cells were seeded into 24-well plates at 1×105 cells/well. LDH levels in the medium and the cell extract were measured as described in MATERIALS AND METHODS. CP, cisplatin; U, U0126. * P<0.05 and ** P<0.01, compared with CP group; ## P<0.01, compared with CP+BSO group.
C. Effects of U0126 on cisplatin-induced apotosis. The procedures for cell and drug treatment were same as (A). Apoptosis was measured as described in MATERIALS AND METHODS. CP, cisplatin; U, U0126. ** P<0.01, compared with CP group; ## P<0.01, compared with CP+BSO group.
D. Annexin-V staining. The procedures for cell and drug treatment were the same as (A). Annexin-V staining was measured as described in MATERIALS AND METHODS.
Apoptotic cells were detected after the cells were fixed and permeabilized followed by PI staining. Condensed or fragmented nuclei were counted as apoptotic cells. As shown in Fig. 6 C, after exposure to 0.25mM of cisplatin, no apoptotic cells were detected at 4 h, but 30% apoptotic cells were detected at 8 h and 45% at 16 h. Pretreatment with U0126 had no effect on cisplatin-induced apoptosis (Fig. 6C). To further confirm this result, we applied Annexin V staining as another method for apoptosis detection. Cells that have bound Annexin V-FITC show green staining in the plasma membrane. Cisplatin alone induced Annexin V staining and U0126 had no effect on this Annexin V staining induced by cisplatin (Fig. 6D).
Cisplatin-induced apoptosis is caspase-dependent (20, 35). Therefore, activities of caspase-3, -8 and -9 were determined in the absence and presence of BSO and/or U0126. The activities of caspase-3, -8, and -9 increased at 4 h, peaked at 8 h and remained elevated at 16 h after cisplatin treatment, as compared to Controls (Fig. 7). U0126 either did not inhibit or actually increased activities of the three caspases in the presence of cisplatin (Fig. 7). Taken as a whole, these results show that cisplatin-induced toxicity in the E47 cells is mainly apoptotic and associated with activation of caspases, and in contrast to the inhibition of cisplatin-induced necrosis by U0126, cisplatin-induced apoptosis or activation of caspases is not inhibited by U0126.
Fig. 7.

Effect of cisplatin with or without BSO or U0126 on the activation of caspase-3, -8, and -9. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin with BSO re-addition. 20 μM of U0126 or DMSO control was added 1 h prior to cisplatin. After incubation at 37°C for 4, 8, or 16 h, cells were collected, washed, and then sonicated. Cell lysates were used to measure activities of caspase-3, -8, and -9 as described in MATERIALS AND METHODS. CP, cisplatin; U, U0126. *P<0.05 and **P<0.01, compared with Control; & P<0.05 and && P<0.01, compared with CP group; # P<0.05 and ## P<0.01, compared with CP+BSO group.
The above experiments were carried out after incubating E47 cells with cisplatin for up to 16 h. In Fig. 1 A, using an MTT assay, we observed cisplatin-induced loss of cell viability after 24 h of treatment which is prevented, in part, by U0126. In order to validate the MTT data, we evaluated apoptosis and necrosis by PI staining after 24 h of treatment of E47 cells with cisplatin. At 24 h, cisplatin alone caused 40% necrosis and 50% apoptosis; U0126 decreased the cell necrosis to 5%, but U0126 had not effect on the cell apoptosis. These data suggest that the partial protection by U0126 against the loss of cell viability observed from MTT reduction (Fig 1A) reflects inhibition of cisplatin-induced necrosis by U0126, whereas the inability of U0126 to completely prevent the loss of cell viability by cisplatin may reflect the inability of U0126 to prevent cisplatin-induced apoptosis.
As mentioned above, cisplatin induced much more necrosis in the presence of BSO (Fig. 6 A and B). We evaluated the effect of BSO on the cisplatin-induced apoptosis. After treatment of E47 cells with BSO plus cisplatin, almost no apoptosis was detected at the three time points studied (Fig. 6 C). In contrast to the Annexin V staining observed with cisplatin alone, no such apoptotic staining was found in BSO plus cisplatin-treated cells (Fig. 6 D). Thus, the BSO treatment switches cisplatin toxicity from an apoptotic mode of cell death to a necrotic mode. This switching may be due to the decrease in caspase activities found in the cisplatin plus BSO treated cells, in contrast to the increase in caspase activities found in cisplatin alone treated cells (Fig. 6). Interestingly, U0126 partially switched necrosis induced by cisplatin plus BSO back to apoptosis, as shown in Figure 6 C and D; no apoptosis was detected after treatment with BSO plus cisplatin, but U0126 pretreatment led to about 10% apoptotic cells being detected at 8 h and 20% at 16 h (Fig 6C). While BSO inhibited the activities of the three caspases, U0126 partially restored these activities at the three time points, especially caspase-3 activity (Fig. 7).
