

Abstract
In recent years the classic view that glucocorticoids, the adrenal steroids secreted during stress, are universally anti-inflammatory has been challenged at a variety of levels. It was first observed that under some circumstances, acute GC exposure could have pro-inflammatory effects on the peripheral immune response. More recently, chronic exposure to GCs has been found to have pro-inflammatory effects on the specialized immune response to injury in the central nervous system. Here we review the evidence that in some cases, glucocorticoids can increase pro-inflammatory cell migration, cytokine production, and even transcription factor activity in the brain. We consider how these unexpected effects of glucocorticoids can co-exist with their well-established anti-inflammatory properties, as well as the considerable clinical implications of these findings.
Keywords: Glucocorticoids, stress, inflammation, LPS, stroke, ischemia, cytokines, NFκB, brain, central nervous system
1. INTRODUCTION
Because life is frequently not easy, simple and complex organisms have developed methods to deal with challenging situations. In humans and other vertebrates, stressful circumstances elicit a wide variety of physiological changes that constitute the “stress-response.” This begins within seconds with the release of the catecholamines of the sympathetic nervous system (epinephrine and norepinephrine). In the ensuing minutes, the hypothalamic-pituitary-adrenal (HPA) axis is activated. CRF produced by the hypothalamus acts on the pituitary to activate the release of ACTH into circulation, resulting in the release of the adrenal steroid glucocorticoids (GCs). The role of GCs is largely to modulate and control the stress response over a longer timeframe in the minutes to hours following a stressor. This response is conducted largely on a genomic level and accordingly the actions of GCs are numerous and varied (Sapolsky et al. 2000).
The pioneering physiological investigations of Walter Cannon and Hans Selye in the early 20th century began to define precisely what is involved in the stress- response. “Fight or flight,” as described by Cannon, reflects the actions of both catecholamines and GCs (although the latter were unknown until the work of Selye, some years later) to immediately increase cardiovascular output and blood flow to the brain and skeletal muscles. This is predominately mediated by catecholamines, although GCs potentiate their effects. Both hormones mobilize energy stores from adipose and hepatic cells, ensuring a supply of energy to exercising muscle. At the same time, GCs decrease less immediately essential activities such as feeding, digestion, growth and reproduction. Of key relevance to this review, the ability of stress and GCs to inhibit the immune system was recognized soon after their discovery. The rationale for GCs diverting stored energy to exercising muscle is clear. It is less clear why it would be adaptive for stress to reduce the activity of the immune system, particularly because a stress response is often accompanied, or even caused by an immune challenge. There is little reproductive value to an injured animal that escapes a predator only to succumb to sepsis soon after.
Several theories were offered early on to help explain the supposed adaptive logic of suppressing immunity during stress (reviewed in (Munck et al. 1984)). The most pervasive of these was the explanation that the anti-inflammatory actions of GCs during stress were a means to conserve energy for a more critical need such as exercising muscle. The strongest blow to this hypothesis was evidence that GCs prevent inflammation via immune cell apoptosis, a type of programmed cell death that is a costly, pro-active enterprise requiring considerable amounts of protein synthesis and cellular remodeling. Moreover, these early theories could not satisfactorily circumvent the problem that dismantling the immune system in response to stress is often quite maladaptive.
In more recent years, the advent of improved assays that allowed for the detection of subtler changes in immune function with better time resolution radically changed the understanding of the effects of stress on the immune system. Specifically, it became clear that early in the stress response, before the stress-induced rise in GC levels can have its effects on target tissues, immune function is activated rather than suppressed (Herbert and Cohen 1993). Munck and colleagues, in a highly influential theoretical paper, posited that the anti-inflammatory effect of elevated concentrations of GCs functions to mediate recovery, after the stressor has abated, from the immune-activating effects of the early phases of the stress response (Munck et al. 1984). Part of this reformulation involved the predication that failure of GCs to mediate this immune recovery would cause a predisposition towards developing autoimmune and inflammatory disorders. Subsequent work has supported these theories, showing that a number of autoimmune disorders, in both experimental animal models and humans, involve a failure of GC actions (Wick et al. 1993). It is these long-term potent anti-inflammatory effects of GCs that are used clinically to counter such autoimmune and inflammatory disorders.
The stimulatory effects of the early phases of stress on immunity are in many cases due to the actions of catecholamines. However, GCs have also been found to be involved in this activation, raising the question of how GCs can both stimulate and suppress the immune response (albeit at different times). To reconcile these observations, Munck suggested that these opposing actions are concentration-dependent (Munck et al. 1984). Basal and low stress levels of GCs are required for the early increases in the immune response. This constitutes a common phenomenon in endocrinology, namely a “permissive” effect. In this case, low GC levels are needed to allow catecholamines to rapidly stimulate immunity with the onset of stress. Only at higher concentrations do GCs begin to exert suppressive effects to prevent autoimmune damage. This explanation fits well into an adaptive understanding of the immune system during the stress response where it needs to be mobilized initially, but must be kept under control in the long term.
More recently, GCs have been found to have pro-inflammatory effects in situations other than the early phase of the stress-response. In particular, the effects of chronic GCs on the immune response in the brain are surprisingly different from the classic picture of suppression in the periphery. Depending on the brain region, chronic exposure to GCs is often not anti-inflammatory and, in greatest contrast to dogma, can actually exacerbate various aspects of inflammation. This is in stark contrast to the effects of high concentrations of GCs in the periphery. The importance of these findings and their application cannot be sufficiently underscored, given the wide use of steroids in clinical neurology for their anti-inflammatory properties.
This review evaluates the evidence that GCs are not universally anti- inflammatory and the situations where they have pro-inflammatory actions. First, we briefly consider the wide range of anti-inflammatory effects of GCs on peripheral inflammation. We then review the more recent studies that suggest GCs enhance peripheral inflammation, particularly in the early phases of the stress response. Finally, we turn to the focus of this review, the injured CNS, where we consider recent evidence that chronic GCs can enhance some facets of inflammation in ways quite different from the cases of the pro-inflammatory effects of acute GCs in the periphery.
2. GLUCOCORTICOID REGULATION OF IMMUNITY OUTSIDE THE NERVOUS SYSTEM
2.1 Classic Anti-Inflammatory Effects of Glucocorticoids
We now review the numerous and well-understood examples of anti-inflammatory GC actions in the periphery. It is critical to this discussion to first define the parameters of an inflammatory response in order to understand the components being increased or decreased and the similarities and differences between a peripheral and a central response (Figure 1). We assume familiarity with the general aspects of these GC actions and review them only briefly as a means for appreciating the contrasting pro-inflammatory effects reviewed in subsequent sections. For more comprehensive review of this literature see (Elenkov and Chrousos 2002; Franchimont 2004).
Figure 1. Comparison of central and peripheral inflammatory responses.
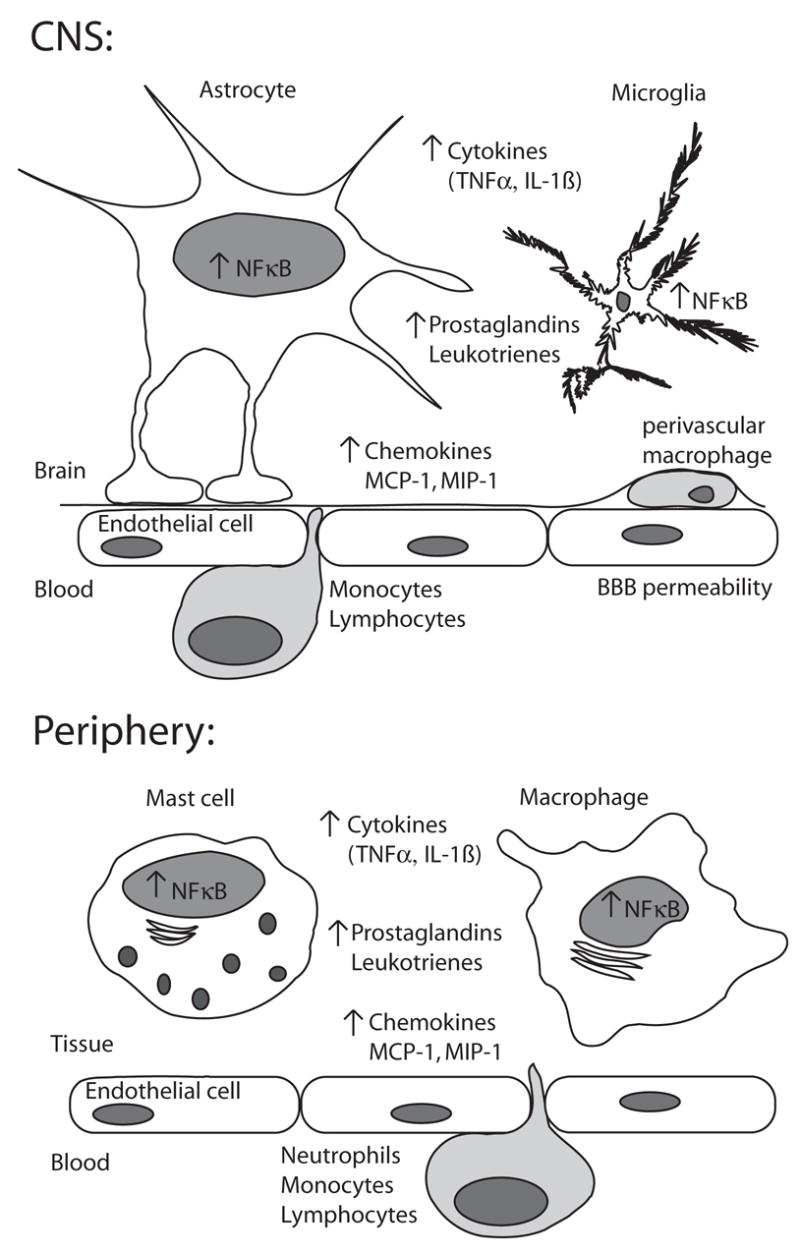
This diagram highlights major similarities and differences between an inflammatory response to injury in the CNS and periphery. The most readily noticeable differences are on a cellular level. Whereas mast cells and tissue resident macrophages are the first lines of response in the periphery, in the brain it is the astrocytes and microglia and microglia are divided between populations in the parenchyma and the perivascular space. The blood brain barrier represents the largest difference between the immune response in the CNS and periphery, with a reduced ability of molecules from the plasma being able to flow across the endothelium during an injury state. Finally, the nature of the cells that extravasate from circulation depends on the nature of the injury, with neutrophils forming less of the immediate response in the CNS. On a molecular level, many of the pro-inflammatory molecules such as cytokines, chemokines, and prostaglandins produced are the same, although as mentioned, the cells producing them are different and there may be differences in the specific profile. Much of this inflammatory response is attributable on a transcriptional level to the activity of the pro-inflammatory transcription factor, NFκB. While active, NFκB affects increases in the molecular response to inflammation as well as eventually causing its own repression.
