

Abstract
The microanatomy of immune clearance of infected brain cells remains poorly understood. Immunological synapses are essential anatomical structures that channel information exchanges between T cell–antigen-presenting cells (APC) during the priming and effector phases of T cells' function, and during natural killer–target cell interactions. The hallmark of immunological synapses established by T cells is the formation of the supramolecular activation clusters (SMACs), in which adhesion molecules such as leukocyte function-associated antigen 1 segregate to the peripheral domain of the immunological synapse (p-SMAC), which surrounds the T cell receptor–rich or central SMAC (c-SMAC). The inability so far to detect SMAC formation in vivo has cast doubts on its functional relevance. Herein, we demonstrate that the in vivo formation of SMAC at immunological synapses between effector CD8+ T cells and target cells precedes and mediates clearance of virally infected brain astrocytes.
Immunological synapses are thought to be the anatomical manifestation of intercellular communication in the immune system (1). Immunological synapses serve as the anatomical substrate of T cell–APC communication during the priming of naive T cells and during the effector phase of T and NK cells' function (1–6). The molecular components of immunological synapses differ between those established by T cells or NK cells. The essential feature of mature immunological synapses formed by T cells is the distinctive “bull's eye” structure, (1, 3–6) a specialized intercellular junction formed by the central supramolecular activation cluster (c-SMAC) containing TCR binding to peptide-MHC, surrounded by a ring (peripheral SMAC [p-SMAC]) containing a high density of adhesion molecules such as leukocyte function-associated antigen 1 (LFA-1), and intercellular adhesion molecule 1 (ICAM-1) (1–8). LFA-1 is associated with Talin in the p-SMAC (6, 9) and activates integrin signaling to link the immunological synapse and the cytoskeleton (7). So far, immunological synapses have only been characterized in culture systems (1–8).
Homogeneous populations of antigen-specific T cells coincubated with epitope-loaded APCs in vitro (6, 8–12) or with artificial planar bilayers (4, 13) have been examined either continuously over time, using live cells imaged with a cooled charge-coupled device camera, or after fixation, using confocal laser scanning microscopy to characterize the structure and kinetics of c- SMAC and p-SMAC at the immunological synaptic interface.
T cell interactions with APCs are dynamic and formation of the mature immunological synapse is the culmination of TCR activation. Immunological synapses are thought to facilitate TCR signaling by concentrating TCRs binding to peptide-MHC. TCR activation stimulates a tyrosine kinase cascade that results in Lck and ZAP-70 phosphorylation, rapid activation of phospholipase Cγ, generation of inositol-polyphosphates, Ca2+ mobilization, and T cell activation (8, 10–12, 14) (for review see reference 1).
However, the physiological relevance of immunological synapses has been challenged by kinetic analyses that show a dissociation between synapse formation from effector function (15–17) and the lack of evidence so far for their in vivo existence during immune responses in a living organism (for a detailed discussion on the status of immunological synapses, see reference 1). Nevertheless, very recent data demonstrate that one of the physiological functions of immunological synapses is to direct cytokine secretion vectorially either directly into the synapse, or in a multidirectional manner outside the synapse (18). This work illustrates how immunological synapses implement the vectorial transfer of information, as neuronal synapses are known to do.
Here, we show that preceding and during the clearance of virally infected cells, effector CD8+ T cells infiltrate specifically the brain area containing infected astrocytes. CD8+ T cells establish mature immunological synapses composed of both the c- and p-SMAC. Immunological synapse formation precedes the clearance of infected astrocytes. In CD8+ T cells contacting infected targets, tyrosine kinases Lck and ZAP-70 became phosphorylated and polarized toward the synaptic interface, a result of TCR activation that leads to T cell activation (8, 10–12, 14) (for review see references 1, 19). Although previous in vivo studies failed to detect mature immunological synapses, our data demonstrate the characteristic segregation of adhesion molecules, TCR, and signaling molecules within effector T cells that adopt the typical structures of mature immunological synapses containing c- and p-SMAC. This proves their immunological significance during the clearance of infected cells in a living organism.
RESULTS
Clearance of infected astrocytes from the brain
To visualize the detailed microanatomy of brain immunological synapses, we set up an experiment in which T cells would selectively target virally infected brain astrocytes. To do so, nonreplicating adenoviral vectors were chosen to infect brain cells within a restricted site within the rat brain (i.e., the striatum) and infected cells were identified through expression of a reporter gene encoded within the vector (e.g., HSV1-thymidine kinase [TK]). More than 85% of infected cells were GFAP-expressing astrocytes (Fig. 1 A). 30 d later, animals were immunized with a systemic injection of RAd-HPRT (20), an adenovirus encoding an unrelated transgene to that expressed in the brain, to stimulate a specific systemic immune response against adenovirus. The systemic administration of adenovirus is needed because the initial delivery of a nonreplicating adenovirus to the brain parenchyma fails to prime a systemic immune response against adenovirus (or other infectious antigens), as a result of the so-called immune privilege of the brain (20–23). A replication-deficient adenovirus was used to avoid any interference from viral replication on cell viability. In this model, cell loss is exclusively immune mediated, rather than as a result of viral replication.
Figure 1.