To evaluate the role of caspases in the cisplatin toxicity, the effect of a pan-caspase inhibitor, Z-VAD-FMK, was studied. Indeed, Z-VAD-FMK inhibited the apoptosis produced by cisplatin alone (Fig. 8 panels 1−2), however, Z-VAD-FMK had no effect on the PI staining produced by cisplatin plus BSO (Fig. 8 panels 3−4), indicating either the latter reflected necrosis, not apoptosis, or caspase-independent apoptosis.
Fig. 8.
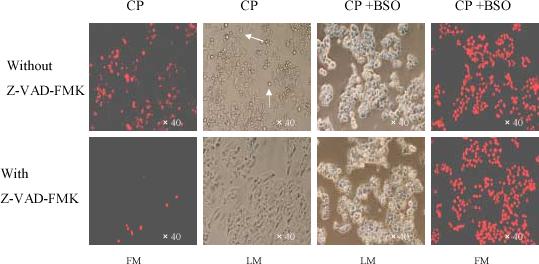
Effect of Z-VAD-FMK on E47 cell necrosis or apoptosis induced by cisplatin or cisplatin plus BSO. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin with BSO re-addition. 50 μM of Z-VAD-FMK or DMSO control was added 1 h prior to cisplatin. After treatment at 37°C for 16 h, 0.5 μM of PI was added into the culture wells for 5 min iincubation, and then the adherent cells were either observed under the fluorescence microscope using the appropriate filter to examine necrotic cells, or under the light microscope to observe cell morphology. Arrows show apoptotic bodies. The morphology shown is magnified 40-fold. CP, cisplatin; FM, fluorescence microscopy; LM, light microscopy.
Caspases are cysteine proteases with a cysteine residue at the active site, and modification of the cysteine residue abrogates their catalytic activities (37). GSH can regulate the redox state within cells by forming reversible mixed disulphides with protein thiols to prevent irreversible oxidation of cysteine residues under oxidative stress conditions (38). Therefore, the possibility was considered that after GSH depletion by BSO, irreversible oxidation of cysteine residues under oxidative stress induced by cisplatin could inactivate caspases, and explain why apoptosis was not detected in the cisplatin plus BSO treated cells and the switch of cell death to necrosis. In contrast, GSHE pretreatment may prevent oxidation of or restore cysteine residues and thereby switch necrosis by cisplatin plus BSO to apoptosis. Indeed, the activities of caspases which were inhibited by cisplatin plus BSO were elevated after GSHE treatment, especially caspase-3 (Fig. 9 A). This recovery of caspase activities resulted in a switch of the cisplatin plus BSO toxicity from necrosis to apoptosis in the presence of GSHE e.g. note the increase in apoptosis by cisplatin plus BSO plus GSHE compared to cisplatin plus BSO (Fig 9B, bar graphs 2 and 6) and the decrease in necrosis found with cisplatin plus BSO plus GSHE compared to cisplatin plus BSO (Fig. 9 C, bar graphs 2 and 6).
Fig. 9.

Effect of GSHE and non-thiol antioxidants on apoptosis or necrosis induced by BSO plus cisplatin. A. Caspase activities: The procedures for cell and drug treatment were similar to Fig.7 except that 5 mM of GSHE instead of U0126 was used. The activities of caspase-3, -8, and -9 were measured as described in MATERIALS AND METHODS. ** P<0.01, compared with CP+BSO group.
B. Apoptosis and C. Necrosis: The procedures for cell and drug treatment were similar to Fig.6 except that 5mM of GSHE, 50 μM of MnTMP, 100 μM of Trolox, or 5mM of DFO instead of U0126 were used. PI staining to assess apoptosis (B) or necrosis (C) was performed as described in MATERIALS AND METHODS. ** P<0.01, compared with BSO+CP group; $$ P<0.01, compared with BSO group.
D. GSH levels: The procedures for cell and drug treatment were similar to Fig.4 A except that 5mM of GSHE, 50 μM of MnTMP, 100 μM of Trolox, or 5mM of DFO were used instead of U0126. Measurement of GSH was performed as described in MATERIALS AND METHODS. Note: Data are shown as GSH levels relative to the BSO group instead of the Control group (GSH levels were about 90% lower after BSO treatment). * P<0.05, compared with BSO group. CP, cisplatin; DFO, deferoxamine mesylate; GSHE, glutathione ethyl ester.
