

Abstract
Background
Complex physiological interactions determine the functional consequences of gene abnormalities and make mechanistic interpretation of phenotypes extremely difficult. A recent example is a single mutation in the C terminus of the cardiac Na+ channel, 1795insD. The mutation causes two distinct clinical syndromes, long QT (LQT) and Brugada, leading to life-threatening cardiac arrhythmias. Coexistence of these syndromes is seemingly paradoxical; LQT is associated with enhanced Na+ channel function, and Brugada with reduced function.
Methods and Results
Using a computational approach, we demonstrate that the 1795insD mutation exerts variable effects depending on the myocardial substrate. We develop Markov models of the wild-type and 1795insD cardiac Na+ channels. By incorporating the models into a virtual transgenic cell, we elucidate the mechanism by which 1795insD differentially disrupts cellular electrical behavior in epicardial and midmyocardial cell types. We provide a cellular mechanistic basis for the ECG abnormalities observed in patients carrying the 1795insD gene mutation.
Conclusions
We demonstrate that the 1795insD mutation can cause both LQT and Brugada syndromes through interaction with the heterogeneous myocardium in a rate-dependent manner. The results highlight the complexity and multiplicity of genotype-phenotype relationships, and the usefulness of computational approaches in establishing a mechanistic link between genetic defects and functional abnormalities.
Keywords: arrhythmia, genes, long-QT syndrome, Brugada syndrome, sodium
A number of defects in the cardiac Na+ channel gene SCN5A have been linked to congenital forms of long-QT (LQT) and Brugada syndromes.1–10 In general, the SCN5A LQT syndrome (LQT3) results from a gain of Na+ channel function that allows for conduction of Na+ ions at depolarized membrane potentials.1,2,7 This leads to persistent INa during the action potential (AP) plateau, which results in prolongation of the AP duration (APD) that can facilitate the development of arrhythmogenic early after depolarizations (EADs).8 The prolonged APD is reflected on the body-surface ECG as increased duration of the QT interval.11
Unlike LQT defects, mutations in SCN5A underlying the Brugada syndrome act to reduce Na+ current. The reduction in INa is expected to result in suppression of the AP plateau in cells with prominent Ito, such as those in the right ventricular epicardium.9,10 Preferential suppression of the AP plateau in epicardial cells will generate a potential gradient that can manifest as ST-segment elevation on the ECG.10
1795insD results from insertion of aspartic acid in the C terminus of SCN5A.12,13 The mutation results in development of both LQT and Brugada syndromes. 1795insD disrupts the Na+ current by enhancing channel inactivation and stabilizing inactivation states, which reduces channel availability. The mutation also promotes channel bursting, resulting in a persistent component of noninactivating current.
Ion channel proteins are expressed nonuniformly within the myocardium, resulting in an intrinsic electrophysiological heterogeneity.14 Here, we use a computational approach to demonstrate that it is precisely this heterogeneity that allows seemingly paradoxical phenotypes associated with 1795insD to coexist. The interplay between the underlying electrophysiological heterogeneity of the myocardium and 1795insD-induced changes in cardiac Na+ channel function provides the substrate for development of both ECG ST-segment elevation (Brugada) and QT interval prolongation (LQT) in a rate-dependent manner.
Methods
The general approach to modeling using the dynamic Luo-Rudy (LRd) model of a ventricular cell15 with recent modifications has been described previously.16–19
Simulations are conducted in isolated epicardial, endocardial, and midmyocardial (M) cells, which are simulated by varying the maximum conductance (density), GKs, of the slowly activating delayed-rectifier potassium current IKs as described previously.18 Ito, the transient outward current, is incorporated into the LRd model using the formulation of Dumaine et al.9 Greenstein et al20 recently investigated the effect of Ito on several dynamic AP models and found qualitatively similar responses in all models. Ito has been included in right ventricular outflow tract (RVOT) epicardial cells, with maximum conductance (Gto) of 1.1 mS/μF.9 The density ratio of IKs to IKr (the rapidly activating delayed rectifier) GKs/GKr = 23.14,18 In M cells, Gto = 0.5 mS/μF9 and GKs/GKr = 17.14,18 In endocardial cells, Gto = 0.059 and GKs/GKr = 19.14,18
The Markov formulation of INa (Figure 1) extends a previously developed model8 and includes several new features, including recently described intermediate inactivation states,4,13 to more accurately simulate channel inactivation and recovery. A fast inactivation state (IF) and two intermediate inactivation states (IM1 and IM2) are required to reproduce the complex fast and slow recovery features of inactivation described recently.3–5,13 The IM1 state acts as a channel “sink” in which the majority of channels reside but are unable to recover and reopen during depolarization. A few channels enter the IM2 state via slow transitions.13 An improved representation of channel closed-state inactivation is achieved via the inclusion of two closed-inactivation states (IC2 and IC3). This allows for channel inactivation from any of the closed available states, allowing for accurate representation of channel availability throughout the physiological voltage range. The wild-type (WT) INa model was developed on the basis of a wide range of data presented in the experimental literature. Model parameters were fit over the physiological range (−140→ + 70 mV). Microscopic reversibility is ensured by fixing the products of the forward and reverse transition rates of closed loops in the model.