Hans Selye in the 1930’s obtained the first evidence for GCs being anti-inflammatory, noting that sustained stress or GC exposure caused involution of the thymus. This was such a dramatic and reliable effect that it was long used as a bioassay for circulating GC concentrations, before the development of radioimmunoassays. In the wake of their establishment as profoundly anti-inflammatory, synthetic GCs came to be used in many domains of clinical pharmacology.
Work since then has examined GC-induced immunosuppression on far more reductive levels than the involution of an organ. On the cellular level, GCs can inhibit both the innate and adaptive immune responses. GCs inhibit lymphocyte proliferation, induce apoptosis in basophils and eosinophils, and cause a redistribution of T-cells. GCs also cause dendritic cells (DCs), the antigen-presenting cells of the adaptive immune system, to maintain an immature state. In addition, GCs can induce T-cell apoptosis and a shift in the population of T cells from mediating cellular immunity with a Th1 phenotype to mediating humoral immunity with a Th2 phenotype.
On the molecular level, GCs limit the capacity of DCs to interact with immature T-cells by inhibiting MHC II expression. Moreover, GCs inhibit or reduce the expression of a variety of chemoattractants and pro-inflammatory cytokines (e.g. IL-1β, IL-6, TNF-α), while enhancing the expression of anti-inflammatory cytokines (e.g. IL-10, TGF-beta). The expression of other pro-inflammatory molecules such as inducible COX-2 and prostaglandins is also inhibited by GCs. The inhibition of cytokines and other pro- inflammatory molecules has been demonstrated at the levels of both protein and mRNA synthesis. In addition, GCs enhance the production of decoy receptors and antagonist molecules such as IL-1R2 and IL-1Ra, resulting in decreased IL-1 signaling (although the latter is disputed).
GCs exert these varied effects through well-established mechanisms of steroid hormone signaling. Upon cellular entry, GCs bind to either the Type I, mineralocorticoid receptor (MR), or the Type II glucocorticoid receptor (GR). MR has a much higher affinity (0.5 nM) for GCs than does GR (5.0 nM), and this difference is a large part of the reason for concentration-dependent differences in GC activity (De Kloet et al. 1998). At basal GC levels, GCs predominantly bind to MR and only slightly occupy GR. In the early stage of a stressor, MR becomes saturated and the occupancy of GR is increased. It is not until a sustained, major stressor that GR occupancy is saturated. Thus, MR is responsible for much of the effects of basal and low-stress levels of GCs (i.e. the permissive effects), whereas GR largely mediates the effects of high stress GC levels. This, combined with the fact that MR and GR signaling often have opposing outcomes, results in a U-shaped curve of GC action where intermediate (basal to low-stress) GC concentrations have the opposite effect of no GCs or high-stress concentrations (Munck et al. 1984; Sapolsky et al. 2000).
Both MR and GR belong to a large class of nuclear receptors which homodimerize and translocate to the nucleus after ligand binding. There, both MR and GR bind to the glucocorticoid response element (GRE) consensus DNA sequence, and modulate the transcriptional activity of downstream genes in a similar manner. Because both MR and GR can bind to the same GRE, their often opposite effects arise from their different patterns of protein-protein interactions with other components of the transcriptional machinery. Unlike MR, GR directly interferes with the transcriptional activity of the pro-inflammatory transcription factors nuclear factor kappa B (NFκB) and activating protein 1 (AP-1), thus decreasing expression of the inflammatory genes they would normally activate. The result is an extensive decrease in inflammatory mediators (reviewed in (De Bosscher et al. 2003)). Interestingly, reports of rapid non-genomic signaling by these nuclear hormone receptors suggest a mechanism by which GCs might be able to mediate temporally distinct effects (Karst et al. 2005; Mulholland et al. 2006).
These well-characterized anti-inflammatory effects of GCs make it seem likely that stress is also anti-inflammatory. However, as discussed next, the effects of stress are not that simple and in many cases can be divided based on the duration of the stressor. Whereas acute stressors frequently enhance the peripheral immune response, chronic stress is suppressive. Surprisingly, the GCs released during stress are involved in both the enhancing and suppressive effects.
2.2 The pro-inflammatory effects of stress outside the nervous system
Stress has been assumed to be anti-inflammatory due to the well-understood anti-inflammatory actions of GCs; however, it turns out that this is not always the case. In particular, acute stress just before an inflammatory challenge often has the adaptive effect of enhancing the immune response, whereas chronic stress has the role of keeping the immune response in check by suppressing it. For example, one type of the delayed type hypersensitivity (DTH) cell-mediated immune response is exacerbated by acute stress but suppressed by chronic stress (Dhabhar and McEwen 1999). Moreover, peripheral and central nervous system (CNS) levels of pro-inflammatory cytokines such as IL-1β are increased following an acute stressor in both rats (O'Connor et al. 2003) and humans (Deinzer et al. 2004). Additionally, the activity of NFκB and expression of NFκB target genes increase following acute stress in human peripheral blood mononuclear cells (Bierhaus et al. 2003) and rat cortex (Madrigal et al. 2001). These studies all concluded that acute stress enhanced the response to a subsequent immune challenge. The timing of the relationship between stressor and immune challenge is critical, in that acute stress after an inflammatory challenge decreases expression of IL-1β , TNF-α, IL-6, and other pro-inflammatory cytokines (Goujon et al. 1995b).
Given the widespread anti-inflammatory actions of GCs, it seems most probable that some other aspect of the stress response is responsible for immune enhancement by acute stress. Most evidence implicates catecholamine release by the sympathetic nervous system (SNS). For example, stress-induced increases in NFκB signaling were modeled in human monocyte THP-1 cells after stimulation with norepinephrine, suggesting that SNS activation may be responsible for stress-induced cytokine increases (Bierhaus et al. 2003). Injection of alpha and beta adrenoceptor antagonists block the stress-mediated increase in IL-1β levels, and adrenoceptor agonists are sufficient to substitute for stress (Johnson et al. 2005). Because the actions of catecholamines are exerted on a shorter timescale than those of GCs, these results fit with the picture of acute and early stages of stress as pro-inflammatory. For an extensive review of the pro-inflammatory actions of stress, see (Black 2002).
2.3 The pro-inflammatory effects of GCs outside the nervous system
While many pro-inflammatory effects of acute stress can be attributed to catecholamines, GCs are not as anti-inflammatory as might be expected. Several recent microarray studies examined the effects of the high-affinity GR agonist dexamethasone on human PBMCs and found that amidst expected anti-inflammatory changes in gene expression, there were many pro-inflammatory systems such as chemokines, compliment proteins, and cytokines with increased expression (Galon et al. 2002; Reyes et al. 2003). The following section reviews several representative situations where acute GCs enhance peripheral inflammation, but is by no means comprehensive. Notably, we do not discuss the relatively extensive stimulatory effects of GCs on the acute phase response to inflammation. See (Sapolsky et al. 2000; Dhabhar 2002; Yeager et al. 2004) for further review of GC effects on peripheral inflammation.
As noted, GCs have profound negative effects on the numbers of circulating leukocytes and can induce apoptosis (Yoshimura et al. 2001). However, a closer look reveals that neutrophil proliferation and survival are enhanced by GCs (Cox 1995; Liles et al. 1995) and that the large-scale depletion of other leukocytes may actually reflect extravasation to a site of injury, rather than cell death (Dhabhar et al. 1996; Dhabhar and McEwen 1997; Viswanathan and Dhabhar 2005). The depletion of circulating leukocytes by GCs has been described metaphorically as a deployment of immune soldiers to the battlefront rather than to the barracks (McEwen et al. 1997). Acute stress-enhanced migration of leukocytes from the bloodstream to wound sites can be mimicked by either a physiological dose of corticosterone or a GR agonist (but not an MR agonist), and is blocked by adrenalectomy or inhibition of GC synthesis (Dhabhar et al. 1996). Notably, chronic stress also fails to have the same effects on leukocyte redistribution and instead decreases baseline leukocyte numbers as well as the magnitude of acute stress effects on leukocyte redistribution (Dhabhar and McEwen 1997). These findings implicate GR occupancy at GC levels typical of an acute stressor as both necessary and sufficient to mediate enhancement of this particular immune response.
Acute stress levels of GCs have also been shown to enhance a model of peripheral inflammation. Acute stress-induced enhancement of the DTH immune response is blocked by adrenalectomy, which removes both GCs and epinephrine and is restored by supplementation with corticosterone (the GC specific to rodents) levels that replicate the early stages of the acute stress-response. This restoration is achieved to a greater extent by the replacement of corticosterone than epinephrine (Dhabhar and McEwen 1999). This surprising result may be due in part to the GC-enhanced leukocyte migration following acute stress. In contrast, chronic exposure to corticosterone or acute exposure to dexamethasone do not have the same effect and are anti-inflammatory (Blecha et al. 1982a; Dhabhar and McEwen 1999). It is an important caveat that in some models of DTH and for some types of stressors, GCs and catecholamines have not been seen to play an important role in stress-mediated augmentation (Blecha et al. 1982a; Blecha et al. 1982b).