Results in Fig 4 showed that cisplatin increased ROS production, and this increase could be further elevated by cisplatin plus BSO treatment. U0126 and GSHE both lowered the elevated ROS produced by cisplatin alone or cisplatin plus BSO (Fig. 4), which may account for their protection against the cisplatin and cisplatin plus BSO toxicity. To more directly evaluate the role of ROS in cisplatin toxicity, especially the mode of cisplatin toxicity, the effect of several antioxidants was evaluated. The necrosis produced by the combination of cisplatin plus BSO was prevented by treatment with a scavenger of superoxide (MnTMP), a synthetic vitamin E analogue which is a powerful inhibitor of lipid peroxidation (Trolox), and by an iron chelator (DFO) (Fig. 9 C). These antioxidants were as effective as GSHE in lowering the cisplatin plus BSO necrosis. Thus oxidative stress plays a key role in the cisplatin-induced necrosis. However, the antioxidants did not mimic GSHE in promoting a switch of cisplatin plus BSO toxicity from necrosis to apoptosis (Fig. 9 B), suggesting that GSHE is not promoting this switch by acting as an antioxidant. The likely reason that GSHE could restore the apoptosis might be related to the increased intracellular glutathione levels by GSHE but not by the other antioxidants (Fig. 9 D).
However, U0126 had no effect on intracellular glutathione content (Fig. 4 A), although it restored or promoted activities of caspases in the presence of cisplatin plus BSO (Fig. 7). It is proposed that there must be other thiol resources which can replace glutathione to adjust the intracellular redox status. We examined the levels of thioredoxin (TRX), a redox active protein which also regulates cellular redox status (39). As shown in Fig. 10 A, TRX expression decreased at 8 h and especially at 16 h after treatment of E47 cells with cisplatin. BSO strikingly decreased TRX expression in the presence of cisplatin even after only 4 h of incubation (Fig 10A). U0126 restored this decreased TRX expression induced by cisplatin alone or in combination with BSO (Fig. 10 A). GSHE could also promote the recovery of the inhibited TRX expression produced by cisplatin alone or in combination with BSO (Fig. 10 B). It is conjectured that in this model, TRX might replace glutathione as a thiol reductant to maintain caspases in their active state when glutathione was depleted by BSO, and that the ability of U0126 to prevent necrosis but not apoptosis is due to the U0126-mediated increase in TRX levels. Note that there is an inverse relationship between ERK activation and levels of TRX i.e. TRX levels are low when ERK is activated by cisplatin and especially cisplatin and BSO, and TRX levels are high when ERK activation is prevented by U0126 or by glutathione (Fig 10 A, B). To further support this hypothesis, TRX siRNA was used to decrease the expression of TRX. As shown in Fig 10 C, TRX siRNA inhibited TRX expression compared to the scrambled siRNA control and the cisplatin plus BSO plus U0126 treated cells. This decrease in TRX levels potentiated the cisplatin loss of cell viability (Fig 10 D). As shown above, CP-induced caspase-3 activity was inhibited by pretreatment with BSO and U0126 restored caspase-3 activity. However, TRX SiRNA partially prevented this restoring of caspase 3 activity (Fig 10 E). Similarly, the partial restoration of apoptosis by U0126 in the cisplatin plus BSO treated cells was lower in the siRNA TRX transfected cells relative to the scrambled siRNA transfected cells (Fig 10 F).
Fig. 10.

A. Effect of U0126 on expression of thioredoxin. Cells were seeded into 6-well plates at 4×105 cells/well and after 2 days' preincubation, 40 μM of BSO or control medium was added for overnight incubation, followed by addition of 0.25 mM of cisplatin alone or with BSO re-addition. 20 μM of U0126 was added 1 h prior to cisplatin. After incubation at 37°C for 4, 8, and 16 h, respectively, cells were collected, washed, and then sonicated. Cell lysates were used for Western blotting analysis to detect either thioredoxin or p-ERK.
B. Effect of GSHE on expression of thioredoxin. The procedures for cell and drug treatment were similar to (A) except that 5mM of GSHE instead of U0126 was added 1 h prior to cisplatin. A total 16 h incubation time was used.
C. Effects of TRX SiRNA on thioredoxin expression. 80 nM SiRNA was added and the E47 cells were incubated for overnight, followed by addition of 0.25 mM of CP for 16 h. Cells were collected, washed, and then sonicated. Cell lysates were used for Western blotting analysis of thioredoxin. CP, cisplatin; CBU, cisplatin+BSO+U0126.
D. Effects of TRX SiRNA on caspase-3 activity. The procedures for cell and SiRNA treatment were the same as (C). Caspase-3 activity was measured as described in MATERIALS AND METHODS. ** P<0.01, compared to Control; && P<0.01, compared with CP group; # P<0.05 and ## P<0.01, compared with CP+BSO group; $ P<0.05, compared with SiRNA scramble group.