Figure 1.
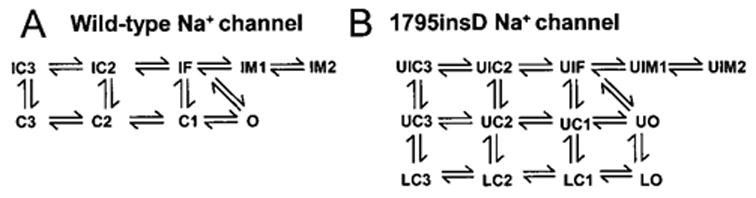
Markovian models for WT (A) and 1795insD (B) cardiac Na+ channels. Mutant model contains two gating modes. U (upper) indicates background mode of gating; L (lower), a small population of bursting channels that fail to inactivate. See text for details.
More information is available in the online Data Supplement, which contains the model equations and additional validation to that provided in Clancy and Rudy.8
Results
Markovian models of WT and 1795insD mutant Na+ channels are shown in Figure 1A and 1B, respectively. 1795insD contains two gating modes. The “background mode” includes the upper (U) nine states and is similar to WT except that entry into the intermediate inactivation states is more favorable (faster transition into UIM1 and UIM2) and rates of recovery from inactivation are reduced. The four lower (L) states of 1795insD (“burst mode”) represent a small channel population (<0.01%) that transiently fails to inactivate. Channels bounce among closed (LC3, LC2, and LC1) and open states (LO), conducting Na+ at potentials at which WT channels normally inactivate, generating late INa during the plateau of the AP.8,13
Figure 2 compares experimentally recorded13 (A, left) and simulated (B, left) recovery from inactivation (protocol, bottom left). The ratio of peak macroscopic INa during a test pulse (P2) to peak current during the conditioning pulse (P1) is shown for WT and 1795insD channels. The rightward shift of the 1795insD recovery curve relative to WT indicates slowed recovery from inactivation. Logarithmic scale (inset) demonstrates that mutants may require >100 ms to recover from inactivation. The majority of channels in both WT and mutant cases (>99%) reside in IM1 and UIM1, respectively, corresponding to a stable channel conformation that acts as a channel sink after fast inactivation. A greater number of mutant channels are “trapped” in the more absorbing UIM2 mutant state (9.1% of channels at 100 ms) than in the WT IM2 state (only 3.5% of channels at 100 ms). By 500 ms, < 1% of channels reside in the WT IM2 state, whereas nearly 3% of channels reside in the mutant UIM2 state.
Figure 2.
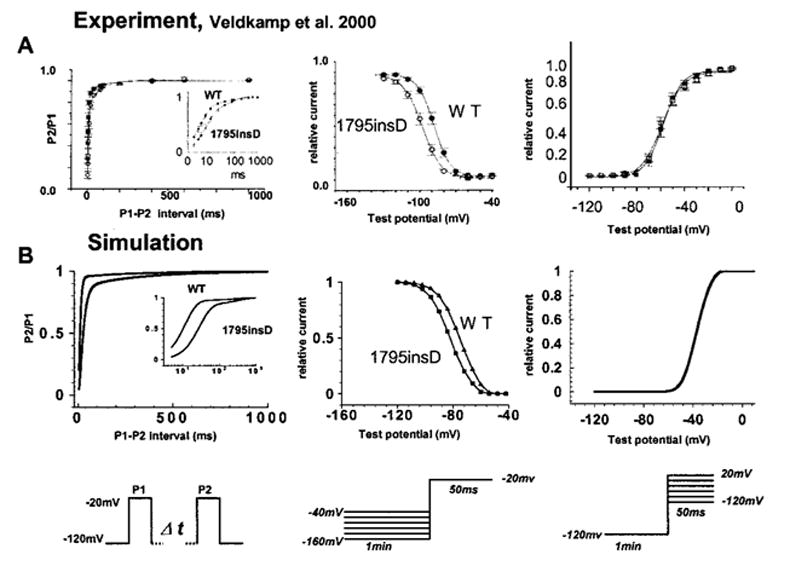
Left, 1795insD mutation reduces rate of recovery from inactivation. Experiment13 and simulation are shown in left panels of A and B, respectively. Mutant channels may take 100 ms to recover from inactivation. Inset illustrates this property on expanded log scale. Protocol is shown at bottom. P1 is a conditioning pulse and P2 is a test pulse. Middle, 1795insD mutation results in reduced channel availability. In both experiment (A) and simulation (B), channel availability curve is shifted leftward because of increased absorbency of inactivation states resulting from the mutation. Protocol is shown beneath middle panels.13 Right, Voltage dependence of activation is unaffected by the 1795insD mutation. WT and mutant steady-state activation curves are indistinguishable in both experiment (A)13 and simulation (B). Protocol is shown beneath right panels.