The systemic immune response clears infected astrocytes and virus genomes from the brain. (A) Virally infected cells are astrocytes. To determine the cellular nature of infected cells, confocal microscopy of triple immunolabeling of TK, a marker of adenovirus infection (green); GFAP, an astrocyte marker (magenta); and NeuN, a neuronal marker (red), was performed in 50-μm thick vibratome sections from the brains of animals studied 30 d after intracranial injection of virus, in the absence or presence, of systemic immunization. The top row illustrates TK, GFAP, and the merging of both images as indicated. The vast majority of the TK-immunoreactive cells were also GFAP+ (i.e., >85%). Some of the cells expressing both markers are indicated by white arrows on all three panels (all the TK-immunoreactive cells illustrated in these panels also expressed GFAP). The bottom row shows the same field, but colocalization was studied between TK and NeuN; no colocalization was detected. This indicates that the virus used expresses its marker gene almost exclusively within astrocytes, but not neurons. Bar, 100 μm. (B) Systemic immunization induces an immune response that clears virally infected expressing astrocytes and viral genomes from the brain. Panels a–e illustrate that after the systemic immunization against adenovirus, the adaptive immune response clears virally infected cells from the striatum. Analysis of brain sections from immunized animals (c, low power, and d, high power) show a reduction in the expression of the virally encoded marker gene, TK, when compared with naive animals (a and b). The decrease in the number of infected cells after immunization was assessed by quantifying adenovirally infected, TK-expressing cells (e), and the decrease in the number of viral genomes was quantified using quantitative real-time TaqMan PCR (f). Decreases in the number of infected cells (e), and adenoviral genomes (f), after the systemic immunization against adenovirus was statistically significant (*, P < 0.05; Student's t test). This indicates that the systemic immune response clears transduced astrocytes and viral genomes from the brain. Bars, 1 mm. (C) Adenovirally infected astrocytes express MHC-I. These panels illustrate a GFAP-immunoreactive astrocyte that has been infected with adenovirus and thus is immunopositive for the marker protein TK, expressing MHC-I. Bar, 40 μm.
Systemic immunization against adenovirus causes a specific infiltration of the brain injection site with T cells, and clearance of TK-expressing astrocytes and adenoviral genomes from the striatum (Figs. 1 B and 2). Astrocytes displayed MHC-I on their plasma membrane, thus constituting a potential target for activated CD8+ T cells (24) (Fig. 1 C). Selective antibody depletion of CD4+ and CD8+ T cells 3 d after the systemic immunization demonstrated that both were necessary for astrocyte clearance (Fig. 3). That each type of T cell makes a separate contribution to clearance was shown by the fact that CD4+ T cells remained confined to the perivascular compartment (Fig. 2), whereas CD8+ T cells did infiltrate the site within the brain parenchyma proper where infected astrocytes were located (Fig. 2). CD8+ T cells also established frequent close anatomical contacts with infected brain cells (Fig. 2, c–f). CD8+ T cells' influx into the central nervous system (CNS) reached its peak at 14 d after immunization, whereas the loss of infected cells occurred between 14 and 30 d (Figs. 1 and 3). The analysis of brain immunological synapses in vivo is based on the detailed study of brains from 25 rats and a total of at least 60 immunological synapses that were thoroughly studied in detail.
Figure 2.

Anatomical localization of brain CD4+ and CD8+ T cells during the clearing of virally infected cells. CD4+ T cells within the brain remained immediately adjacent to brain blood vessels in the striatum (a and b), whereas CD8+ T cells (red) infiltrated the brain parenchyma proper (c and d). Time course studies (see Fig. 3) indicated that peak brain numbers of T cells were reached at 30 d for CD4+ T cells (b), and at 14 d for CD8+ T cells (d). Panels e and f illustrate that brain-infiltrating CD8+ T cells (red) within the rat striatum 7 and 14 d after immunization concentrate within the immediate vicinity of virally infected cells (i.e., TK-expressing cells [green]). Bars, 100 μm. CD8+ T cells establish close intercellular contacts with virally infected cells. CD8+ T cells were found surrounding virally infected (TK expressing) brain cells establishing multiple close contacts with them, especially at 14 d after immunization. Here, we illustrate two examples taken at 14 d after immunization. e and f represent confocal images throughout the thickness of the brain sections. Boxed areas in e and f are shown magnified to the right, but only a single 0.5-μm-thick optical section through the stacks shown in e and f is shown to demonstrate the close anatomical apposition between both cell types; xz and yz side views are illustrated too. White arrows indicate areas of close anatomical apposition of CD8+ T cells and infected no or very few brain cells. No or very few brain parenchyma–infiltrating T cells were detected in the hemisphere not infected with adenovirus or in the brains of injected animals that had not been immunized. Bar, 25 μm.
Figure 3.

CD4+ and CD8+ T cells are necessary for viral clearance from the brain. (A) CD4+ T cells are necessary for clearance of virally infected cells from the brain parenchyma. There was no loss of virally infected cells in animals in which CD4+ T cells had been depleted during the systemic immunization against adenovirus (panel a; CD4+ T cells depleted rats: immunized + CD4-depleting monoclonal antibody OX34. Control nondepleted rats: immunized animals + control nonimmune antibody isotype). A peak in the number of CD4+ T cells within the striatum was detected at 30 d after immunization in nondepleted immunized animals, but not in immunized rats depleted of CD4+ T cells (b). As shown in Fig. 2, CD4+ T cells remained close to brain blood vessels. *, P < 0.05 compared with nondepleted controls (nonparametric Kruskal-Wallis). (B) CD8+ T cells are necessary for clearance of virally infected cells from the brain parenchyma. No loss of virally infected cells could be detected in animals in which CD8+ T cells had been depleted during the systemic immunization against adenovirus (a; CD8+ T cell–depleted rats: immunized + CD8-depleting monoclonal antibody OX8. Control nondepleted rats: immunized animals + control nonimmune antibody isotype). A peak in the number of CD8+ T cells entering the brain parenchyma was detected at 14 d after immunization in nondepleted immunized animals, but not in immunized rats depleted of CD8+ T cells (b). As shown in Fig. 2, CD8+ T cells enter in the brain parenchyma proper and establish close appositions with virally infected cells. *, P < 0.05 compared with nondepleted controls (nonparametric Kruskal-Wallis). Rats that were injected with adenovirus in the brain, but were neither immunized nor injected with antibodies, were used as further controls and are illustrated in B. Notice that levels of infected, TK-immunoreactive cells in these animals did not differ from T cell–depleted rats (a).