E. Effects of TRX SiRNA on apoptosis. The procedures for cell and SiRNA treatment were the same as (C). Apoptosis was measured as described in MATERIALS AND METHODS. ** P<0.01, compared to Control; && P<0.01, compared with CP group; # P<0.05 and ## P<0.01, compared with CP+BSO group; $$ P<0.01, compared with SiRNA scramble group.
F. Effects of TRX SiRNA on cell viability. The procedures for cell and SiRNA treatment were similar to (C) except that cells were seeded in 24-well plates. MTT assays were performed to measure cell viability. ** P<0.01, compared with Control; P<0.05, # compared with SiRNA-scramble group.
Discussion
Cisplatin induces activation of MAPK such as P38 MAPK, JNK, and ERK in different cell models (17-20). In E47 cells, cisplatin induced ERK activation but not activation of P38 MAPK and JNK. Although many studies show that ERK serves as a surviving signaling pathway, recent evidence suggests that the activation of ERK can also contribute to cell death under certain conditions. For example, ERK is activated in neuronal and renal epithelial cells upon exposure to oxidative stress and toxicants, and inhibition of the ERK pathway blocks apoptosis (40). In cisplatin-induced cytotoxicity models, a death-promoting role for ERK was also reported. For example, Wang et al (17) have shown that only ERK activation is the most important factor for cisplatin-induced apoptosis, although all three MAPK members are activated. Other MAPK e,g, JNK or P38 MAPK were found to be important for cisplatin toxicity in other models (19, 20). It has been suggested that the precise pattern of ERK activation ultimately determines whether ERK protects or promotes cell death, e.g. the sustained activation of ERK promotes cell death in a HT22 hippocampal cell line (41,42). The kinetics and duration of ERK activation may direct ERK toward downstream targets that will either promote or limit neuronal survival (43,44). In the present study, we found that cisplatin led to sustained activation of ERK in both C34 and E47 cells, but activation occurred earlier in E47 cells than C34 cells. Depletion of glutathione with BSO not only enhanced cisplatin cytotoxicity, but also enhanced ERK activation, and the inhibition of ERK by U0126 or MEK1/2 SiRNA protected, at least in part, cells against cytotoxicity induced by cisplatin alone or in combination with BSO. GSHE inhibited cisplatin alone or cisplatin plus BSO cytotoxicity as well as ERK activation. It is suggested that activation of ERK contributed to cisplatin-induced cytotoxicity in CYP2E1-overexpressing HepG2 (E47) cells.
ERK has been implicated in apoptotic events upstream of caspase-3 activation, or contributes to apoptosis through the suppression of the anti-apoptotic signaling molecule AKT (40). In Hela cells, ERK activation blocks cytochrome c release and subsequent activation of caspase 3 in cisplatin-induced apoptosis (17). Most previous studies have focused on cisplatin-induced apoptosis. In E47 cells, however, activation of ERK contributed to cisplatin-induced necrosis but not apoptosis, as U0126 blocked cisplatin or cisplatin plus BSO induced necrosis but did not inhibit cisplatin-induced apoptosis. Cisplatin-induced apoptosis is mediated by caspases. In addition to an antioxidant action, glutathione can regulate redox state within cells by forming reversible mixed disulphides with protein thiols to prevent irreversible oxidation of cysteine residues under oxidative stress (38). In E47 cells, cisplatin caused a 25−60% loss of glutathione, however, the mode of toxicity was mainly apoptotic suggesting that the remaining glutathione still could prevent irreversible oxidation of caspase cysteine residues. But after nearly complete glutathione depletion by BSO (about 90%), the low remaining glutathione could not prevent irreversible oxidation of cysteine residues, hence caspases could not be activated (Fig. 7), and cisplatin toxicity switched from an apoptotic mode of cell death to a necrotic mode.