1795insD channels are more likely to enter inactivation states from closed states than WT, and less likely to recover from these inactivation states because of slower recovery transition rates.13 Together with the increased absorbency of UIM2, this increases 1795insD residency in inactivation states, thereby reducing channel availability and shifting the mutant steady-state channel availability curve by −10 mV in the experiment (Figure 2A, middle) and −8 mV in the simulation (Figure 2,B middle). Interestingly, this result suggests that reduced rates of recovery are sufficient to explain the leftward shift of the availability curve. The mutation does not affect the voltage dependence of channel activation in either the experiment13 (Figure 2A, right) or the simulation (Figure 2B, right).
WT and 1795insD INa display a progressive reduction of peak current at fast stimulation rates. This phenomenon is amplified in the mutant as a result of the increased absorbency of inactivation states. Figure 3 illustrates this rate-dependent reduction in experimental13 (A) and simulated (B) currents. At a stimulation interval of 2.5 s, a long recovery period (2.0 s at −100 mV) after a 500-ms depolarization to 0 mV allows for complete recovery from inactivation of both WT (Figure 3A and 3B, top left) and 1795insD mutant currents (Figure 3A and 3B, top right). Normalized peak currents in both the experiment13 and simulation (Figure 3C) show little to no reduction during stimulation at 2.5-s intervals (WT, solid squares; 1795insD, open circles). In contrast, at a stimulation interval of 0.52 s, a short recovery period (20 ms) after a 500-ms depolarization results in a progressive loss of current during periodic stimulation. The current reduction is more pronounced in the 1795insD mutant (Figure 3A and 3B, bottom right) compared with WT (Figure 3A and 3B, bottom left). The larger rate-dependent reduction in mutant current is shown in Figure 3C (open squares indicate WT; solid circles, 1795insD) in both experiment (top) and simulation (bottom).
Figure 3.
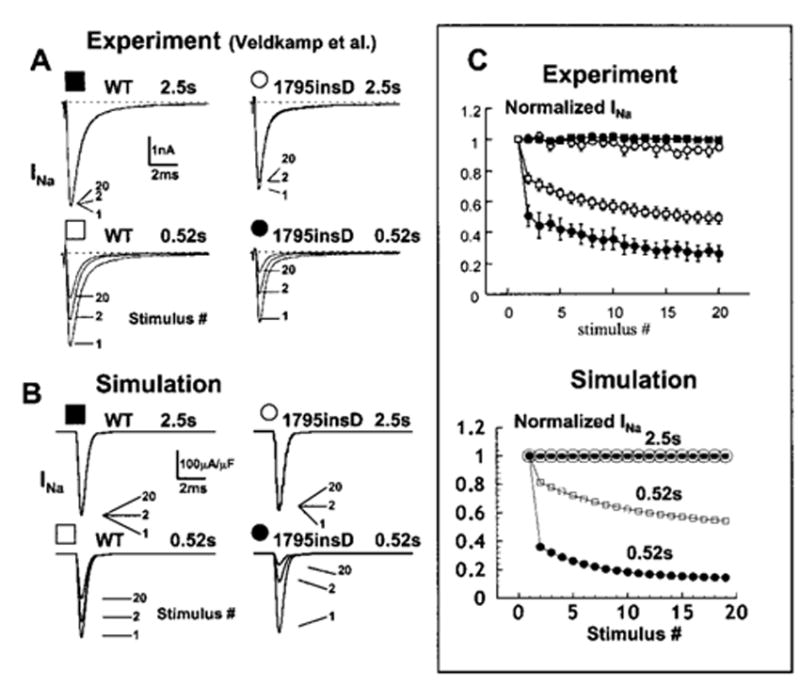
1795insD causes a rate-dependent reduction in experimental13 (A) and simulated INa (B). A train of 500-ms depolarizing pulses is applied with 2000-ms (2.5-s interval) and 20-ms (0.52-s interval) recovery periods shown in top and bottom panels, respectively, for WT (left) and mutant (right). INa is shown for 1, 2, and 20 stimuli. At 2.5 s, WT and mutant channels recover from inactivation between stimuli. At 0.52 s, both exhibit a rate-dependent current reduction. Reduction in 1795insD INa is greater than WT. Normalized peak INa is shown in panel C in experiment13 (top) and simulation (bottom). 1795insD mutation (●) increases rate-dependent current reduction at fast rates (0.52-s interval) compared with WT (□).