The differences between the effects of acute and chronic stress on immune function described by Dhabhar and McEwen (Dhabhar and McEwen 1997) cannot be explained by the concentration-dependent effects of MR and GR signaling. As noted, the pro-inflammatory migration of leukocytes to wound cites is GR- rather than MR-mediated. Thus, the most meaningful way of understanding the dichotomy between the pro- and anti-inflammatory actions is not so much the contrast between MR and GR occupancy, as the contrast between acute and major/prolonged stress-responses. Presumably, these differences are manifested on the molecular level through the duration and extent of GR occupancy.
GCs can have permissive and suppressive actions through mechanisms other than activation of the opposing effects of MR and GR signaling. While GCs reduce the expression of most pro-inflammatory cytokines, they often simultaneously increase the expression of the corresponding cytokine receptor. This means that at low GC levels, cytokines are in abundance, but their receptors are not and signaling is low. Conversely, at high GC levels, receptors are in abundance, but cytokines are suppressed so signaling is low. Thus, cytokine signaling is minimal at extreme GC concentrations and peaks at an intermediate GC concentration (Munck and Naray-Fejes-Toth 1992). This observation has been shown to have functional importance in the proliferative effect of IL-2 signaling on T cells. GCs decrease IL-2 production, but they expedite expression of the IL-2 receptor. The result is that acute stress-levels of GCs enhance T cell proliferation by accelerating the ability of IL-2 to signal through its receptor (Wiegers et al. 1995; Almawi et al. 1999). Notably, exposure to chronic, high levels of corticosterone prevented this effect (Sterzer et al. 2004). The applicability of this model to other cytokines and their receptors remains to be investigated. Weigers and Reul have suggested that these effects of GCs on cytokine signaling might serve to optimize the course of inflammation (Wiegers and Reul 1998).
Although GCs inhibit many pro-inflammatory cytokines, they increase expression of at least one potent pro-inflammatory mediator. The macrophage migration inhibitory factor (MIF) was one of the earliest discovered pro-inflammatory cytokines. Its release from intracellular pools in macrophages and T cells results in the production of nitric oxide and TNF-α in an autocrine stimulatory manner (Bernhagen et al. 1994). MIF also has been implicated in DTH, as MIF-blocking antibodies attenuate the DTH immune response (Bernhagen et al. 1996). In humans, circadian plasma levels of GCs correlate with a circadian pattern of MIF concentrations (Petrovsky et al. 2003). Moreover, basal concentrations of GCs increase MIF production in T cells and macrophages. Strikingly, physiologically relevant concentrations of MIF can completely overcome the anti-inflammatory effects of GCs on other aspects of inflammation in LPS stimulated monocytes (Calandra et al. 1995). Furthermore, in a high dose in vivo LPS challenge where GC treatment reduces mortality, concomitant treatment with MIF negates the protection conferred by GCs. Taken together, these findings suggest a physiological role for MIF in countering the anti-inflammatory effects of GCs (Donnelly and Bucala 1997).
GC enhanced MIF production is dose-dependent and fits the model that low-levels of GCs are pro-inflammatory and high doses are anti-inflammatory. Low physiological doses of cortisol, the species typical GC of humans and most primates, stimulate MIF secretion in murine monocytes, while high doses suppress its production (Calandra et al. 1995). Surprisingly, dexamethasone was as stimulatory as cortisol at low doses and became anti-inflammatory at approximately the same dose. Given the striking difference in GR binding affinity between these two GCs, this result suggests that these effects of GCs on MIF are not necessarily controlled by the degree to which GR is bound. For the most part, the actions of GCs on MIF fit our working model where low doses suppress, and high doses enhance inflammation.
The timing of GC exposure relative to an immune challenge has additional importance in determining their action. When GCs are administered prior to an inflammatory challenge, they have actually been found to augment the subsequent cytokine response. Treatment with GCs twelve hours prior to LPS and IFN gamma stimulation increases NFκB signaling and IL-6, TNF-α, and nitrite production in a murine macrophage cell line (Smyth et al. 2004). In a similar manner, exposure to GCs twelve hours before LPS stimulation increases plasma levels of TNF-α and IL-6 in humans (Barber et al. 1993). This enhanced response is not necessarily due to preparative cytokine receptor upregulation because similar increases are not observed when GCs are given six hours prior to inflammatory challenge. Finally, acute stress twenty-four hours prior to LPS increases plasma levels of TNF-α, IL-6, and IL-1β in the rat; however the role of GCs in this outcome was not tested (Johnson et al. 2002a). In each instance of GC pre-treatment, GCs had been removed from the media at the time of the inflammatory stimulation in vitro, and in the in vivo model, GC levels had returned to baseline at the time of LPS exposure. Thus, these effects are not due to the concentration of GCs at the time of the inflammatory response. Instead, Maier et al described stress as having a preparative “priming” effect on the subsequent inflammatory response to an immune challenge (Johnson et al. 2002a; Johnson et al. 2002b; Johnson et al. 2003).
It is clear that much work remains to be done to fully understand what governs when GCs are pro- or anti-inflammatory in the periphery. GC type and concentration, time course of exposure, and the activation state of the immune system are all significant. Several trends emerge among the diverse GC enhancing effects on inflammation: low-doses, acute-exposure, and GC presence prior to inflammation all tend to augment it, whereas high-doses, chronic-exposure, and GC presence after inflammation tend to suppress it. The evidence suggests that in the context of an acute stressor and immune challenge, GCs play a complex role in orchestrating an optimal and timely response that maintains control over the potentially damaging effects of an over-extended immune response. In the next section we will examine whether GCs have similar effects on inflammation in the brain.
3. GLUCOCORTICOID REGULATION OF IMMUNITY IN THE NERVOUS SYSTEM
3.1 Normative glucocorticoid effects in the brain
Glucocorticoids have an enormous and varied array of effects upon the brain. In particular, hippocampal cells express high levels of MR and GR and are a primary target of GC actions. Such actions include various salutary outcomes mediated by MR occupancy and/or the small increase in GR occupancy seen in the early phase of the stress-response. These basal and permissive effects include prevention of neuron death in the dentate gyrus, enhancement of synaptic plasticity, and facilitation of hippocampal-dependent cognition. However, GC actions in the hippocampus show the classic inverse-U pattern, in that heightened and prolonged GC exposure has an array of deleterious effects, mediated by high GR occupancy. These include impaired cognition and synaptic plasticity, inhibition of neurogenesis, atrophy of dendritic arbors, and a reduction in spine density (McEwen and Magarinos 2001).
3.2 Glucocorticoid effects on the injured brain
The adverse consequences of prolonged elevation of GC concentrations are particularly dramatic when coincident with neuronal injury. An extensive literature now demonstrates that chronic stress and GCs, signaling through GR, can compromise the ability of neurons in the hippocampus, cortex and striatum to survive a variety of neurological insults, including hypoxia-ischemia, seizure, hypoglycemia, anti-metabolites, oxygen radical generators, the beta-amyloid peptide, and the gp120 glycoprotein of HIV. These “endangering” actions are exacerbated by the fact that many of these neurological insults stimulate additional GC secretion. A fair amount is now understood about the mechanisms underlying this endangerment. Specifically, GCs inhibit glucose uptake throughout these brain structures, an effect that is not sufficient to be energetically disruptive by itself, but it exacerbates the declines in ATP concentrations and mitochondrial potential caused by these insults. As a result, affected neurons and astrocytes have less energy available for the costly tasks of high-affinity reuptake of the potentially excitotoxic glutamate from the synapse. In addition, affected neurons are compromised in their ability to sequester and/or extrude free cytosolic calcium from post-synaptic neurons, and to contain and quench oxygen radicals, all of which imperil neurons (Sapolsky 1999; Reagan 2002).
These endangering effects of chronic GCs in the injured brain are difficult to reconcile with their ability to decrease inflammation. To understand this apparent contradiction we must first consider the nature and consequences of the inflammation caused by the types of neurological insults just discussed. When neurons die necrotically, they release ATP, cytokines, prostaglandins, calcium and glutamate, and other cellular debris. This activates microglia and astrocyte proliferation and causes leukocytes such as granulocytes and monocytes to extravasate from the blood stream and migrate to the site of damage (Figure 1). Whether this inflammatory response is beneficial or detrimental to the neurons that survive the initial insult is important for understanding how to appropriately treat neurological insults. At least some attributes of the inflammation appear to be beneficial because TNF-α receptor knockout mice have increased lesion size following neuronal insult (Bruce et al. 1996). Similarly, inhibition of astrocyte proliferation exacerbates neuron loss in focal brain injury models (Myer et al. 2006). On the other hand, peripheral immune cells that respond release additional pro-inflammatory cytokines, proteases, and oxygen radicals and these factors are responsible for much of the damage that occurs (Dinkel et al. 2004). Broadly, it appears as if the early stages of a moderate inflammatory response in the injured brain can be beneficial, whereas more extensive inflammation may adversely affect surviving neurons (del Zoppo et al. 2001).
Therefore, this suggests that chronic GCs should exert two opposing effects in the injured brain. On the one hand, GCs directly compromise the ability of various neuron types to survive a number of neurological insults, augmenting damage. In contrast, by decreasing the extensive inflammation caused by these insults, GCs should decrease damage (and in support of this, non-steroidal anti-inflammatory agents that blunt inflammation are neuroprotective (Patel et al. 1993; Hara et al. 1998), however, see (Blais et al. 2005)). This raises the issue of whether the endangering GC effect upon neurons, or their protective inhibition of extensive inflammation is more dominant. The fact that exposure to high stress concentrations of GCs augments the extent of neurotoxicity and inflammation in response to these insults in vivo suggests that the endangering effects prevail (Sapolsky 1999).