It has been reported that BSO was able to change the mode of cell death caused by H2O2 or anti-tumor drugs from apoptosis to necrosis (35, 36). In the E47 cells, adding GSHE switched cisplatin plus BSO-induced necrosis to apoptosis, because intracellular glutathione levels were increased. Is this switch an antioxidant action of glutathione? However, despite their anti-necrosis effect, other antioxidants could not switch cisplatin plus BSO-induced necrosis to apoptosis, because they could not increase intracellular glutathione levels (Fig. 8). Therefore, maintaining the intracellular glutathione levels appears crucial for onset of apoptosis in this model. However, although U0126 increased apoptosis caused by BSO plus cisplatin, U0126 did not increase the intracellular glutathione levels. Another redox active protein, thioredoxin (TRX) decreased dramatically after treatment with cisplatin plus BSO, but pretreatment with U0126 restored the decreased TRX (Fig. 10 A). The TRXs are a family of small redox active proteins (12 kD) that undergo reversible oxidation/reduction and help to maintain the redox state of cells; TRX acts in a manner similar to glutathione in thiotransferase reactions (39). TRX is maintained in a reduced state, under conditions resulting in glutathione depletion and oxidation (45). Besides maintaining the cellular redox state, TRX has an antioxidant effect. By reducing oxidized thioredoxin peroxidase, TRX restores the enzyme to its monomeric form, thus allowing it to continue its ROS scavenging action (39). In fact, addition of GSHE also restored TRX (Fig. 10 B); inhibition of TRX by SiRNA lowered the elevation of caspases and the restoration of apoptosis by U0126 (Fig 10 E, 10F). This suggests that, in the absence of glutathione, TRX can also maintain the redox state of cisplatin-treated cells thereby promoting caspase activity and apoptosis, and by restoring TRX expression, U0126 can activate the activities of caspases. The mechanisms by which U0126 increases TRX and the reciprocal regulation of TRX levels by ERK will require further studies. In addition, these results do not rule out possible roles of other cellular reductants which help maintain the cellular redox state since TRX siRNA was only partially effective in enhancing cisplatin toxicity and preventing restoration of caspase-3 activity and apoptosis.
Oxidative stress is closely associated with cisplatin-induced cytotoxicity (46). Glutathione depletion induces accumulation of ROS, leading to cell death (8). Intracellular glutahione can be depleted by cisplatin through formation of cisplatin GSH conjugates, and subsequent accumulation of ROS leads to cell death. Cisplatin-induced cytotoxicity was inhibited by GSHE pretreatment and exacerbated by depleting intracellular glutathione with BSO. U0126 protected against cytotoxicity induced by cisplatin plus BSO, but U0126 increased TRX expression instead of maintaining or restoring the glutathione levels. By restoring TRX expression, U0126 inhibited ROS accumulation, and protected against necrosis. Cisplatin lowered TRX levels in the E47 cells (Fig 10), and human TRX was shown to protect against cisplatin cytotoxicity (46, 47). Thus, inhibition of ERK by U0126 in E47 cells caused dual effects: inhibition of ERK protected against necrosis, probably via TRX's antioxidant action (other antioxidants also protected against the cisplatin plus BSO induced necrosis); on the other hand, inhibition of ERK restored activities of caspases and switched necrosis to apoptosis, probably via maintaining the redox state of cells by TRX. Further studies such as overexpression of TRX by transfection will be helpful to further evaluate the role of TRX in the necrotic and apoptotic modes of cell death produced by cisplatin in the E47 cells.
Many reports in the literature support a role for ROS production as an early mediator of cell signaling, with ERK being a downstream effector. ERK stimulation by ROS has been reported widely (31-33, 48, 49). Our data suggest a reciprocal relationship between ROS production and ERK activation. GSHE inhibited but BSO enhanced cisplatin-induced ERK activation as well as ROS accumulation; this suggests that ROS accumulation caused activation of ERK. On the other hand, at earlier times after addition of cisplatin, ERK had been activated but ROS production had not yet been elevated; moreover, inhibition of ERK by U0126 inhibited ROS production, suggesting that ERK activation caused ROS accumulation. This suggests a possible feedback mechanism between ERK activation and ROS production. ERK activation induced ROS that in turn further activated ERK. This role of ERK activation as a moderator of ROS production has also been found in lung endothelial cells, where hyperoxia-induced ERK activation was necessary for hyperoxia-induced NADPH oxidase activation which can result in ROS production (50), and in pulmonary epithelial cells, where inhibition of ROS production by N-acetyl-cysteine pretreatment blocked mechanical strain-induced ERK activation (51). However, the mechanisms by which ERK regulates ROS production require further studies. A model for interactions between glutathione, ROS production, ERK activation and TRX expression, and cisplatin toxicity, and the mode and switch of cell death is shown in Fig. 11 and discussed in the legend to Fig. 11.
Fig. 11.

Model for cisplatin toxicity in E47 cells. Cisplatin causes moderate glutathione depletion, ROS accumulation, ERK activation and lowers TRX. This results in elevated activity of caspases so cell death is mainly apoptosis. However, cisplatin plus BSO causes severe glutathione depletion, ROS accumulation, ERK activation and very low levels of TRX. This results in inhibition of caspase activities, so cell death is mainly necrosis, and antioxidants can block this cell death. When glutathione is elevated by adding GSHE or when TRX is restored by U0126, caspases are reactivated and necrosis is switched to apoptosis. CP, cisplatin; U, U0126; TRX; thioredoxin; GSH, glutathione; GSHE, glutathione ethyl ester.