Initial stages in the formation of immunological synapses in vivo
Polarization of phosphorylated tyrosine kinases, the initial stages in the formation of immunological synapses in vivo, can be detected preceding and throughout the clearance of infected astrocytes by CD8+ T cells. To further characterize immune–target cell interactions and the activation of T cells, we studied the distribution of phosphorylated Lck and ZAP-70 in CD8+ T cells contacting target astrocytes. Engagement of the TCR on CD8+ T cells by MHC-I peptide complexes present on the surface of target APCs activates effector T cells and stimulates the T cell tyrosine kinase signaling cascade (10, 11, 14). Tyrosine kinase signaling activation was assessed by immunocytochemistry using antibodies recognizing specifically active, phosphorylated-Lck (p-Lck) (recognizing pY394), or active, phosphorylated-ZAP-70 (p-ZAP-70) (recognizing pY319) (10, 11, 14, 25).
Phosphorylated-Lck and p-ZAP-70 were polarized in CD8+ T cells to sites of close membrane apposition between CD8+ T cells and target astrocytes only in the virally injected hemisphere (Figs. 4 and 5). Phosphorylation of tyrosine kinases precedes the establishment of mature immunological synapses. Thus, we studied the anatomical arrangements of these early sites of T cell–astrocyte interactions through serial reconstruction appositions identified with the confocal microscope, and three-dimensional artistic rendering of the confocal images (Figs. 4 B and 5 B). Most CD8+ T cells displayed a single site of close membrane apposition with astrocytes (Fig. 4, A and B), although in some cases a CD8+ T cell would make up to three such polarized contacts with a single target astrocyte (Fig. 5). CD8+ T cells formed complex close interactions with target astrocytes. T cells either surrounded the target cell's processes (Fig. 5 B [b, d, and e]) or displayed cytoplasmic evaginations that appeared to press into the target cell body (Fig. 5 B [c, f, and g]). Thus, brain-infiltrating CD8+ T cells increased TK cascade phosphorylation induced by TCR signaling (i.e., Lck and ZAP-70), indicating that the CD8+ T cells were activated through interaction with antigenic peptides on MHC-I expressed on astrocytes (Fig. 1 C). Polarization of phosphorylated TKs at contacts between CD8+ T cells and target astrocytes strongly implied the visualization of immunological synapse formation in progress.
Figure 4.

Polarized expression of p-Lck and p-ZAP-70 in CD8+ T cells tightly apposed to astrocytes during clearance of virally infected cells from the brain. Animals were infected unilaterally in the striatum with an adenovirus, and 30 d later they were immunized against adenovirus systemically. The systemic immunization induces a selective infiltration of CD8+ T cells into the injected, but not contralateral, hemisphere, which eventually clears infected astrocytes and viral genomes. All images shown represent 0.5-μm-thick confocal optical sections. Panels in A illustrate an activated lymphocyte in close anatomical apposition of the process of an astrocyte. Activation was established by p-Lck (green) immunoreactivity (a) within CD8+ (red) lymphocytes (b). Only CD8+ T cells that contacted astrocytes directly (GFAP+ processes) were immunoreactive for p-Lck, indicating the selective activation of T cells (a and b). White arrows indicate that the CD8+ T cell is contacting a GFAP-immunoreactive process (magenta) (c and merged images). DAPI nuclear staining (blue) was used as a cellular counterstain. Minimal T cells were observed within the noninjected hemisphere (not depicted). P-Lck within the CD8+ T cells was polarized to the T cell's membrane in close contact with the astrocyte's process (white arrow in merged images). B represents a three-dimensional reconstruction of the contact illustrated in A; the confocal image of the contact area is shown in a; b illustrates the color scales for A and B, including colors of areas of overlap between fluorophores. C illustrates the polarized expression of phosphorylated ZAP-70 in several CD8+ T cells during the clearance of infected GFAP+ astrocytes from the brain; p-ZAP-70 (green), CD8 (red), GFAP (magenta or white), and DAPI (nuclear marker in blue). Note that ZAP-70 is polarized to the site of contact between the CD8+ T cell and the astrocytes' processes. Bars, 15 μm.
Figure 5.