These findings come as somewhat of a surprise given the anti-inflammatory potential of chronic GCs. However, it turns out that at least part of the explanation is that the anti-inflammatory effects of chronic GCs in the brain are actually less consistent and potent than in the periphery.
3.3 Just how anti-inflammatory are GCs in the brain?
Anti-inflammatory GC actions in the CNS have been demonstrated in numerous studies. However, as will be seen, there are also circumstances where GCs fail to have anti-inflammatory actions and where they even augment inflammation. We review these cases, and then try to discern the circumstances predicting when GCs stimulate or suppress inflammation.
3.3.1 Anti- and pro-inflammatory GC effects at the histological and cellular levels
GCs are quite effective at reducing certain types of inflammatory cytotoxicity in the brain. For example, treatment with dexamethasone decreases iNOS-mediated toxicity of microglia (Golde et al. 2003). Direct infusion of LPS into the prefrontal cortex causes necrotic cell death accompanied by an extensive inflammatory response and is commonly used as a model for inflammatory neurological disorders. Nadeau and Rivest found that acute-stress levels of GCs were both necessary and sufficient to lessen damage due to direct LPS infusion into the brain (Nadeau and Rivest 2002; Nadeau and Rivest 2003). Thus, basal GC levels and the physiological response to acute stress are anti-inflammatory and reduce inflammatory cytotoxicity (Rivest 2003).
In contrast, there are a number of circumstances where GCs fail to have these anti-inflammatory properties. For example, dexamethasone fails to reduce inflammation in tuberculosis meningitis, although it does correspond with an improved prognosis (Simmons et al. 2005) and fails to decrease cytotoxic edema following cerebral ischemia. For a recent synthesis of clinical applications where GCs do and do not have theraputic benefit see (Gomes et al. 2005).
Most strikingly, long-term or high stress levels of GCs appear to exacerbate similar measures of cellular inflammation. Thus, chronic high dose corticosterone treatment worsens excitotoxin-induced or ischemic hippocampal damage (Sapolsky and Pulsinelli 1985; Dinkel et al. 2003). The chronic unpredictable stress (CUS) paradigm exposes animals to a different stressor each day and animals subjected to CUS after direct LPS infusion into the prefrontal cortex have markedly increased neuron and astroglia death. This effect of CUS is reversed by blocking GR, suggesting that GC signaling is the critical component of CUS involved (de Pablos et al. 2006). These findings are in contrast to the aforementioned protective effects of basal to low-stress levels of corticosterone on LPS toxicity. Use of the same neurological insult highlights the concentration-dependent effects of GCs on neurological inflammation. Notably, GCs also dose-dependently inhibit astrocyte proliferation (Crossin et al. 1997).
The pro-inflammatory GC effects extend to the extravasation and migration of circulating inflammatory cells to a site of neurological injury. This may not come as a surprise given the previously mentioned effects of GCs on these cells in the periphery. GC concentrations positively correlate with the degree of inflammatory infiltration of granulocytes and macrophages to the site of hippocampal infusion of excitotoxin (Figure 2). Blocking GC synthesis reduces the migration and abundance of these cells at the injury site. Furthermore, rats exposed to chronic high stress levels of corticosterone show accelerated and augmented microglial activation and migration of peripheral cells to the injury site (Dinkel et al. 2003). Similarly, GCs released during CUS enhance microglial activation following direct LPS injection to the prefrontal cortex (de Pablos et al. 2006). These findings implicate chronic stress levels of GCs in enhanced activation of the immune cell response to neurological cytotoxicity.
Figure 2. Glucocorticoid-increased immune cell response to CNS injury.
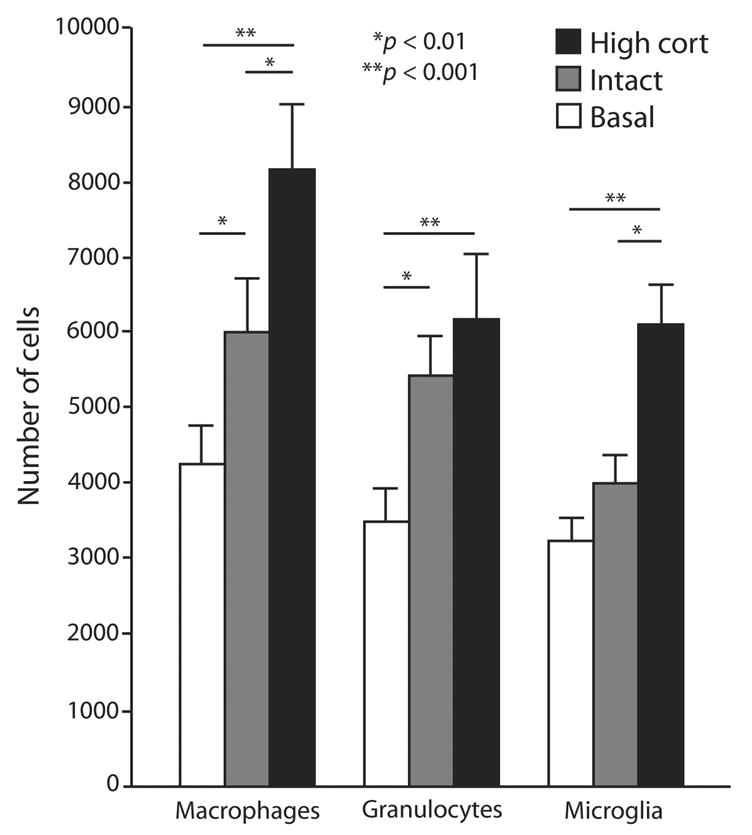
Rats were exposed to different amounts of GCs for three days, given a unilateral infusion of kainic acid in the CA3 region of the hippocampus and the abundance of different immune cells was quantified over 72 hours post kainic acid. This graph shows an increase in the abundance of different immune cells with increasing exposure to corticosterone. Rats in the basal group (white) were adrenalectomized and given a low-dose subcutaneous corticosterone pellet, which locks circulating corticosterone levels at a basal level. Rats in the intact group (gray) experienced a stress response from a sham adrenalectomy as well as a physiological stress response to kainic acid, but were otherwise unmanipulated. Rats in the high cort group (black) were injected daily with corticosterone in order to induce chronic high exposure. [From Dinkel et al. 2003 Blackwell publishing, with permission]
3.3.2 Anti- and pro-inflammtory GC effects at the messenger level
Levels of pro-inflammatory cytokines are an index of the inflammatory signaling cascades activated by injury. GCs potently decrease the production of these molecules in the periphery and also promote secretion of anti-inflammatory cytokines. In many cases, this is also true in the brain. Adrenalectomized or GR antagonist-treated animals have been found to have increased production of many cytokines in many different models (Goujon et al. 1995a; Goujon et al. 1996; Nadeau and Rivest 2003). The studies that have looked at the effects of chronic GC exposure have also found similar results for basal GCs: locking GCs at basal levels augments excitotoxin-induced upregulation of IL-1β and TNF-α expression and protein levels in the hippocampus (Dinkel et al. 2003). Pharmacological inhibition of GR increases TNF-α, IL-1 beta, and iNOS protein levels in rats given LPS peripherally (Munhoz et al. 2006). Moreover, in hippocampal cultures, low concentrations of corticosterone inhibit excitotoxin-induced expression of IL-1β and TNF-α (MacPherson et al. 2005). These findings suggest that basal levels of GCs have anti-inflammatory properties on these pro-inflammatory cytokines during necrotic cell death.
However, in the context of sustained stress, or stress levels of GCs, a very different picture emerges. CUS fails to suppress the increased expression of IL-1β and TNF-α following infusion of LPS into the prefrontal cortex (de Pablos et al. 2006). Even more striking, CUS augments TNF-α, IL-1β , and iNOS protein levels in hippocampus and cortex when LPS is administered peripherally, an effect that is GR-mediated (Munhoz et al. 2006). Similarly, high stress concentrations of corticosterone exacerbate excitotoxin-induced increases in IL-1β and TNF-α mRNA and protein levels both in vivo and in vitro (Dinkel et al. 2003; MacPherson et al. 2005). A more detailed analysis of GC concentration reveals inverse-U properties where intermediate concentrations enhance inflammation the most. Mild stress levels of corticosterone are even more effective than high stress levels in augmenting excitotoxin-induced expression of TNF-α and iNOS in vitro and in vivo (MacPherson et al. 2005; Munhoz in prep). Dexamethasone causes a dose-and time-dependent increase in expression and activity of prostaglandin D2 synthase in neuronal cultures (Garcia-Fernandez et al. 2000) and dexamethasone or corticosterone treatment increases the pro-inflammatory leukotriene synthesis enzyme 5-lipoxygenase in both the hippocampus (Uz et al. 1999) and macrophages (Riddick et al. 1997). Finally, dexamethasone alone or more robustly in conjunction with retinoic acid, causes increased COX-1 expression in neuroblastoma cell lines (Schneider et al. 2001).
3.3.3 Anti- and pro-inflammatory GC effects at the transcription factor level
As discussed, a classic anti-inflammatory GC action in the periphery is to decrease the activity of NFκB. Commensurate with that, in unstressed rats, GR antagonists increase LPS-induced NFκB DNA binding activity in the frontal cortex and hippocampus (Munhoz et al. 2006). Moreover, dexamethasone treatment decreases basal NFκB DNA binding activity in cortex and hippocampus (Unlap and Jope 1995; Unlap and Jope 1997) and also following LPS treatment (Wang et al. 2006) (however there are some caveats to this interpretation that will be discussed later.)
In contrast, CUS increases LPS-induced NFκB DNA binding and protein levels in these brain structures (Figure 3), a GR-mediated effect (Munhoz et al. 2006). Moreover, CUS following direct LPS infusion into the prefrontal cortex increases components of the MAPK signaling pathway involved in inflammatory pathogenesis including phosphorylated JNK, p38, and ERK (de Pablos et al. 2006). These effects of CUS are also GR-mediated. Taken together, these findings suggest that as for other aspects of neuroinflammation, basal GC levels decrease pro-inflammatory transcription factor signaling while high GC levels enhance it.