To study the cellular electrophysiological consequences of the mutation, WT and 1795insD channel models are inserted into the LRd model of the cardiac ventricular cell.8,15–18 The ventricular myocardium is heterogeneous, comprising epicardial cells, M cells, and endocardial cells with distinct electrophysiological properties.11,14 Using models of these different cell types, we show that 1795insD affects the epicardial and M-cell AP differently, explaining the seemingly paradoxical coexistence of Brugada and LQT phenotypes.
Effects of 1795insD mutation on RVOT epicardial cells are shown in Figure 4 for three cycle lengths (CLs) of 300 ms (A), 750 ms (B), and 1000 ms (C). The WT AP (thin line) exhibits a characteristic spike-and-dome morphology as a result of a large Ito and a short APD due to a large IKs. In contrast, the 1795insD APs (thick line) exhibit distinctive rate-dependent morphologies. At fast rates (300 ms), there is alternation between loss of the AP dome and a “coved dome”21 morphology (Figure 4A). At slower rates (750 ms [Figure 4B] and 1000 ms [Figure 4C]), epicardial cell APs exhibit a coved dome morphology that becomes less pronounced as pacing is slowed. Figure 4D compares 1795insD AP morphologies in endocardial and epicardial cells. The left panel shows an epicardial AP displaying loss of the dome. The right panel shows a coved-dome epicardial AP. Notably, the coved-dome morphology is associated with a prolonged epicardial APD resulting in a crossover of epicardial and endocardial APs.
Figure 4.
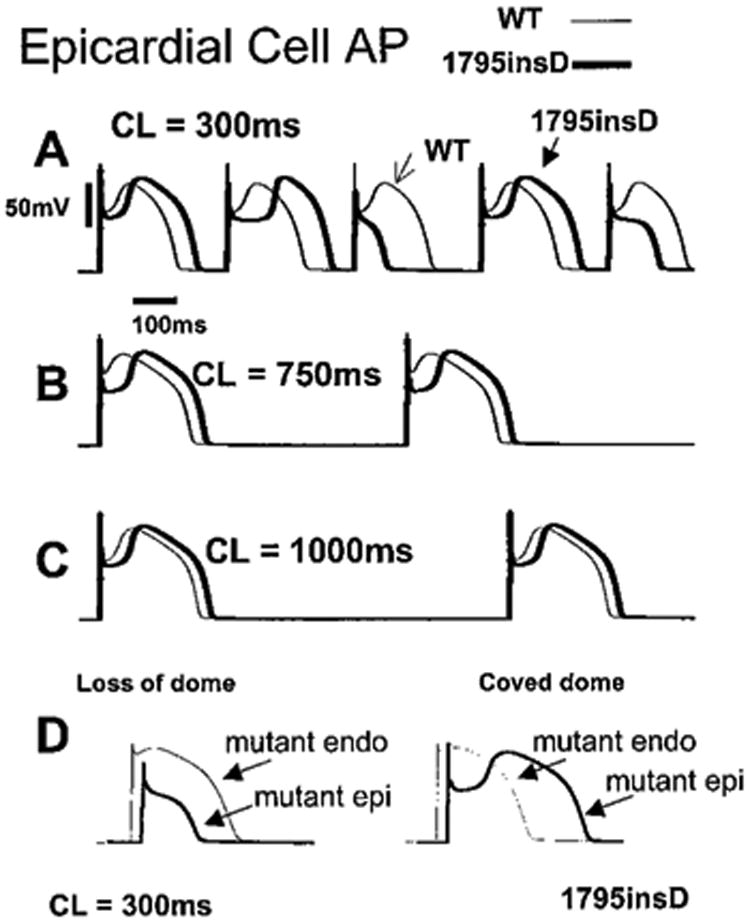
A, 1795insD affects AP of epicardial cells in a rate-dependent manner. A simulated epicardial cell is paced for 100 beats from resting steady state at three pacing rates. At fast rate (300 ms; A), mutant AP morphology alternates between spike and dome with a notch (“coved dome”) and loss of dome. At intermediate rate (750 ms; B), mutant is coved dome on every beat. At slow rate (1000 ms; C), WT and mutant morphologies are similar. D, Comparison of 1795insD effects on epicardial (thick line) and endocardial (thin line) APs at a fast rate (CL = 300 ms).