Multiple sites of polarized accumulation of p-Lck in a CD8+ T cell establishing several contacts with a target astrocyte. (A) Confocal images of a CD8+ T cell that displays three areas of close membrane apposition to an astrocyte. (a–c) A stack of sections representing approximately a total of 20 μm. (a) The immunoreactivity of the CD8+ T cell for p-Lck indicating three areas of p-Lck polarization (white arrows 1, 2, and 3 in a). b illustrates CD8-immunoreactivity and c shows the immunoreactivity of the target astrocyte for GFAP. The merged image (d) represents a single 0.5-μm optical section in which all three individual channels shown in a–c have been merged, with the associated xz and yz lateral views, to illustrate two intercellular contacts that are indicated by the white arrows labeled 1 and 2. Bar, 25 μm (B) The three-dimensional reconstruction, in detail, of the immunological synapses shown in A. (a) The merged image of the three individual stacks shown individually in A. Panel a has been rotated ∼35° to the left to clarify the view of two of the close appositions; these have been reconstructed in three dimensions and are shown at higher magnification in b–g. Within the three-dimensional reconstruction, the location of two close contacts is indicated by the broken circles 1 and 2. Close contact 1 is shown in b, d, and e in more detail. Panel b is the contact 1 of the three-dimensional reconstruction, from which a slice of the image has been removed to demonstrate the polarization of p-Lck (green) to the area of close apposition between both cells. Notice how the CD8+ T cell appears to engulf the GFAP process (magenta). An enlarged confocal image of the close contact is illustrated in d, whereas e is a contour map of each of the immunoreactivities illustrated in d. Panels c, f, and g illustrate the same analysis for contact number 2. Notice how the CD8+ T cell appears to form protrusions into the GFAP process of the astrocyte (magenta) (c, f, and g).
SMAC formation during the maturation of immunological synapses in vivo
To determine whether mature immunological synapses containing c-SMAC and p-SMAC develop at the CD8+ T cell–astrocyte junctions previously shown to contain polarized tyrosine kinases, we studied the distribution of LFA-1 and TCR on T cells participating potentially in the formation of immunological synapses; we used TK as a marker of virally infected cells. The analysis of LFA-1 and TCR expression in T cells not in contact with infected cells showed a homogeneous, nonpolarized distribution (Fig. 6). The distribution of LFA-1 and TCR at the T cell membrane closely apposed to infected target astrocytes clearly indicated the formation of p-SMAC (LFA-1 rich, TCR poor) and c-SMAC (LFA-1 poor, TCR rich) in vivo (Figs. 7 and 8). This indicates that T cell–astrocyte junctions mature to form proper immunological synapses with SMAC formation, concomitantly with the influx of CD8+ T cells into the brain, and preceding the clearance of virally infected astrocytes.
Figure 6.

Homogenous distribution of LFA-1 and TCR immunoreactivity along the plasma membrane of T cells not in contact with infected target cells. LFA-1 and TCR show homogeneous distribution along the plasma membrane of T cells neither in close apposition of target infected cells, nor involved in the formation of immunological synapses. Top panels (A–E) show a LFA-1 positive cell (B) not in contact with a TK-immunoreactive process (D), showing a uniform distribution of LFA-1. Panels F-J show a T cell with no LFA-1 expression (G) with a homogeneous distribution of TCR (H). Panels K-O show a T cell expressing LFA-1 (L) and TCR (M) not contacting a TK positive cell (N) showing no specific polarization and distribution in clusters. Panels P–T illustrate a CD8+ T cell expressing LFA-1 in a nonpolarized fashion in a cell that does not contact any infected cell. Bar, 25 μm.
Figure 7.

Formation of c-SMAC and p-SMAC in vivo. TCR polarization and clustering within c-SMAC (A) and LFA-1 distribution to the p-SMAC (B). TCR staining is polarized to membrane areas displaying close contacts between T cells and infected brain cells, and clusters at sites where immunological synapses form (two different immunological synapses are shown in A and C). Three-dimensional reconstructions allow visualization of the formation of c-SMAC at the interface of lymphocytes and infected cells (A, under “Interface”); the quantification of the clustering of the TCR at the c-SMAC of the immunological synapses shownin A is illustrated in C (yellow arrow and intensity graph). LFA-1 forms a ring (p-SMAC) that surrounds c-SMAC, but is reduced within c-SMAC itself (two different immunological synapses are shown in B and D). Three-dimensional reconstructions show the LFA-1 immunoreactive p-SMAC ring at the interface between the T cells and target infected brain cells (B, under “Interface”); the quantification of the distribution of LFA-1 immunoreactivity at the p-SMAC of immunological synapses shown in B is illustrated in D (yellow arrow and intensity graph). Bars, 25 μm.
Figure 8.

SMAC formation at immunological synapses in vivo, between T cells and infected astrocytes in the brain. Two mature immunological synapses displaying typical and characteristic SMAC formations, are shown in A and B. The row of upper panels in A and B illustrate images captured in the given channels, from left to right: DAPI (blue), LFA-1 (red), TCR (green), LFA1+TCR, LFA1+TCR+DAPI (MERGE 1), the virally infected cell (TK; in white), and LFA1+TCR+DAPI+TK (MERGE 2). Notice that these mature immunological synapses are characterized by the specific distribution of LFA-1 and TCR contacting the virally infected cell. In the views shown in the top panels (side views of the immunological synapses), LFA-1 immunoreactivity displays high relative fluorescence density within areas of the membrane lateral to the close apposition of membranes of the lymphocyte with the infected cell, thus displaying the typical central region of lower density (see arrows in red channel) at the site of highest density of immunoreactivity for TCR (see arrows in the green channel and in the MERGE 1 and 2 images). The yellow asterisk indicates, in the white channel, the anatomical location of the T cell. The row of bottom panels in A and B illustrate, respectively, low (a) and high (b) magnifications of the close apposition between the T cell and the infected cell. These images were reconstructed using three-dimensional visualization software to illustrate the characteristic structure of the p-SMAC (LFA-1 ring) and c-SMAC (TCR cluster) at the interface of the immunological synapses seen in en face views (c and d). To produce the three-dimensional image of the interface shown in c and d, the three-dimensional reconstruction was rotated so that the plane of the interface of the immunological synapse (b, broken arrow) could be observed from above (white arrow in b shows the angle of vision of c and d). At the interface, typical “bull's eye” structures, characteristic of p-SMAC (LFA-1 outer ring) and c-SMAC (central TCR cluster) can be clearly recognized (c and d). In C, the intensity of fluorescence (I) was measured at the interface (yellow line) of the immunological synapse in a 0.5-μm confocal layer taken through the interface. The small graphs to the right of each synapse show the relative intensity values of fluorescence of LFA-1 (in red) and TCR (in green). Note the maximum of intensity for LFA-1 was in the periphery of the synapse (p-SMAC), whereas the TCR is clustered toward the center (c-SMAC). (D) A diagrammatic view of a T cell contacting an antigen-expressing astrocyte illustrating the localization of molecules involved in the immunological synapse and the polarization and phosphorylation of key tyrosine kinases. (E) A characteristic activated T cell nucleus displaying the polarized arched conformation opening toward the immunological synaptic interface. Bars, 15 μm.