Due to being poorly coupled with NADPH-cytochrome P-450 reductase, CYP2E1 exhibits enhanced NADPH oxidase activity and elevated rates of production of superoxide and hydrogen peroxide (21).This CYP2E1-dependent production of ROS occurs in the absence of added substrate (52,53). CYP2E1-mediated oxidative stress is thought to play an important role in alcoholic liver disease and it can produce synergistic toxic effects with other hepatotoxins such as endotoxin (22 ,23). We have previously reported that the E47 cells were more sensitive to cisplatin than C34 cells (11). CYP2E1 expression was not affected by cisplatin. Compared with C34 cells, ERK was activated earlier in E47 cells. U0126 when added at 16 h after cisplatin treatment prevented cytotoxicity in C34 cells but not in E47 cells. U0126 when added even only 1 h after cisplatin prevented cytotoxicity indicating early activation of ERK sets events in action which subsequently cause loss of cell viability. It is proposed that, in E47 cells, CYP2E1 along with activated ERK produce high levels of ROS, and the elevated ROS further activates ERK which in turn further increases ROS production, ultimately causing greater cell death, compared with C34 cells.
Acknowledgement:
This work is supported by NIH Grant AA03312.
Glossary
Abbreviations:
- BSO
L-Buthionine-[R,S]-sulfoximine
- ERK
extracellular signal-regulated kinases
- GSH
reduced glutathione
- GSHE
glutathione ethyl ester
- JNK
c-JUN NH2-terminal protein kinase
- MAPK
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI
propidium iodide
- ROS
reactive oxygen species
- TRX
thioredoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Referencces:
- 1.Chu G. Cellular response to cisplatin. J. Biol. Chem. 1994;269:787–790. [PubMed] [Google Scholar]
- 2.Timmer-Bosscha H, Mulder NH, Vries E.G.E. de. Modulation of cis-diamminedichloroplatinum (II) resistance: a review. Br. J. Cancer. 1992;66:227–238. doi: 10.1038/bjc.1992.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGinness JE, Proctor PH, De mopoulos HB, Hokanson JA, Kirkpatrick DS. Amelioration of cis-platinum nephrotoxicity by Orgotein (superoxide dismutase). Physiol. Chem. Phys. 1978;10:267–277. [PubMed] [Google Scholar]
- 4.Sugihara K, Gemba M. Modification of cisplatin toxicity by antioxidants. Jpn. J. Pharmacol. 1986;40:353–355. doi: 10.1254/jjp.40.353. [DOI] [PubMed] [Google Scholar]
- 5.Hannemann J, Baumann K. Cisplatin-induced lipid peroxidation and decrease of gluconeogenesis in rat kidney cortex: different effects of antioxidants and radical scavengers. Toxicology. 1988;51:119–132. doi: 10.1016/0300-483x(88)90143-6. [DOI] [PubMed] [Google Scholar]
- 6.Sodhi A, Gupta P. Increased release of hydrogen peroxide (H2O2) and superoxide anion (O2—) by murine macrophages in vitro after cisplatin treatment. Int. J. Immunopharmacol. 1986;8:709–714. doi: 10.1016/0192-0561(86)90006-8. [DOI] [PubMed] [Google Scholar]
- 7.Masuda H, Tanaka T, Takahama U. Cisplatin generates superoxide anion by interaction with DNA in a cell-free system. Biochem. Biophys. Res. Commun. 1994;203:1175–1180. doi: 10.1006/bbrc.1994.2306. [DOI] [PubMed] [Google Scholar]
- 8.Murphy ME, Scholich H, Sies H. Protection by glutathione and other thiol compounds against the loss of protein thiols and tocopherol homologs during microsomal lipid peroxidation. Eur. J. Biochem. 1992;210:139–146. doi: 10.1111/j.1432-1033.1992.tb17401.x. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y, Kawashima A, Horii I, Zhong L. Effects of BSO and L-cysteine on drug-induced cytotoxicity in primary cell cultures: drug-, cell type-, and species-specific difference. Drug Chem.Toxicol. 2004;27:269–280. doi: 10.1081/dct-120037748. [DOI] [PubMed] [Google Scholar]
- 10.Lu Y, Kawashima A, Horii I, Zhong L. Cisplatin-induced cytotoxicity in BSO-exposed renal proximal tubular epithelial cells: sex, age, and species. Ren. Fail. 2005;27:629–633. doi: 10.1080/08860220500200668. [DOI] [PubMed] [Google Scholar]
- 11.Lu Y, Cederbaum AI. Cisplatin-induced hepatotoxicity is enhanced by elevated expression of cytochrome P450 2E1. Toxicol. Sci. 2005;89:515–523. doi: 10.1093/toxsci/kfj031. [DOI] [PubMed] [Google Scholar]