The mechanism underlying the AP morphologies in epicardial cells during fast pacing is shown in Figure 5. Simulated 96th through 100th paced beats are shown, with AP in the top panel and corresponding INa and ICaL (the L-type calcium current) in the middle and bottom panels, respectively. Mutant peak INa (right) alternates as a result of the mutation-induced slowing of channel recovery kinetics. WT channels recover between beats even at the fast rate (left). The alternating peak INa interacts with large outward Ito and IKs present in RVOT epicardial cells.9,10,14,18 When INa is reduced because of incomplete recovery (arrows), Ito and IKs tilt the balance of currents in the outward direction, causing premature repolarization before significant activation of plateau ICaL. This leads to suppression of the AP plateau and premature repolarization with loss of the AP dome. On alternate beats, Na+ channels may recover more completely, thereby allowing for a larger INa. The resulting higher membrane potential and shallower AP notch allow for substantial activation of plateau ICaL, which generates the AP dome.
Figure 5.
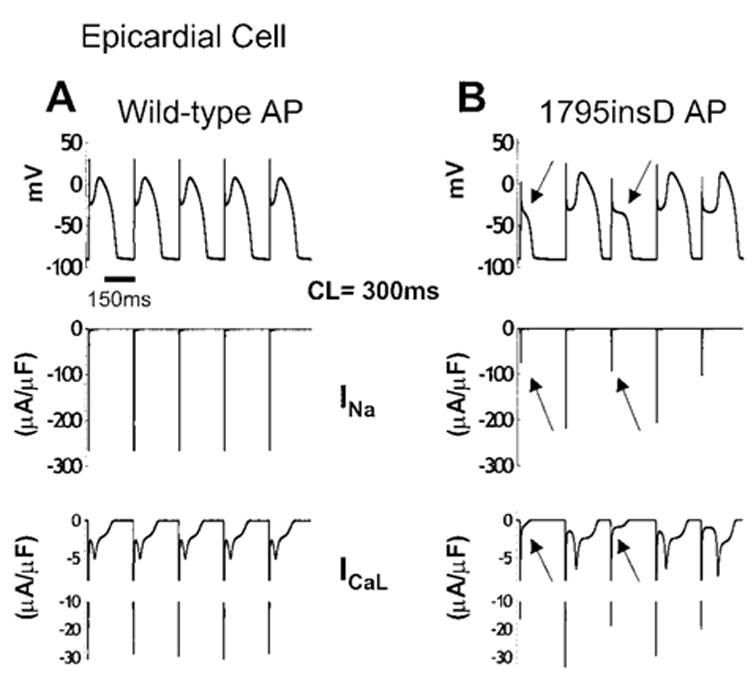
AP morphology changes in 1795insD epicardial cells. The 96th through 100th beats (CL = 300 ms) are shown for a simulated epicardial cell. A, WT APs (top) with INa (middle) and ICaL (bottom). B, 1795insD APs (top) with corresponding INa and ICaL. Beat-dependent reduction in mutant INa (middle, arrows) on the background of large Ito and IKs present in epicardial cells acts to deepen the AP notch, which suppresses plateau ICaL activation (bottom, arrows), resulting in loss of the AP dome (top, arrows).
In M cells, effects of 1795insD are also rate dependent. An M cell paced at three different rates is shown in Figure 6. WT (thin line) and mutant (thick line) APs are superimposed. At a fast rate (Figure 6A, 300 ms), the 1795insD had little effect on the M-cell AP morphology (WT and mutant APs are practically superimposed). At 850 ms (Figure 6B), the mutation results in a prolongation of APD by ≈60 ms. Slow pacing (Figure 6C, 1000 ms) leads to a markedly prolonged APD and arrhythmogenic EADs.17
Figure 6.
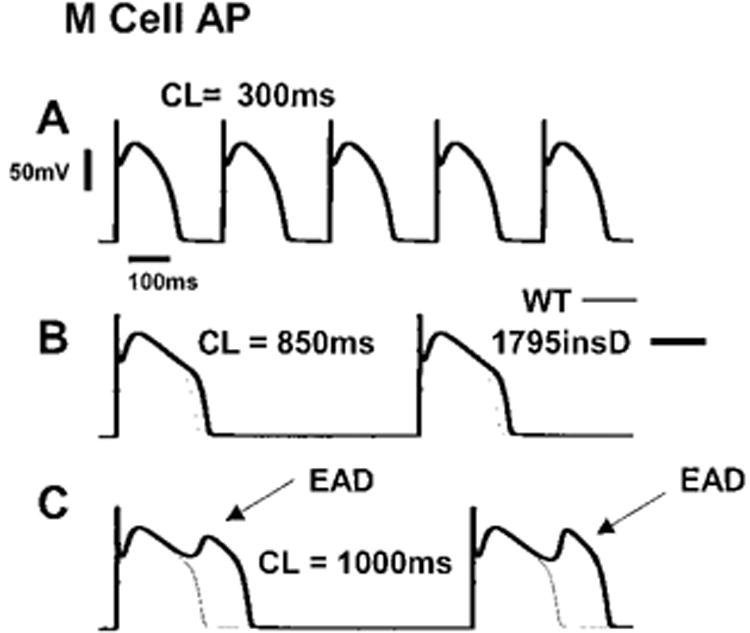
1795insD affects M cells in a rate-dependent manner. At fast rates (300 ms, A), M-cell APs are unaffected. At a rate of 850 ms, AP is prolonged by the mutation (B). At slow rates (1000 ms; C), arrhythmogenic EADs develop.