Figure 3. Stress enhances NFκB activation in the CNS.
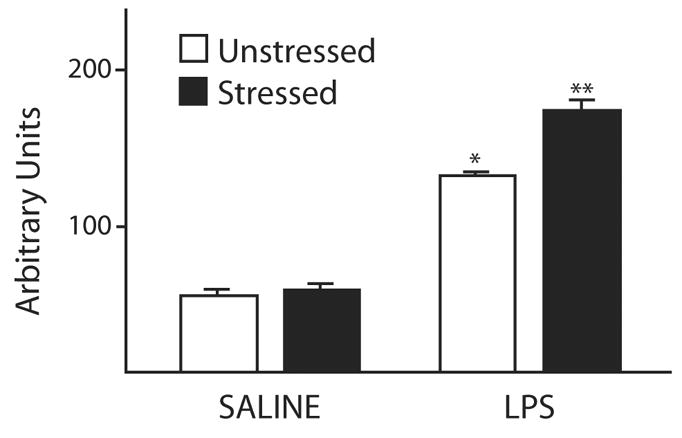
The DNA-binding activity of nuclear NFκB is increased in the frontal cortex of animals exposed to peripheral LPS and this effect is enhanced by stress. Administration of the GR antagonist RU-486 30 minutes prior to LPS completely attenuates the stress-induced increase (not shown). Animals were exposed to a chronic unpredictable stress paradigm (CUS, black bars), which consisted of varying daily stressors for two weeks or control (white bars). They then received i.v. saline or LPS and the DNA-binding activity of NFκB isolated from frontal cortical nuclei was assessed via electrophoretic mobility shift assay (EMSA). * p < 0.001 LPS relative to saline and ** p < 0.05 relative to LPS and no stress. [Based on data from Munhoz et al. 2006 Elsevier publishing, with permission]
4. DISCUSSION
This emerging literature suggests that it is no longer tenable to consider GCs to be universally anti-inflammatory. Moreover, the rules that dictate how GCs affect inflammation appear to be different between the CNS and periphery. Before we discuss the conditions that lead to increased or decreased inflammation, we first address several potential confounds in these studies.
4.1 Is GC-enhanced inflammation the result of GC resistance?
One possible explanation is that GC-mediated increases in inflammation might be due to the formation of GC resistance from exposure to chronic high doses. Indeed, many of the described hallmarks of the increased inflammatory response are similar to the increases seen from GC resistance. Resistance to GCs, however, is characterized by a decrease in GR function. In many of the studies reviewed, treatment with GR antagonists reverses the increased inflammation. This suggests that it is not a deficit in GR function, but rather a hyperactivity of GR signaling that is responsible for the observed increases in inflammation following chronic exposure to GCs.
4.2 Is GC-enhanced inflammation merely due to the neurotoxic effects of GCs?
As reviewed, stress and GCs can compromise the ability of hippocampal, cortical and striatal neurons to survive a variety of insults. This endangerment involves direct GC actions upon neurons themselves, resulting in exacerbation of the damaging cascade of glutamate/calcium/oxygen radical excess. Insults such as hypoxia-ischemia, seizure and hypoglycemia overwhelmingly result in necrotic, rather than apoptotic neuron death, and stress levels of GCs increase the incidence of necrotic, rather than apoptotic neurotoxicity (Sapolsky 1999; Roy and Sapolsky 2003). Given the inflammation triggered by necrotic neuron death, are the observed pro-inflammatory effects of GCs merely a secondary consequence of the hormone causing more necrotic neuron death?
This possibility cannot be completely discounted for every instance of GC enhanced inflammation, however, several lines of evidence weaken this interpretation. First, the pro-inflammatory effects of GCs have been observed prior to the emergence of neuron death (Dinkel et al. 2003). This suggests that the enhanced inflammation is not the result of GC-increased damage. Moreover, a correlative analysis of those data indicate that the extent of enhanced inflammatory cell migration at a particular time point post-insult predicts the extent of augmented neurotoxicity at a later time point. This finding is compatible with the idea that GC-enhanced inflammation may, in part, contribute to the increased neuron death. In contrast, the extent to which GCs augment neurotoxicity at a particular time point does not predict the subsequent extent of inflammation (Dinkel et al. 2003). Finally, and perhaps most compellingly, GC-enhanced neuroinflammation can occur in the absence of neuron death (i.e. from peripheral LPS administration) (Munhoz et al. 2006; Munhoz in prep).
4.3 Is GC enhanced inflammation due to a decrease in local GC exposure?
There are often functional differences between synthetic and endogenous GCs. As noted, the synthetic GC dexamethasone differs from corticosterone in that the former only binds to GR. But an important additional difference is that dexamethasone crosses the blood-brain barrier (BBB) poorly due to the activity of the multi-drug resistance transporter p-glycoprotein (De Kloet et al. 1998). As a result, dexamethasone exerts a disproportionate percentage of its neuroendocrine effects at the pituitary, rather than the brain. A consequence of a strong dexamethasone signal at the pituitary is a negative feedback inhibition of the adrenocortical axis, resulting in decreased secretion of ACTH and corticosterone. The consequence is that little dexamethasone penetrates the brain parenchyma and systemic corticosterone secretion drops. Given that the total amount of GCs (either endogenous or synthetic) reaching the injured brain will decrease, are the pro-inflammatory effects of dexamethasone merely an artifact of a net decrease in GC anti-inflammatory actions?
This possibility has been raised (Glezer and Rivest 2004) as a possible explanation of observed pro-inflammatory effects of dexamethasone (Bruccoleri et al. 1999), but it cannot be universally applicable. First, GCs can augment pro-inflammatory cytokine expression in primary brain tissue culture, a setting where issues of BBB permeability and adrenocortical negative feedback regulation are irrelevant (MacPherson et al. 2005). Second, a similar question has been raised concerning the effects of dexamethasone on memory consolidation and for this topic there exist many lines of evidence that support the conclusion that like corticosterone, dexamethasone is capable of exerting its effects centrally (Roozendaal 2000). Finally, as reviewed, corticosterone is also capable of increasing inflammation in vivo and antagonism of GR signaling blocks this effect (de Pablos et al. 2006; Munhoz et al. 2006; Munhoz in prep).
In the following sections we consider circumstances that favor pro- and anti-inflammatory GC effects in the brain.
4.4 Basal/permissive levels of GCs versus stress levels
As reviewed, the more than half-century old view of the effects of stress and GCs on peripheral immunity has been modified in two important ways. The first is the recognition that the primary effect of stress on inflammation is not suppression, but rather stimulation; this reflects the immunostimulatory effects of sympathetic catecholamines in the first few minutes of the stress-response. These findings lead to the revisionist view that GC anti-inflammatory actions are not mediating the stress-response, but rather constraining it and facilitating recovery from it.
The second modification is the recognition that the early, immunostimulatory effects of stress are not merely due to catecholamines, but also to basal GC levels. These findings led to the revisionist view that at the beginning of the stress-response, basal levels (or even the earliest phases of stress-induced increases in GC secretion) have permissive immunostimulatory actions; this is the inverse-U of GC function. Importantly, this revisionist view of the stimulation of immunity by GCs does not challenge the traditional view that stress levels of GCs, and the pharmacological levels of synthetic GCs used clinically, are robustly immunosuppressive.
The finding that GCs can have pro-inflammatory effects in the brain would initially seem to fit comfortably in this revisionist picture, in that these GC actions would “merely” be basal, permissive effects, predominately mediated by MR occupancy at the beginning of a stress-response. The studies reviewed, however, make clear that this is not the case. It is basal levels of GCs or an acute stressor which inhibit TNF-α , IL-1β and iNOS expression and protein levels in the face of a neurological challenge, both in vivo and in cell culture (Goujon et al. 1996; Nadeau and Rivest 2003; MacPherson et al. 2005; Munhoz et al. 2006). Instead, sustained stress, stress levels of GCs, or heavy and selective occupancy of GR all have pro-inflammatory effects on these endpoints (Figures 2 and 3) (Uz et al. 1999; Garcia-Fernandez et al. 2000; Dinkel et al. 2003; MacPherson et al. 2005; de Pablos et al. 2006; Munhoz et al. 2006; Munhoz in prep). Collectively, the pro-inflammatory effects of GCs reported in the brain reflect effects of chronic stress far more than basal or acute GCs.
4.5 Synthetic versus endogenous GCs
Because of their clinical relevance, the properties of synthetic GCs (most commonly dexamethasone) are pervasively explored in the literature. Care must be taken when considering the actions of these compounds, as they are frequently quite different than the effects of endogenous GCs. Besides the aforementioned poor BBB permeability of dexamethasone, this GC also exhibits greatly reduced binding to normal GC carrier molecules such as the corticosteroid-binding globulin, and an enhanced in vivo affinity for GR as opposed to MR (McEwen et al. 1997; De Kloet et al. 1998). As a result, dexamethasone permeates tissues and activates nuclear receptors in a way that is completely different than endogenous GCs. A concerted effort is needed to explore the differences between how endogenous and synthetic GCs affect inflammation in the CNS.
4.6 GCs prior to versus during or after inflammation
As reviewed, an observed trend in the periphery is that GC exposure far enough prior to an inflammatory stimulus results in an enhanced response to subsequent inflammation (Barber et al. 1993; Smyth et al. 2004). This phenomenon appears to be separate from the effects of GCs given at the same time as an ongoing inflammatory response as studies that administer GCs concurrent with, or following an inflammatory challenge are more likely to have classic anti-inflammatory effects (depending on dose, as discussed) (Goujon et al. 1995b; Goujon et al. 1996; Nadeau and Rivest 2003). A number of these investigations have looked at the effects of acute stress but not GCs. Nonetheless, many of the effects of prior acute stress are similar to those of prior GC treatment.