- 12.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q, Olashaw N, Wu J. Participation of reactive oxygen species in the lysophosphatidic acid-stimulated mitogen-activated protein kinase kinase activation pathway. J. Biol. Chem. 1995;270:28499–28502. doi: 10.1074/jbc.270.48.28499. [DOI] [PubMed] [Google Scholar]
- 14.Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- 15.Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J. Neurochem. 1999;72:112–119. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell. Res. 2003;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J. Biol. Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 18.Choi BK, Choi CH, Oh HL, Kim YK. Role of ERK activation in cisplatin-induced apoptosis in A172 human glioma cells. Neurotoxicology. 2004;25:915–924. doi: 10.1016/j.neuro.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J. Biol. Chem. 2003;278:19245–19556. doi: 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- 20.Wu YJ, Muldoon LL, Neuwelt EA. The chemoprotective agent N-acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J Pharmacol Exp Ther. 2005;312:424–431. doi: 10.1124/jpet.104.075119. [DOI] [PubMed] [Google Scholar]
- 21.Ekstrom G, Ingelman-Sundberg M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem. Pharmacol. 1989;38:1313–1319. doi: 10.1016/0006-2952(89)90338-9. [DOI] [PubMed] [Google Scholar]
- 22.Lu Y, Wang X, Cederbaum AI. Lipopolysaccharide-induced liver injury in rats treated with the CYP2E1 inducer pyrazole. Am. J. Physiol. Gastrointest. Liver Physiol. 2005;289:G308–G319. doi: 10.1152/ajpgi.00054.2005. [DOI] [PubMed] [Google Scholar]
- 23.Lu Y, Cederbaum AI. Enhancement by pyrazole of lipopolysaccharide-induced liver injury in mice: role of cytochrome P450 2E1 and 2A5. Hepatology. 2006;44:263–274. doi: 10.1002/hep.21241. [DOI] [PubMed] [Google Scholar]
- 24.Wu D, Cederbaum AI. Removal of glutathione produces apoptosis and necrosis in HepG2 cells overexpressing CYP2E1. Alcohol. Clin. Exp. Res. 2001;25:619–628. [PubMed] [Google Scholar]
- 25.Wu D, Cederbaum AI. Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. J. Biol. Chem. 1996;271:23914–23919. doi: 10.1074/jbc.271.39.23914. [DOI] [PubMed] [Google Scholar]
- 26.Cederbaum AI, Wu D, Mari M, Bai J. CYP2E1-dependent toxicity and oxidative stress in HepG2 cells. Free Radic. Biol. Med. 2001;31:1539–1543. doi: 10.1016/s0891-5849(01)00743-2. [DOI] [PubMed] [Google Scholar]
- 27.Chen Q, Cederbaum AI. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in Hep G2 cells. Mol. Pharmacol. 1998;53:638–648. doi: 10.1124/mol.53.4.638. [DOI] [PubMed] [Google Scholar]
- 28.Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J. Biol. Chem. 1999;274:26217–26224. doi: 10.1074/jbc.274.37.26217. [DOI] [PubMed] [Google Scholar]
- 29.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 30.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 32.Dursun B, He Z, Somerset H, Oh J, Faubel S, Edelstein CL. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. Am. J. Physiol. Renal Physiol. 2006;291:F578 – F587. doi: 10.1152/ajprenal.00455.2005. [DOI] [PubMed] [Google Scholar]
- 33.Mitsui H, Takuwa N, Maruyama T, Maekawa H, Hirayama M, Sawatari T, Hashimoto N, Takuwa Y, Kimura S. The MEK1-ERK map kinase pathway and the PI 3-kinase-Akt pathway independently mediate anti-apoptotic signals in HepG2 liver cancer cells. Int. J. Cancer. 2001;92:55–62. [PubMed] [Google Scholar]
- 34.Mari M, Cederbaum AI. CYP2E1 overexpression in HepG2 cells induces glutathione synthesis by transcriptional activation of gamma-gluta mylcysteine synthetase. J. Biol. Chem. 2000;275:15563–15571. doi: 10.1074/jbc.M907022199. [DOI] [PubMed] [Google Scholar]
- 35.Troyano A, Sancho P, Fernandez C, de Blas E, Bernardi P, Aller P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell. Death Differ. 2003;10:889–898. doi: 10.1038/sj.cdd.4401249. [DOI] [PubMed] [Google Scholar]
- 36.Troyano A, Fernandez C, Sancho P, de Blas E, Aller P. Effect of glutathione depletion on antitumor drug toxicity (apoptosis and necrosis) in U-937 human promonocytic cells. The role of intracellular oxidation. J. Biol. Chem. 2001;276:47107–47115. doi: 10.1074/jbc.M104516200. [DOI] [PubMed] [Google Scholar]