The optical images obtained with the confocal microscope were further analyzed using custom-made three-dimensional reconstruction software. Images were α blended to perform a three-dimensional reconstruction from the two-dimensional serial images. This model was rotated in three dimensions, and reconstructions were used to examine in detail the microanatomy of mature brain immunological synapses at the interface plane (Figs. 7–9 ).
Figure 9.

Criteria used in the selection of optical planes from three-dimensional reconstructions of mature immunological synapses, for the illustration of SMAC formation in vivo. 90° rotations of three-dimensional reconstructions were made to observe the xz optical planes 1, 2, and 3 through axis y. Images A1, A2, and A3 show xz optical planes from image A. The immunological synaptic interface seen in optical plane xz (1) is illustrated in A1; this optical plane was selected as the one in which the T cell's nucleus could not be visualized. Optical planes 2 and 3 (illustrated in A2 and A3) show the T cell's nucleus, demonstrating that these optical planes are at a distance from the synaptic interface and thus do not allow observation of the SMAC. Note that the optical plane 1 is the only one that shows the SMAC. yz optical planes 4, 5, and 6 were obtained from a 90° rotation of image B through the x axis. In plane 5 (illustrated in A5) the TCR is closely apposed to the infected cell without any intervening LFA-1; this plane is taken through the center of the c-SMAC. In images of planes 4 and 6 (illustrated in A4 and A6) LFA-1 immunoreactivity, rather than TCR, is in close apposition of the infected cell, indicating that these planes are taken through the p-SMAC. Note that this detailed three-dimensional reconstruction analysis was performed on the synapse illustrated in Fig. 8 A.
At the interface plane, the p-SMAC is characterized by an LFA-1–immunoreactive ring (Figs. 7–9 ) surrounding a central area of increased TCR immunoreactivity (Figs. 7–9 ) and low or absent LFA-1 (Figs. 7, 8, and 10). Note that in most cases LFA-1 immunoreactivity increases within the p-SMAC (Fig. 10, D, G, and I), whereas in others there is a drop in c-SMAC LFA-1 (Fig. 10, F, H, and E); both distributions provide a p-SMAC–rich/c-SMAC–poor LFA-1 pattern. Finally, in those T cells forming mature immunological synapses, we observed that T cells' nuclei displayed a polarized notch open toward the immunological synapse, indicating that the whole structure of T cells polarized toward the immunological synapse (Fig. 8, A and E).
Figure 10.

Quantitative distribution of LFA-1 through the plasma membrane of T cells involved in formation of mature immunological synapses. The relative fluorescence of LFA-1 was measured in six T cells forming mature immunological synapses contacting virally infected cells (A). The red LFA-1 channel was selected [B] and the complete distribution of LFA-1 throughout the extent of the plasma membrane was delineated measuring the intensity of fluorescence in a linear graph (illustrated schematically in C). Panels D–I show the quantification of LFA-1 in six different cells forming mature immunological synapses. Small white arrows ‘1’ and ‘3’ indicate the p-SMAC and arrow ‘2’ the c-SMAC. Note that the level of LFA-1 is reduced (or absent) within the c-SMAC of all cells. Further LFA-1 levels are increased in the p-SMAC in some cells (illustrated in D, G, and I), while it does not increase in the p-SMAC of others [F, H, E]. Note that for clarity the target virally infected cells, found in apposition of the immunological synapses illustrated, are not shown; synapses shown in (D) and (E) are illustrated in further detail in Fig. 7, and synapse (F) in Fig. 8 A. Synapses H, G, and I are not illustrated further.
DISCUSSION
In this study, we have shown that CD8+ T cells form typical immunological synapses in vivo displaying p- and c-SMAC at the interface with virally infected astrocytes, preceding and during the clearance of infected cells from the brain. We do so in a model of immune-mediated clearance of infected astrocytes, and demonstrate that both CD8+ T cells and CD4+ T cells are necessary parts of the effector arm of the immune response, possibly as CD4+ Th1 cells. However, detailed morphological analysis demonstrated that CD4+ T cells remain circumscribed to the perivascular compartment, whereas CD8+ T cells enter the brain parenchyma and form close anatomical contacts with infected cells. A likely explanation is that CD4+ T cells may aid in the entry of CD8+ T cells into the brain; this is similar to the strategies adopted by both CD4+ and CD8+ T cells during the clearance of MHV from the brain (26).