Figure 7 demonstrates the mechanism of AP prolongation in M cells containing the 1795insD mutation at slow rates. Unlike WT APs (Figure 7A), mutant M-cell APs (Figure 7B) are prolonged and plagued with EADs (arrows). The diastolic interval is sufficiently long to allow for complete recovery of Na+ channels between beats. Recovered channels open, resulting in the AP upstroke. In mutant cells, a small number of bursting channels conduct Na+ during the AP plateau, generating late INa (arrow). This current prolongs the plateau, allowing for the recovery and reactivation of ICaL that depolarizes the membrane and generates the EADs.17 This behavior is characteristic of the LQT syndrome.1,8
Figure 7.
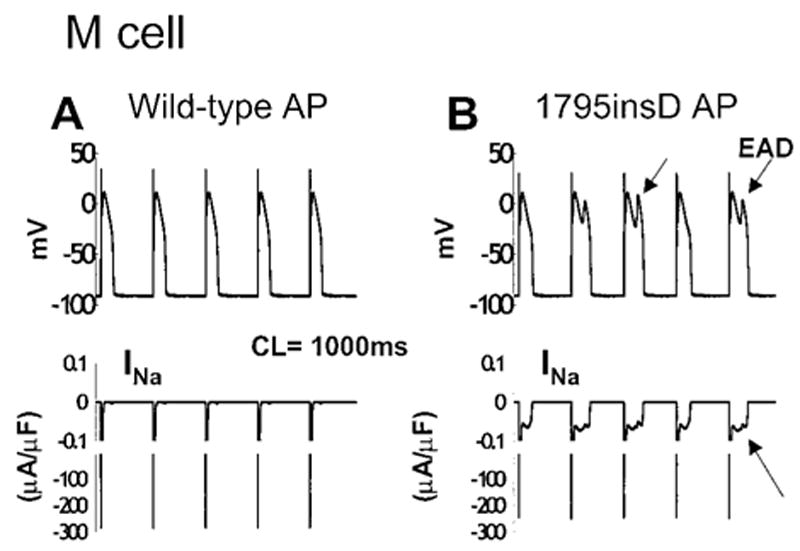
Mechanism of AP prolongation in 1795insD M cells. The 96th through 100th APs (top) are shown in WT (A) and mutant (B) simulated cells with corresponding INa (bottom). At a slow rate (1000 ms), mutants recover from inactivation between beats and burst, generating late INa (lower arrow), which prolongs APD and causes EAD generation (upper arrows).
The simulations in Figure 8 demonstrate the cellular basis of the ECG changes associated with the 1795insD mutation and their rate dependence. The effects of AP changes caused by the 1795insD mutation on the transmural potential gradient are shown at fast (Figure 8A, 300 ms) and slow (Figure 8B, 850 ms) rates. The selective variation in AP morphology in epicardial cells (Figure 8A, thick line) and not in M cells (Figure 8A, thin line) at fast rates results in dispersion of plateau potentials across the ventricular wall. During the plateau phase, a potential gradient (▽Vm) is established from the epicardial to the M-cell AP. Because ECG potentials are proportional to −▽Vm,22 this gradient generates ST-segment elevation on the ECG, the hallmark of Brugada syndrome.9,10,21 The coved-dome AP in Figure 8A has a prolonged APD that extends beyond the APD of the M cell at this fast heart rate. This “crossover” reverses the transmural potential gradient during the repolarization phase and explains the inversion of the T wave that is seen in patients with the 1795insD mutation.12 In Figure 8B, the cellular mechanism of QT prolongation is shown at a slow CL of 850 ms. In both WT and mutant simulated cells, APD is increased as pacing is slowed as a result of adaptation.16,18 The 1795insD mutant M cell (Figure 8B, thick line) exhibits a larger APD prolongation (ΔAPD ≈60 ms) compared with WT (Figure 8B, thin line). The longer diastolic interval at a slow pacing frequency allows for more channel recovery from inactivation (UI states → UC states) and increased channel availability. Available channels have the opportunity to enter the burst mode via UC → LC transitions. This results in an increased bursting channel population that acts to prolong the APD. APD prolongation is reflected in a prolonged QT interval on the ECG, the hallmark of the LQT syndrome.