- 37.Shin JN, Seo YW, Kim M, Park SY, Lee MJ, Lee BR, Oh JW, Seol DW, Kim TH. Cisplatin inactivation of caspases inhibits death ligand-induced cell death in vitro and fulminant liver damage in mice. J. Biol. Chem. 2005;280:10509–10515. doi: 10.1074/jbc.M413865200. [DOI] [PubMed] [Google Scholar]
- 38.Cotgreave IA, Gerdes RG. Recent trends in glutathione biochemistry--glutathione-protein interactions: a molecular link between oxidative stress and cell proliferation? Biochem. Biophys. Res. Commun. 1998;242:1–9. doi: 10.1006/bbrc.1997.7812. [DOI] [PubMed] [Google Scholar]
- 39.Biaglow JE, Miller RA. The thioredoxin reductase/thioredoxin system: novel redox targets for cancer therapy. Cancer Biol. Ther. 2005;4:6–13. doi: 10.4161/cbt.4.1.1434. [DOI] [PubMed] [Google Scholar]
- 40.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 41.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12 200–12 206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 42.Stanciu M, DeFranco DB. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J. Biol. Chem. 2002;277:4010–4017. doi: 10.1074/jbc.M104479200. [DOI] [PubMed] [Google Scholar]
- 43.Chu CT, Levinthal DJ, Kulich SM, Chalovich EM, DeFranco DB. Oxidative neuronal injury. The dark side of ERK1/2. Eur. J. Biochem. 2004;271:2060–2066. doi: 10.1111/j.1432-1033.2004.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kulich SM, Chu CT. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson's disease. J. Neurochem. 2001;77:1058–1066. doi: 10.1046/j.1471-4159.2001.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watson WH, Yang X, Choi YE, Jones DP, Kehrer JP. Thioredoxin and its role in toxicology. Toxicol. Sci. 2004;78:3–14. doi: 10.1093/toxsci/kfh050. [DOI] [PubMed] [Google Scholar]
- 46.Sasada T, Iwata S, Sato N, Kitaoka Y, Hirota K, Nakamura K, Nishiyama A, Taniguchi Y, Takabayashi A, Yodoi J. Redox control of resistance to cis-diamminedichloroplatinum (II) (CDDP): protective effect of human thioredoxin against CDDP-induced cytotoxicity. J. Clin. Invest. 1996;97:2268–2276. doi: 10.1172/JCI118668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arner ES, Nakamura H, Sasada T, Yodoi J, Holmgren A, Spyrou G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic. Biol. Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 48.Schweyer S, Soruri A, Heintze A, Radzun HJ, Fayyazi A. The role of reactive oxygen species in cisplatin-induced apoptosis in human malignant testicular germ cell lines. Int. J. Oncol. 2004;25:1671–1676. doi: 10.3892/ijo.25.6.1671. [DOI] [PubMed] [Google Scholar]
- 49.Wu WS, Tsai RK, Chang CH, Wang S, Wu JR, Chang YX. Reactive oxygen species mediated sustained activation of protein kinase C alpha and extracellular signal-regulated kinase for migration of human hepatoma cell HepG2. Mol. Cancer Res. 2006;4:747–758. doi: 10.1158/1541-7786.MCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 50.Parinandi NL, Kleinberg MA, Usatyuk PV, Cummings RJ, Pennathur A, Cardounel AJ, Zweier JL, Garcia JG, Natarajan V. Hyperoxia-induced NAD(P)H oxidase activation and regulation by MAP kinases in human lung endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003;284:L26–L38. doi: 10.1152/ajplung.00123.2002. [DOI] [PubMed] [Google Scholar]
- 51.Chess PR, O'Reilly MA, Sachs F, Finkelstein JN. Reactive oxidant and p42/44 MAP kinase signaling is necessary for mechanical strain-induced proliferation in pulmonary epithelial cells. J. Appl. Physiol. 2005;99:1226–1232. doi: 10.1152/japplphysiol.01105.2004. [DOI] [PubMed] [Google Scholar]
- 52.Gorsky LD, Koop DR, Coon MJ. On the stoichiometry of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. Products of oxygen reduction. J Biol Chem. 1984;259:6812–6817. [PubMed] [Google Scholar]
- 53.Dai Y, Rashba-Step J, Cederbaum AI. Stable expression of human cytochrome P4502E1 in HepG2 cells: characterization of catalytic activities and production of reactive oxygen intermediates. Biochemistry. 1993;32:6928–6937. doi: 10.1021/bi00078a017. [DOI] [PubMed] [Google Scholar]