Although both cell types peak at an early time point in the lymph nodes, it is likely that differential chemokine and adhesion molecule expression by CD4+ T cells determines their delayed migration to the CNS and their selective perivascular accumulation when compared with CD8+ T cells (27–30). The delayed peak of intracranial CD4+ T cells may serve to facilitate the entry of macrophages into the CNS (31).
Initial TCR activation leads to the specific stimulations of the TK signaling pathway; e.g., phosphorylated-Lck and phosphorylated-ZAP-70. These become polarized to areas of close membrane apposition with target cells where mature immunological synapses will form later. Membrane junctions between CD8+ T cells with target astrocytes display complex three-dimensional morphology; at times, T cells' membranes completely surrounds individual processes of target-infected cells, or even insinuate their cell body directly into the target astrocyte's soma.
Our experiments demonstrate that CD8+ T cells establish SMAC at immunological synapses with infected brain astrocytes that express MHC-I. The data shown herein demonstrate that the mature immunological synapses between T cells–APCs in vivo precede and mediate the clearing of virally infected astrocytes. In addition, we determined that nuclei of T cells formed an open arch toward the immunological synapse, a finding compatible with the known polarization of the microtubule organization center and Golgi apparatus of T cells toward the immunological synapse (32).
The previous use of homogeneous populations of cloned T cells, APCs, and time-lapse confocal microscopy has allowed the detailed characterization of the kinetics of T cell–APC interaction and immunological synapse assembly and disassembly in culture (6, 8–12). Previous studies of T cell–APC interactions during in vivo immune responses could not demonstrate the formation of p- and c-SMAC, as the result of limited resolution of microscopical techniques used (25, 33). Multiphoton laser scanning microscopy and confocal microscopy and other advanced imaging methodologies have made major contributions to our dynamic understanding of T cell–APC interactions in lymph nodes and other tissues in vivo (34). Although Kawakami et al. (33) used an ex vivo model to study the influx into the spinal cord of antimyelin CD4+ T cells and McGavern et al. (25) studied entry of CD8+ T cells into the meninges of lymphocytic choriomeningitis virus (LCMV)–infected animals, the anatomical resolution of these studies precluded the morphological identification of SMAC formation in vivo (25, 33) (for a discussion on the status of visualization of immunological synapses in vivo, see reference 1, Section 7, page 407).
That SMACs indeed form at immunological synapses during in vivo immune responses within the brain parenchyma during clearing of viral infections has hereby been demonstrated. Even if potential contacts between T cells and local APCs were described, the techniques used were unable to demonstrate the existence of SMAC formation during natural immune responses in the brain in vivo (1), thus not resolving the existence of SMAC formation as part of in vivo immunological synapse formation.
We believe that the following factors aided in our capacity to detect SMAC formation in vivo in the context of a model antiviral immune response. First, we used an established model in which the kinetics of T cell influx contact with potential targets and elimination of transduced cells were all characterized in much detail. This allowed us to look for formation of immunological synapses at the time of peak T cell entry, but preceding the loss of transduced cells. Second, we used a replication-defective virus expressing a marker gene. This allowed us to identify potential target cells by their expression of a marker gene, without viral replication compromising the survival of infected cells. Third, we optimized the perfusion of animals and our immunocytochemical protocol, in such a way to achieve best preservation of cellular structures through careful perfusion of experimental animals, and full and homogenous antibody penetration throughout the 50-μm-thick vibratome section, through careful improvements to the immunocytochemical protocols used. This allowed us to unravel the three-dimensional structure of interactions between T cells and the complex morphology of target brain astrocytes, and uncover sites of membrane apposition that would not have been found in thinner sections, or in the absence of complete antibody penetration. Fourth, we studied simultaneously the distribution of the essential markers that characterize SMAC formation in immunological synapses, and markers of the target cells, in combination within single sections using four-color immunostaining. Finally, the custom-made three-dimensional reconstruction software for confocal images allowed us to rotate cells in close anatomical appositions and observe the distribution of the several markers at different optical planes in a way that provides a complete picture of the distribution of immunological synaptic proteins in three full dimensions. This resulted in the typical “bull's eye” images of the mature immunological synaptic interfaces illustrated in Fig. 8.
Although the functional consequences of molecular segregation in the immunological synapse remain under investigation (16, 35), recent work has demonstrated that immunological synapses serve to channel cytokines and effector molecules toward target cells (18, 36). That mature immunological synapses were found in the brain preceding the clearance of viral infected cells, suggests that the presence of SMAC-containing immunological synapses in a physiological context in vivo may be necessary for clearance of virally infected cells to occur. However, the ultimate proof of this hypothesis will have to wait the development of compounds that selectively inhibit immunological synapse formation.
The demonstration of mature immunological synapses in vivo during antiviral immune responses will allow further experimental exploration of immunological synaptic function during normal and pathological immune responses in vivo. Furthermore, this work should help contribute to settling the controversy over the existence and functional significance of mature immunological synapses in vivo during antiviral immune responses. In summary, we propose that mature, SMAC-containing immunological synapses are the anatomical substrate that mediates the complex sequence of activation and effector function of T cells in vivo, as CD8+ T cells clear virally infected cells from the CNS.
MATERIALS AND METHODS
Animals, surgical procedures, viruses.