Figure 8.
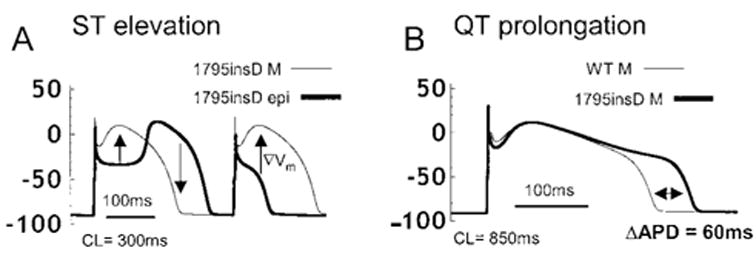
Effects of1795insD on transmural voltage gradients and APD. A, At fast rates, mutation-induced changes in epicardial AP morphologies (thick line) cause dispersion of plateau potentials and a voltage gradient (▽Vm, arrows) from epicardial to M cell (thin line). This gradient will manifest on the ECG as ST-segment elevation, indicative of Brugada syndrome. For coved-dome morphology of the epicardial AP, ▽Vm is reversed during phase 3 repolarization, which can cause T-wave inversion on the ECG. B, At a slow rate (CL = 850 ms), mutation prolongs APD in M cells (thick line) compared with WT (thin line). Delay in repolarization (ΔAPD ≈60 ms) is reflected as QT prolongation on the ECG, a hallmark of LQT syndrome.
Discussion
This study demonstrates that a single gene defect, through its interaction with a heterogeneous physiological substrate, can give rise to multiple distinct phenotypes with different pathological and clinical manifestations. Here, the single mutation actually results in opposite phenotypes, as follows: LQT, associated with a gain of Na+ channel function and QT prolongation on the ECG, and Brugada with loss of Na+ channel function and ST-segment elevation. Our theoretical approach provides a possible mechanistic explanation for this paradoxical duality. 1795insD INa exhibits a rate-dependent reduction in peak current amplitude and contains a late component during the AP plateau. In epicardial cells, the balance of reduced INa on the background of large repolarizing currents (Ito and IKs) results in alternation between premature plateau repolarization and coved AP domes that can cause ST-segment elevation and T-wave inversion on the ECG (Brugada phenotype). Clinically, ST-segment elevation is observed in right precordial leads of Brugada patients, consistent with the large Ito density in right ventricular epicardium.9,10 In M cells, in which Ito and IKs are smaller, premature repolarization does not occur and late INa acts to prolong the AP plateau, which increases the QT interval on the ECG (LQT phenotype).
This study provides insight into the role of changes in cardiac Na+ channel biophysical properties in the complex behavior of the heterogeneous myocardium. We develop new models of the cardiac WT and 1795insD mutant Na+ channel that allow study of complex features of closed- and open-state inactivation. We demonstrate that the measured reduction in the rate of Na+ channel recovery from inactivation is sufficient to account for the negative shift in the steady-state inactivation curve, reflecting a reduction in channel availability. The altered recovery kinetics arising from the 1795insD mutation are the basis for the severe dynamic AP changes observed at fast rates in simulated epicardial cells. These changes, relative to a much smaller effect on M-cell AP, provide a mechanistic basis for ST-segment elevation and T-wave inversion seen in patients.
This study also provides insight into the rate dependence of channel bursting. At slow rates, the long diastolic interval allows mutant channels to fully recover from inactivation to closed available states (UI states → UC states) at which they have the opportunity to enter the burst mode (UC states → LC states) and conduct Na+ at plateau potentials at which WT channels are normally inactivated, leading to APD prolongation. At fast heart rates, the short diastolic interval results in reduced channel recovery from inactivation (UI states → UC states), resulting in a reduced likelihood of channel bursting. Consequently, the sustained INa is smaller, which can facilitate the rate-dependent loss of the AP dome in epicardial cells.
β-Agonist therapy has been proposed as a remedy for ST-segment elevation in Brugada patients. Presumably through action on ICaL, isoproterenol restores the AP plateau10 and normalizes ST elevation.23 Our study suggests that β-agonist therapy would be ill advised in patients with 1795insD. An increase in ICaL would likely restore the AP plateau in epicardial cells and reduce ST elevation. However, it would also increase the severity of AP prolongation in M cells. At slow heart rates, this could lead to a large dispersion of repolarization and EADs that might trigger severe life-threatening arrhythmias. One should consider though, that β-agonist exposure has multiple effects,24 including enhancing IKs, which will counteract the APD prolongation effect of ICaL. Therefore, the outcome of β-agonist intervention will depend on its effects on various interactive processes within the complex dynamic system of the cell.