Prior acute stress also has measurable effects on subsequent inflammation in the CNS. If animals are exposed to an acute stressor 24 hours prior to LPS treatment, the result is an enhanced inflammatory response across several measures, including plasma and brain levels of IL-1β and TNF-α (Johnson et al. 2002a). More specifically, prior acute stress was found to enhance LPS-induced production of IL-1β in microglia (Frank et al. 2006). The enhanced response in the brain is relatively long-lived, being observable for at least four days following the stressor (Johnson et al. 2002b). Injection of IL-1 receptor antagonist completely blocked stress-enhancement of IL-1β production, illustrating that pro-inflammatory signaling by IL-1β is necessary to observe its own potentiation by stress. In addition, centrally injected human recombinant IL-1β was sufficient to reproduce the effects of an acute stressor (Johnson et al. 2004).
4.7 Regional differences in GC effects in the brain
To date, virtually all the reports of pro-inflammatory GC effects have examined the hippocampus or cortex (or cultures derived from them) (Dinkel et al. 2003; MacPherson et al. 2005; de Pablos et al. 2006; Munhoz et al. 2006; Munhoz in prep). One study found that LPS-induced NFκB activation, iNOS, and TNF-α levels were more pronounced in the cortex than the hippocampus across the full physiological range of corticosterone concentrations (Munhoz in prep). Another demonstrated simultaneous pro- and anti-inflammatory GC effects in different brain regions; specifically CUS stress exacerbated LPS-induced NFκB activation in the hippocampus and cortex, while inhibiting activation in the hypothalamus (Munhoz et al. 2006). One can readily imagine an array of mechanisms to explain these regional differences including, for example, differing concentrations of GR and MR in neurons, microglia or the vascular endothelium. Understanding the mechanisms that drive these regional differences is an important subject of future research.
5. SUMMARY AND CONCLUSIONS
The abundance of evidence in diverse experimental systems makes an unconfounded interpretation difficult, however, when summarized (Table I), several trends emerge. Based on these studies, the classic picture of GCs as anti-inflammatory must be modified in a number of ways:
Table I.
Experimental conditions where GCs have been found to increase inflammation
| PERIPHERAL | |||||
|---|---|---|---|---|---|
| GC dose | GC treatment time | Peripheral Location | Pro-inflammatory effect | Inflammatory stimulus | Reference |
| Acute stressor | Concurrent | Human PBMCs ex vivo, THP-1 cell line | NFkB activity | None | (Bierhaus et al. 2003) |
| Basal cort levels | Concurrent | Mouse macrophage cell line | MIF secretion | None | (Calandra et al. 1995) |
| Dexamethasone | Concurrent | Human PBMCs ex vivo | Cytokines, complement, chemokines | None | (Galon et al. 2002) |
| Dexamethasone | Concurrent | Human PBMCs ex vivo, THP-1 cell line | 5-lipoxygenase | None | (Riddick et al. 1997) |
| Dexamethasone | Concurrent | THP-1 cell line | IL-1 beta | PMA | (Wang et al. 1997) |
| Dexamethasone | Concurrent | THP-1 cell line | NFkB activity | PMA | (Wang et al. 1997) |
| Dexamethasone or prednisolone | >16 hours prior | Human lymphocyte cultures | Proliferation | Mitogens, x- linking Abs, or PMA+ionomycin | (Almawi et al. 1999) |
| Low stress cort levels | Acute 2hrs prior | Rat leukocytes in vivo | Leukocyte migration | DTH | (Dhabhar et al. 1996) |
| Low stress cort levels | Concurrent | Rat lymphocyte cultures | Proliferation | IL-2 | (Wiegers et al. 1995) |
| Low stress cort levels | >6 hours prior | Mouse macrophage cell line | NFkB activity | LPS and IFNγ | (Smyth et al. 2004) |
| Low stress cort levels | >6 hours prior | Mouse macrophage cell line | TNF-alpha, IL-6, nitrite | LPS and IFNγ | (Smyth et al. 2004) |
| Low stress cort levels | >12 hours prior | Human plasma | TNF-alpha, IL-6 | LPS | (Barber et al. 1993) |
| CENTRAL | |||||
| GC treatment | GC treatment time | Central Location | Pro-inflammatory effect | Inflammatory stimulus | Reference |
| Acute stressor | Concurrent | Rat cortex | NFkB activity | None | (Madrigal et al. 2001) |
| Acute stressor | 24 hours prior | Rat brain | IL-1 beta | Peripheral LPS | (Johnson et al. 2002) |
| Acute stressor | 24 hours prior | Rat hippocampal microglia | IL-1 beta | Peripheral LPS | (Frank et al. 2006) |
| CUS | Chronic 9 days after | Rat prefrontal cortex | Microglia activation | Central LPS | (de Pablos et al. 2006) |
| CUS | Chronic 9 days after | Rat prefrontal cortex | TNF-alpha | Central LPS | (de Pablos et al. 2006) |
| CUS | Chronic 9 days after | Rat prefrontal cortex | MAPK signaling | Central LPS | (de Pablos et al. 2006) |
| CUS | Chronic 14 days prior | Rat hippocampus, frontal cortex | TNF-alpha, IL-1 beta | Peripheral LPS | (Munhoz et al. 2006) |
| CUS | Chronic 14 days prior | Rat hippocampus, frontal cortex | NFkB activity | Peripheral LPS | (Munhoz et al. 2006) |
| Dexamethasone | Concurrent | Mouse neuronal cell line | PGD2 synthase | None | (Garcia-Fernandez et al. 2000) |
| Dexamethasone* | One hour prior | Mouse hippocampus | TNF-alpha, TNF-beta, IL-1 beta | TMT | (Bruccoleri et al. 1999) |
| High stress cort levels | Chronic 3 days prior | Rat hippocampus | Leukocyte migration | Kainic acid | (Dinkel et al. 2003) |
| High stress cort levels | Chronic 3 days prior | Rat hippocampus | Microglia activation | Kainic acid | (Dinkel et al. 2003) |
| High stress cort levels | Chronic 24 hours prior | Rat hippocampal cultures | TNF-alpha, IL-1 beta | Kainic acid | (MacPherson et al. 2005) |
| Intermediate stress cort levels | Chronic 3 days prior | Rat hippocampus, frontal cortex | TNF-alpha, IL-1 beta | Peripheral LPS | (Munhoz in prep) |
| Intermediate stress cort levels | Chronic 3 days prior | Rat hippocampus, frontal cortex | NFkB activity | Peripheral LPS | (Munhoz in prep) |
| Low stress cort levels | Chronic 10 days | Rat hippocampus, cerebellum | 5-lipoxygenase | None | (Uz et al. 1999) |
The variety of experimental conditions in which GC enhanced inflammation has been observed makes definitive conclusions difficult. The top table is a non-comprehensive list of pro-inflammatory effects of GCs on peripheral immunity. The bottom table lists conditions where GCs have been found to enhance signs of central inflammation and is more extensive. The type of GC treatment varies between experimental groups; however broad categorizations have been assigned. Basal stress cort levels are between 1-10μg/dL, low stress cort levels are between 10–25μg/dL, and high stress cort levels are above 25μg/dL. Dexamethasone doses are roughly equivalent to high stress cort doses at 10−7M. Importantly, enhanced inflammation due to acute stress is not necessarily attributable to GCs. GC treatment time refers to the time of exposure to GCs relative to a subsequent inflammatory stimulus or measurement. See text for a more detailed description of many of these investigations.
study potentially affected by poor BBB permeability of dexamethasone (see text).
Ab = antibody; cort = corticosterone (in rodent studies) or cortisol (in primate studies); CUS = chronic unpredictable stress; IL = interleukin; LPS = lipopolysaccharride; MAPK = mitogen-activated protein kinase;, MIF = macrophage migration initiation factor; NFkB = nuclear factor kappa B; PBMC = peripheral blood mononuclear cells; PGD2= prostaglandin D2; PMA is the NFkB activator phorbol 12-myristate 13-acetate; THP-1 = human monocyte cell line; TMT = trimethyltin; TNF = tumor necrosis factor
The immunosuppressive effects of GCs during chronic stress do not so much represent mediation of the immune stress-response as containment of and recovery from it. This conclusion rests heavily on the fact that the earliest phases of the stress-response involve enhancement of immunity in the periphery.
While much of that enhancement appears to be mediated by catecholamines, GCs do play a role. As such, in the periphery, basal GC concentrations and those in the low stress range have permissive pro-inflammatory effects, while high stress levels of GCs remain anti-inflammatory in the periphery.
These opposing GC actions fit well with other examples of inverse-U GC actions; moreover, a number of molecular mechanisms exist to explain such inverse-U GC effects.
In both the periphery and CNS, prior exposure to stress or GCs can result in a “priming” of the immune response to a subsequent inflammatory challenge. The involvement of GCs in this phenomenon remains to be more rigorously tested.
In the CNS, chronic GCs and stress are not uniformly anti-inflammatory and there are circumstances where they can actually increase inflammation; this has been observed across inflammatory cell extravasation and migration, inflammatory messenger levels, and transcription factor activation.
The picture of pro-inflammatory GC effects in the brain is quite different from that in the periphery. In the injured nervous system, it is high, stress levels of GCs that are pro-inflammatory, while basal or low stress levels have traditional anti-inflammatory effects; this is the opposite of what is seen in the periphery.
Whether pro-inflammatory GC effects occur in the brain can depend upon the brain region in question, the use of synthetic versus endogenous GCs, and the timing of GC exposure with respect to the inflammatory challenge.
These findings produce two challenges. The first is in the realm of basic science, namely to understand the mechanisms underlying the contrasting anti- and pro-inflammatory GC actions. As one example of the magnitude of this challenge, GCs can have diametrically opposite effects simultaneously in the injured cortex and hypothalamus (Munhoz et al. 2006). Contrasts such as these could well arise from differences in effects of GC/GR complexes upon GREs, from GC/GR interactions with other components of gene transcriptional machinery, or from mechanisms completely independent of the genome.