Adult male Sprague-Dawley rats (250 g body weight) (Charles River) were used according to Cedars- Sinai Medical Center's Institutional Animal Care and Use Committee–approved protocols. Adenoviruses used in this study were first-generation E1/E3-deleted recombinant adenovirus vectors based on adenovirus type 5. The construction of RAdTK (expressing herpes simplex virus type I thymidine kinase, HSV1-TK) and RAdHPRT (expressing hypoxanthine-guanine phosphoribosyl-transferase), all contain the hCMV promoter and have all been described in detail elsewhere (37). Animals were injected unilaterally in the left striatum with 107 infectious units (i.u.) of RAdTK in a volume of 1 μl and immunized 30 d later with 5 × 108 infectious units of RAdHPRT injected subcutaneously. Animals were killed for analysis at different time points after immunization as described in more detail in the following sections.
Role of CD4+ and CD8+ T cells in clearing of adenovirally infected cells from the brain.
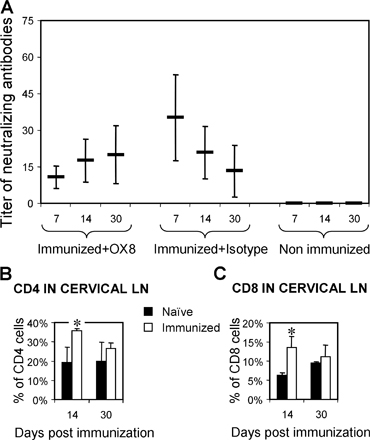
75 animals were injected with 107 i.u. of RAdTK into the striatum at day 0. 1 mo later, rats were anaesthetized briefly and immunized subcutaneously in the back with 100 μl of either sterile saline (n = 15) or 5 × 108 i.u. of RAdHPRT (n = 60). 3 d after systemic immunization against adenovirus, one group of animals (n = 15) was injected weekly with 0.5 mg of OX8 monoclonal antibody i.p. to deplete CD8+ T cells. Another group (n = 15) was injected i.p. with 1 mg of OX34 monoclonal antibody every 2 wk to deplete CD4+ T cells, and two groups of animals were injected with mouse monoclonal irrelevant isotype antibodies as appropriate controls. The number of CD4+ and CD8+ T cells were quantified in draining cervical lymph nodes (Fig. S1). Results of the depletion studies are shown in Fig. 3. 7, 14, and 30 d after the immunization, five animals from each experimental group were killed via anesthetic overdose, transcardially perfused with 200–500 ml of oxygenated Tyrode solution, and spleen and cervical lymph nodes were removed for flow cytometry. Immediately afterward, animals were perfused/fixed with 4% paraformaldehyde to fix the brain. Brains were postfixed in 4% paraformaldehyde for up to 48 h, after which they were washed in phosphate buffer, cut into smaller tissue pieces, and sectioned as described in the following paragraph. This procedure provides excellent quality preservation of brain tissue for further analysis. For the CD8 depletion studies, postfixed brains were sectioned on a vibratome (Leica Instruments) at 50-μm section thickness. In the CD4 depletion studies, brains were cryoprotected in 20% sucrose and 16-μm sections were cut on the cryostat (Leica Instruments).
Immunocytochemical procedures and confocal analysis.
50-μm coronal brain sections were cut serially through the striatum on a Leica vibratome, and immunofluorescence or DAB detection was performed as described previously (20), using the following primary antibodies recognizing: CD8 (1:500, mouse, Serotec), CD4 (1:100, mouse, Serotec), TK (1:10,000, rabbit, custom made), NeuN (1:1,000, mouse, Chemicon), GFAP (1:500, guinea pig, Advanced Immunochemical), phosphorylated Lck (1:50, rabbit, Cell Signaling), LFA-1 (1:500, mouse, IgG2a, BD Biosciences), TCR (1:100, mouse, IgG1, BD Biosciences), phosphorylated ZAP-70 (1:100, rabbit, Cell Signaling), and MHC-I (1:1,000, mouse, Serotec). Sections were examined using a Leica DMIRE2 confocal microscope (Leica Microsystems). Three-dimensional reconstructions to allow rotation of the images were rendered with α-blending software (custom made by K. Wawrowsky). Striatal sections from 25 animals were screened and analyzed in their whole extent searching for T cells interactions; a total of at least 60 immunological synapses (a likely underestimation of the total number of synapses present in the brain) at various stages of development were recorded and analyzed in detail.
Note that given the complexity of the confocal analysis, the number of total immunological synapses illustrated throughout the manuscript in detail demonstrates the existence of immunological synapses in vivo, but obviously cannot be considered a faithful estimation of their total number. Currently, it remains technically impossible to record accurately the exact number of mature immunological synapses present in vivo.
To determine the approximate number of activated T cells over the total number of T cells, the expression of phosphorylated ZAP-70, a marker of T cell activation after TCR engagement, and thus, a close surrogate of potential immunological synaptic engagement, was quantified. This analysis indicated that 75.4% of all CD8+ T cells within the injected striatum express phosphorylated ZAP-70. This provides a quantitative estimate of the potential frequency of immunological synapses being established by T cells.
Confocal analysis.
The brain sections were examined using a Leica DMIRE2 confocal microscope with the 63x oil objective and Confocal Software (Leica Microsystems). A series range for each section was determined by setting an upper and lower threshold using the Z/Y Position for Spatial Image Series setting, and confocal microscope settings were established and maintained by Leica and local technicians for optimal resolution. Contacts were defined as areas where colocalization of both markers occurs between two cells in at least two 0.5-μm-thick optical sections. Contacts can also be illustrated as they appear throughout the stack of sections as a simple 0.5-μm layer or as a transparency of all layers merged together. Relative fluorescence intensity along the plane of the immunological synaptic interface was measured with the Leica confocal software and is illustrated in the figures with corresponding arrows traversing the measured optical planes.