Supplemental Data
Acknowledgments
This work was supported by grants from the National Institutes of Health, National Heart, Lung, and Blood Institute (R01-HL49054 and R37-HL33343 to Dr Rudy), and a Whitaker Foundation Development Award. Dr Clancy also was supported by National Institutes of Health grant T32 HL07887-03.
Footnotes
The online version of this article, along with updated information and services, is located on the World Wide Web at: http://circ.ahajournals.org/cgi/content/full/105/10/1208
References
- 1.Bennett PB, Yazawa K, Makita N, et al. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 2.Dumaine R, Wang Q, Keating MT, et al. Multiple mechanisms of Na+ channel-linked Long-QT syndrome. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 3.Abriel H, Cabo C, Wehrens XH, et al. Novel arrhythmogenic mechanism revealed by a long-QT syndrome mutation in the cardiac Na(+) channel. Circ Res. 2001;88:740–745. doi: 10.1161/hh0701.089668. [DOI] [PubMed] [Google Scholar]
- 4.Wang DW, Makita N, Kitabatake A, et al. Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ Res. 2000;87:E37–E43. doi: 10.1161/01.res.87.8.e37. [DOI] [PubMed] [Google Scholar]
- 5.Wehrens XH, Abriel H, Cabo C, et al. Arrhythmogenic mechanism of an LQT-3 mutation of the human heart Na(+) channel α-subunit: a computational analysis. Circulation. 2000;102:584–590. doi: 10.1161/01.cir.102.5.584. [DOI] [PubMed] [Google Scholar]
- 6.Wang DW, Yazawa K, George AL, Jr, Bennett PB. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci U S A. 1996;93:13200–13205. doi: 10.1073/pnas.93.23.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandra R, Starmer CF, Grant AO. Multiple effects of the KPQ deletion on gating of human cardiac Na+ channels expressed in mammalian cells. Am J Physiol. 1998;274:H1643–H1654. doi: 10.1152/ajpheart.1998.274.5.H1643. [DOI] [PubMed] [Google Scholar]
- 8.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature. 1999;400:566–569. doi: 10.1038/23034. [DOI] [PubMed] [Google Scholar]
- 9.Dumaine R, Towbin JA, Brugada P, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 10.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 11.Antzelevitch C. Electrical heterogeneity, the ECG, and cardiac arrhythmias. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, Pa: WB Saunders Co Inc; 2000. pp. 222–238. [Google Scholar]
- 12.Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–1213. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 13.Veldkamp MW, Viswanathan PC, Bezzina C, et al. Two distinct congenital arrhythmias evoked by a multidysfunctional Na(+) channel. Circ Res. 2000;86:E91–E97. doi: 10.1161/01.res.86.9.e91. [DOI] [PubMed] [Google Scholar]
- 14.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 15.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential, I: simulations of ionic currents and concentration changes. Circ Res. 1994;74:1071–1096. doi: 10.1161/01.res.74.6.1071. [DOI] [PubMed] [Google Scholar]
- 16.Zeng J, Laurita KR, Rosenbaum DS, Rudy Y. Two components of the delayed rectifier K+ current in ventricular myocytes of the guinea pig type: theoretical formulation and their role in repolarization. Circ Res. 1995;77:140–152. doi: 10.1161/01.res.77.1.140. [DOI] [PubMed] [Google Scholar]
- 17.Viswanathan PC, Rudy Y. Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc Res. 1999;42:530–542. doi: 10.1016/s0008-6363(99)00035-8. [DOI] [PubMed] [Google Scholar]
- 18.Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 19.Clancy CE, Rudy Y. Cellular consequences of HERG mutations in the long-QT syndrome: precursors to sudden cardiac death. Cardiovasc Res. 2001;50:301–313. doi: 10.1016/s0008-6363(00)00293-5. [DOI] [PubMed] [Google Scholar]
- 20.Greenstein JL, Wu R, Po S, et al. Role of the calcium independent transient outward current Ito 1 in shaping action potential morphology and duration. Circ Res. 2000;87:1026–1033. doi: 10.1161/01.res.87.11.1026. [DOI] [PubMed] [Google Scholar]
- 21.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. J Cardiovasc Electrophysiol. 2001;12:268–272. doi: 10.1046/j.1540-8167.2001.00268.x. [DOI] [PubMed] [Google Scholar]
- 22.Rudy Y. The electrocardiogram and cardiac excitation. In: Sperelakis N, Kurachi Y, Terzic A, et al., editors. Heart Physiology and Pathophysiology. San Diego, Calif: Academic Press; 2000. pp. 133–148. [Google Scholar]
- 23.Miyazaki T, Mitamura H, Miyoshi S, et al. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol. 1996;27:1061–1070. doi: 10.1016/0735-1097(95)00613-3. [DOI] [PubMed] [Google Scholar]
- 24.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.