The second challenge is one in the realm of clinical neurology. There has often been a dogma about the uniformly beneficial effects of GCs in the face of brain inflammation. Yet, as long emphasized by leaders in the field, GCs do not lessen post-stroke edema and, if anything, worsen the outcome (Fishman 1982); furthermore, the same seemingly paradoxical worsening is seen when GCs are used after traumatic brain injury (Roberts et al. 2004). Finally, while there is increasing evidence that chronic low-grade brain inflammation can increase the likelihood of late-onset Alzheimer’s Disease, and that long-term NSAID treatment can buffer against this, long-term GC treatment has no such beneficial effects (Aisen et al. 2000). Thus, the basic science findings considered in this review may well have considerable clinical implications.
Acknowledgments
Manuscript assistance was provided by Jessie Ansari, Firdaus Dhabhar, Angela Lee, Carol Munhoz, Norman Ruby, and Sara Brownell; funding for some of the studies described was provided by the Adler Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, Thal LJ. A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000;54:588–593. doi: 10.1212/wnl.54.3.588. [DOI] [PubMed] [Google Scholar]
- Almawi WY, Hess DA, Assi JW, Chudzik DM, Rieder MJ. Pretreatment with glucocorticoids enhances T-cell effector function: possible implication for immune rebound accompanying glucocorticoid withdrawal. Cell Transplant. 1999;8:637–647. doi: 10.1177/096368979900800610. [DOI] [PubMed] [Google Scholar]
- Barber AE, Coyle SM, Marano MA, Fischer E, Calvano SE, Fong Y, Moldawer LL, Lowry SF. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol. 1993;150:1999–2006. [PubMed] [Google Scholar]
- Bernhagen J, Bacher M, Calandra T, Metz CN, Doty SB, Donnelly T, Bucala R. An essential role for macrophage migration inhibitory factor in the tuberculin delayed-type hypersensitivity reaction. J Exp Med. 1996;183:277–282. doi: 10.1084/jem.183.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci U S A. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black PH. Stress and the inflammatory response: a review of neurogenic inflammation. Brain Behav Immun. 2002;16:622–653. doi: 10.1016/s0889-1591(02)00021-1. [DOI] [PubMed] [Google Scholar]
- Blais V, Turrin NP, Rivest S. Cyclooxygenase 2 (COX-2) inhibition increases the inflammatory response in the brain during systemic immune stimuli. J Neurochem. 2005;95:1563–1574. doi: 10.1111/j.1471-4159.2005.03480.x. [DOI] [PubMed] [Google Scholar]
- Blecha F, Barry RA, Kelley KW. Stress-induced alterations in delayed-type hypersensitivity to SRBC and contact sensitivity to DNFB in mice. Proc Soc Exp Biol Med. 1982a;169:239–246. doi: 10.3181/00379727-169-41338. [DOI] [PubMed] [Google Scholar]
- Blecha F, Kelley KW, Satterlee DG. Adrenal involvement in the expression of delayed-type hypersensitivity to SRBC and contact sensitivity to DNFB in stressed mice. Proc Soc Exp Biol Med. 1982b;169:247–252. doi: 10.3181/00379727-169-41339. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Pennypacker KR, Harry GJ. Effect of dexamethasone on elevated cytokine mRNA levels in chemical-induced hippocampal injury. J Neurosci Res. 1999;57:916–926. [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377:68–71. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- Cox G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- Crossin KL, Tai MH, Krushel LA, Mauro VP, Edelman GM. Glucocorticoid receptor pathways are involved in the inhibition of astrocyte proliferation. Proc Natl Acad Sci U S A. 1997;94:2687–2692. doi: 10.1073/pnas.94.6.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- de Pablos RM, Villaran RF, Arguelles S, Herrera AJ, Venero JL, Ayala A, Cano J, Machado A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006;26:5709–5719. doi: 10.1523/JNEUROSCI.0802-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinzer R, Granrath N, Stuhl H, Twork L, Idel H, Waschul B, Herforth A. Acute stress effects on local Il-1beta responses to pathogens in a human in vivo model. Brain Behav Immun. 2004;18:458–467. doi: 10.1016/j.bbi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- del Zoppo GJ, Becker KJ, Hallenbeck JM. Inflammation after stroke: is it harmful? Arch Neurol. 2001;58:669–672. doi: 10.1001/archneur.58.4.669. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS. Stress-induced augmentation of immune function--the role of stress hormones, leukocyte trafficking, and cytokines. Brain Behav Immun. 2002;16:785–798. doi: 10.1016/s0889-1591(02)00036-3. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11:286–306. doi: 10.1006/brbi.1997.0508. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS, McEwen BS. Enhancing versus suppressive effects of stress hormones on skin immune function. Proc Natl Acad Sci U S A. 1999;96:1059–1064. doi: 10.1073/pnas.96.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhabhar FS, Miller AH, McEwen BS, Spencer RL. Stress-induced changes in blood leukocyte distribution. Role of adrenal steroid hormones. J Immunol. 1996;157:1638–1644. [PubMed] [Google Scholar]
- Dinkel K, Dhabhar FS, Sapolsky RM. Neurotoxic effects of polymorphonuclear granulocytes on hippocampal primary cultures. Proc Natl Acad Sci U S A. 2004;101:331–336. doi: 10.1073/pnas.0303510101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel K, MacPherson A, Sapolsky RM. Novel glucocorticoid effects on acute inflammation in the CNS. J Neurochem. 2003;84:705–716. doi: 10.1046/j.1471-4159.2003.01604.x. [DOI] [PubMed] [Google Scholar]
- Donnelly SC, Bucala R. Macrophage migration inhibitory factor: a regulator of glucocorticoid activity with a critical role in inflammatory disease. Mol Med Today. 1997;3:502–507. doi: 10.1016/S1357-4310(97)01133-7. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Chrousos GP. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966:290–303. doi: 10.1111/j.1749-6632.2002.tb04229.x. [DOI] [PubMed] [Google Scholar]
- Fishman RA. Steroids in the treatment of brain edema. N Engl J Med. 1982;306:359–360. doi: 10.1056/NEJM198202113060609. [DOI] [PubMed] [Google Scholar]
- Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci. 2004;1024:124–137. doi: 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2006 doi: 10.1016/j.bbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. Faseb J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- Garcia-Fernandez LF, Iniguez MA, Eguchi N, Fresno M, Urade Y, Munoz A. Dexamethasone induces lipocalin-type prostaglandin D synthase gene expression in mouse neuronal cells. J Neurochem. 2000;75:460–470. doi: 10.1046/j.1471-4159.2000.0750460.x. [DOI] [PubMed] [Google Scholar]
- Glezer I, Rivest S. Glucocorticoids: protectors of the brain during innate immune responses. Neuroscientist. 2004;10:538–552. doi: 10.1177/1073858404263494. [DOI] [PubMed] [Google Scholar]
- Golde S, Coles A, Lindquist JA, Compston A. Decreased iNOS synthesis mediates dexamethasone-induced protection of neurons from inflammatory injury in vitro. Eur J Neurosci. 2003;18:2527–2537. doi: 10.1046/j.1460-9568.2003.02917.x. [DOI] [PubMed] [Google Scholar]
- Gomes JA, Stevens RD, Lewin JJ, 3rd, Mirski MA, Bhardwaj A. Glucocorticoid therapy in neurologic critical care. Crit Care Med. 2005;33:1214–1224. doi: 10.1097/01.ccm.0000166389.85273.38. [DOI] [PubMed] [Google Scholar]
- Goujon E, Parnet P, Cremona S, Dantzer R. Endogenous glucocorticoids down regulate central effects of interleukin-1 beta on body temperature and behaviour in mice. Brain Res. 1995a;702:173–180. doi: 10.1016/0006-8993(95)01041-9. [DOI] [PubMed] [Google Scholar]
- Goujon E, Parnet P, Laye S, Combe C, Dantzer R. Adrenalectomy enhances pro-inflammatory cytokines gene expression, in the spleen, pituitary and brain of mice in response to lipopolysaccharide. Brain Res Mol Brain Res. 1996;36:53–62. doi: 10.1016/0169-328x(95)00242-k. [DOI] [PubMed] [Google Scholar]
- Goujon E, Parnet P, Laye S, Combe C, Kelley KW, Dantzer R. Stress downregulates lipopolysaccharide-induced expression of proinflammatory cytokines in the spleen, pituitary, and brain of mice. Brain Behav Immun. 1995b;9:292–303. doi: 10.1006/brbi.1995.1028. [DOI] [PubMed] [Google Scholar]
- Hara K, Kong DL, Sharp FR, Weinstein PR. Effect of selective inhibition of cyclooxygenase 2 on temporary focal cerebral ischemia in rats. Neurosci Lett. 1998;256:53–56. doi: 10.1016/s0304-3940(98)00755-1. [DOI] [PubMed] [Google Scholar]
- Herbert TB, Cohen S. Stress and immunity in humans: a meta-analytic review. Psychosom Med. 1993;55:364–379. doi: 10.1097/00006842-199307000-00004. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, Fleshner M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience. 2005;135:1295–1307. doi: 10.1016/j.neuroscience.2005.06.090. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Deak T, Spencer RL, Watkins LR, Maier SF. Prior stressor exposure primes the HPA axis. Psychoneuroendocrinology. 2002a;27:353–365. doi: 10.1016/s0306-4530(01)00057-9. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun. 2002b;16:461–476. doi: 10.1006/brbi.2001.0638. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Hansen MK, Watkins LR, Maier SF. Effects of prior stress on LPS-induced cytokine and sickness responses. Am J Physiol Regul Integr Comp Physiol. 2003;284:R422–432. doi: 10.1152/ajpregu.00230.2002. [DOI] [PubMed] [Google Scholar]
- Johnson JD, O'Connor KA, Watkins LR, Maier SF. The role of IL-1beta in stress-induced sensitization of proinflammatory cytokine and corticosterone responses. Neuroscience. 2004;127:569–577. doi: 10.1016/j.neuroscience.2004.05.046. [DOI] [PubMed] [Google Scholar]
- Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liles WC, Dale DC, Klebanoff SJ. Glucocorticoids inhibit apoptosis of human neutrophils. Blood. 1995;86:3181–3188. [PubMed] [Google Scholar]
- MacPherson A, Dinkel K, Sapolsky R. Glucocorticoids worsen excitotoxin-induced expression of pro-inflammatory cytokines in hippocampal cultures. Exp Neurol. 2005;194:376–383. doi: 10.1016/j.expneurol.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Madrigal JL, Moro MA, Lizasoain I, Lorenzo P, Castrillo A, Bosca L, Leza JC. Inducible nitric oxide synthase expression in brain cortex after acute restraint stress is regulated by nuclear factor kappaB-mediated mechanisms. J Neurochem. 2001;76:532–538. doi: 10.1046/j.1471-4159.2001.00108.x. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, Goldfarb RH, Kitson RP, Miller AH, Spencer RL, Weiss JM. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Magarinos AM. Stress and hippocampal plasticity: implications for the pathophysiology of affective disorders. Hum Psychopharmacol. 2001;16:S7–S19. doi: 10.1002/hup.266. [DOI] [PubMed] [Google Scholar]
- Mulholland PJ, Self RL, Hensley AK, Little HJ, Littleton JM, Prendergast MA. A 24 h corticosterone exposure exacerbates excitotoxic insult in rat hippocampal slice cultures independently of glucocorticoid receptor activation or protein synthesis. Brain Res. 2006;1082:165–172. doi: 10.1016/j.brainres.2006.01.069. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Munck A, Naray-Fejes-Toth A. The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol Cell Endocrinol. 1992;90:C1–4. doi: 10.1016/0303-7207(92)90091-j. [DOI] [PubMed] [Google Scholar]
- Munhoz CD. (in prep) [Google Scholar]
- Munhoz CD, Lepsch LB, Kawamoto EM, Malta MB, Lima Lde S, Avellar MC, Sapolsky RM, Scavone C. Chronic unpredictable stress exacerbates lipopolysaccharide-induced activation of nuclear factor-kappaB in the frontal cortex and hippocampus via glucocorticoid secretion. J Neurosci. 2006;26:3813–3820. doi: 10.1523/JNEUROSCI.4398-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Endotoxemia prevents the cerebral inflammatory wave induced by intraparenchymal lipopolysaccharide injection: role of glucocorticoids and CD14. J Immunol. 2002;169:3370–3381. doi: 10.4049/jimmunol.169.6.3370. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003;23:5536–5544. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor KA, Johnson JD, Hansen MK, Wieseler Frank JL, Maksimova E, Watkins LR, Maier SF. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res. 2003;991:123–132. doi: 10.1016/j.brainres.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Patel PM, Drummond JC, Sano T, Cole DJ, Kalkman CJ, Yaksh TL. Effect of ibuprofen on regional eicosanoid production and neuronal injury after forebrain ischemia in rats. Brain Res. 1993;614:315–324. doi: 10.1016/0006-8993(93)91050-3. [DOI] [PubMed] [Google Scholar]
- Petrovsky N, Socha L, Silva D, Grossman AB, Metz C, Bucala R. Macrophage migration inhibitory factor exhibits a pronounced circadian rhythm relevant to its role as a glucocorticoid counter-regulator. Immunol Cell Biol. 2003;81:137–143. doi: 10.1046/j.0818-9641.2002.01148.x. [DOI] [PubMed] [Google Scholar]
- Reagan LP. Glucose, stress, and hippocampal neuronal vulnerability. Int Rev Neurobiol. 2002;51:289–324. doi: 10.1016/s0074-7742(02)51009-6. [DOI] [PubMed] [Google Scholar]
- Reyes TM, Walker JR, DeCino C, Hogenesch JB, Sawchenko PE. Categorically distinct acute stressors elicit dissimilar transcriptional profiles in the paraventricular nucleus of the hypothalamus. J Neurosci. 2003;23:5607–5616. doi: 10.1523/JNEUROSCI.23-13-05607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddick CA, Ring WL, Baker JR, Hodulik CR, Bigby TD. Dexamethasone increases expression of 5-lipoxygenase and its activating protein in human monocytes and THP-1 cells. Eur J Biochem. 1997;246:112–118. doi: 10.1111/j.1432-1033.1997.00112.x. [DOI] [PubMed] [Google Scholar]
- Rivest S. Molecular insights on the cerebral innate immune system. Brain Behav Immun. 2003;17:13–19. doi: 10.1016/s0889-1591(02)00055-7. [DOI] [PubMed] [Google Scholar]
- Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, Laloe V, Munoz-Sanchez A, Arango M, Hartzenberg B, Khamis H, Yutthakasemsunt S, Komolafe E, Olldashi F, Yadav Y, Murillo-Cabezas F, Shakur H, Edwards P. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. 2004;364:1321–1328. doi: 10.1016/S0140-6736(04)17188-2. [DOI] [PubMed] [Google Scholar]
- Roozendaal B. 1999 Curt P. Richter award. Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology. 2000;25:213–238. doi: 10.1016/s0306-4530(99)00058-x. [DOI] [PubMed] [Google Scholar]
- Roy M, Sapolsky RM. The exacerbation of hippocampal excitotoxicity by glucocorticoids is not mediated by apoptosis. Neuroendocrinology. 2003;77:24–31. doi: 10.1159/000068337. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol. 1999;34:721–732. doi: 10.1016/s0531-5565(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Pulsinelli WA. Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications. Science. 1985;229:1397–1400. doi: 10.1126/science.4035356. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- Schneider N, Lanz S, Ramer R, Schaefer D, Goppelt-Struebe M. Up-regulation of cyclooxygenase-1 in neuroblastoma cell lines by retinoic acid and corticosteroids. J Neurochem. 2001;77:416–424. doi: 10.1046/j.1471-4159.2001.00264.x. [DOI] [PubMed] [Google Scholar]
- Simmons CP, Thwaites GE, Quyen NT, Chau TT, Mai PP, Dung NT, Stepniewska K, White NJ, Hien TT, Farrar J. The clinical benefit of adjunctive dexamethasone in tuberculous meningitis is not associated with measurable attenuation of peripheral or local immune responses. J Immunol. 2005;175:579–590. doi: 10.4049/jimmunol.175.1.579. [DOI] [PubMed] [Google Scholar]
- Smyth GP, Stapleton PP, Freeman TA, Concannon EM, Mestre JR, Duff M, Maddali S, Daly JM. Glucocorticoid pretreatment induces cytokine overexpression and nuclear factor-kappaB activation in macrophages. J Surg Res. 2004;116:253–261. doi: 10.1016/S0022-4804(03)00300-7. [DOI] [PubMed] [Google Scholar]
- Sterzer P, Wiegers GJ, Reul JM. Long-term in vivo administration of glucocorticoid hormones attenuates their capacity to accelerate in vitro proliferation of rat splenic T cells. Endocrinology. 2004;145:3630–3638. doi: 10.1210/en.2003-1578. [DOI] [PubMed] [Google Scholar]
- Unlap MT, Jope RS. Dexamethasone attenuates NF-kappa B DNA binding activity without inducing I kappa B levels in rat brain in vivo. Brain Res Mol Brain Res. 1997;45:83–89. doi: 10.1016/s0169-328x(96)00240-9. [DOI] [PubMed] [Google Scholar]
- Unlap T, Jope RS. Inhibition of NFkB DNA binding activity by glucocorticoids in rat brain. Neurosci Lett. 1995;198:41–44. doi: 10.1016/0304-3940(95)11963-w. [DOI] [PubMed] [Google Scholar]
- Uz T, Dwivedi Y, Savani PD, Impagnatiello F, Pandey G, Manev H. Glucocorticoids stimulate inflammatory 5-lipoxygenase gene expression and protein translocation in the brain. J Neurochem. 1999;73:693–699. doi: 10.1046/j.1471-4159.1999.0730693.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan K, Dhabhar FS. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc Natl Acad Sci U S A. 2005;102:5808–5813. doi: 10.1073/pnas.0501650102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kang JS, Li Y, Yuan ZX, Liu SS, Sun LK. The effects of dexamethasone on rat brain cortical nuclear factor kappa B (NF-kappaB) in endotoxic shock. Toxicol Appl Pharmacol. 2006;214:263–269. doi: 10.1016/j.taap.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Wick G, Hu Y, Schwarz S, Kroemer G. Immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in autoimmune diseases. Endocr Rev. 1993;14:539–563. doi: 10.1210/edrv-14-5-539. [DOI] [PubMed] [Google Scholar]
- Wiegers GJ, Labeur MS, Stec IE, Klinkert WE, Holsboer F, Reul JM. Glucocorticoids accelerate anti-T cell receptor-induced T cell growth. J Immunol. 1995;155:1893–1902. [PubMed] [Google Scholar]
- Wiegers GJ, Reul JM. Induction of cytokine receptors by glucocorticoids: functional and pathological significance. Trends Pharmacol Sci. 1998;19:317–321. doi: 10.1016/s0165-6147(98)01229-2. [DOI] [PubMed] [Google Scholar]
- Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand. 2004;48:799–813. doi: 10.1111/j.1399-6576.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- Yoshimura C, Miyamasu M, Nagase H, Iikura M, Yamaguchi M, Kawanami O, Morita Y, Iwata T, Yamamoto K, Hirai K. Glucocorticoids induce basophil apoptosis. J Allergy Clin Immunol. 2001;108:215–220. doi: 10.1067/mai.2001.116575. [DOI] [PubMed] [Google Scholar]