Three-dimensional reconstructions where generated with a custom-made software. Images were α-blended to perform a three-dimensional reconstruction from the two-dimensional serial images. The images were rotated in three-dimensional perspective to get the optical plane of the interface. The criteria of choosing the interface are illustrated in detail in Fig. 9. Artistic renderings (made by C. Barcia) were produced based on the three- dimensional analysis for Figs. 4 B and 5 B.
Neutralizing antibody assay.
Adenovirus neutralizing antibody titers were measured in serum samples as described by us (20, 38) (Fig. S1).
Real-time quantitative PCR analysis.
12 rats were injected bilaterally into the striatum with 107 infectious units of RAdTK. 1 mo later, six animals were control immunized with saline, and six animals were immunized intradermally with RAdHPRT. Rats were killed 60 d after the immunization by neck dislocation and decapitation, after an anesthetic overdose. Brains were removed, cut into five blocks (left and right forebrain, left and right midbrain, and cerebellum), snap frozen in liquid nitrogen, and stored at −80°C. Frozen blocks of the left or right forebrain (containing the striatum) were ground to a fine powder under liquid nitrogen. DNA was extracted from 50 mg of ground tissue using spin columns for DNA purification (QIAGEN). 250 ng of DNA, or a quantity of cDNA corresponding to 100 ng of total RNA was amplified by PCR using TKf 5′-CGAGCCGATGAC TTACTGGC-3′ and TKr 5′-CCCCGGCGGATATCTCAC-3′ primers and the FAM-labeled TKp 5′-TACACCCAACACCGCCTCGACC-3′ TaqMan probe (PerkinElmer). RNA control was performed using two primers and a VIC-labeled 18S ribosomal probe (PerkinElmer). The TK copy number standard curve was obtained using dilutions of the previously described EpTK plasmid. The final concentration was 0.2 μM for each primer, probe 0.2 μM, dNTP 0.2 mM (except for dUTP 0.4mM), MgCl2 5 mM, UNG 0.5U in PCR buffer in the presence of 1.25 U of AmpliTaq Gold polymerase (PCR core reagents, PerkinElmer). DNA determinations were made in triplicate. Amplifications used a ABI Prism 7700 sequence detection system (PerkinElmer) with an initial step at 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of the following: denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min. Raw data were analyzed using the SDS v1.6.3. software (PerkinElmer).
Statistical analysis.
Viability data were expressed as mean ± SEM and evaluated by two- or one-way analysis of variance (followed by Dunnet or Tukey multiple comparisons tests). Differences were considered significant if P < 0.05. When significance testing using analysis of variance was not applicable, the Kruskal-Wallis nonparametric test was used instead.
Online supplemental material.
Fig. S1 illustrates neutralizing antiadenoviral antibody titers in immunized and nonimmunized rats, and levels of CD4+ and CD8+ T cells in cervical lymph nodes of naive and immunized rats. Fig. S2 illustrates the effects of specific depleting antibodies on levels of CD8+ and CD4+ T cells in the spleen. The supplemental Materials and methods section includes detailed descriptions of immunocytochemistry and its stereological quantification. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20060420/DC1.
Supplemental Material
Acknowledgments
We thank the support and academic leadership of S. Melmed, R. Katzman, and D. Meyer for their superb administrative and organizational support.
This work was supported by grants from the National Institutes of Health/National Institute of Neurological Disorders and Stroke grant nos. 1 RO1 NS 42893.01, U54 NS045309-01, and 1R21 NS047298-01 (to P.R. Lowenstein), and The Bram and Elaine Goldsmith Chair In Gene Therapeutics (P.R. Lowenstein); National Institutes of Health/National Institute of Neurological Disorders and Stroke grant no. 1R01 NS44556.01, National Institute of Diabetes and Digestive and Kidney Diseases grant no. 1 RO3 TW006273-01 (to M.G. Castro), and The Medallion Chair in Gene Therapeutics (M.G. Castro); as well as The Linda Tallen and David Paul Kane Annual Fellowship (to M.G. Castro and P.R. Lowenstein). We also thank the generous funding our Institute receives from the Board of Governors at Cedars Sinai Medical Center.
The authors have no conflicting financial interests.
Note added in proof. In addition to the data illustrated in Figs. 4 and 5, using novel custom-made chicken anti–thymidine kinase antibodies, we have now detected polarized expression of p-Lck and p-ZAP-70 in CD8+ T cells tightly apposed to adenovirus-infected cells expressing thymidine kinase.
Abbreviations used: CNS, central nervous system; c-SMAC, central SMAC; ICAM-1, intercellular adhesion molecule 1; LFA-1, leukocyte function-associated antigen 1; c-SMAC, central SMAC; p-SMAC, peripheral SMAC; SMAC, supramolecular activation cluster; TK, thymidine kinase; i.u., infectious units.
References
- 1.Dustin, M.L. 2005. A dynamic view of the immunological synapse. Semin. Immunol. 17:400–410. [DOI] [PubMed] [Google Scholar]
- 2.Davis, D.M., I. Chiu, M. Fassett, G.B. Cohen, O. Mandelboim, and J.L. Strominger. 1999. The human natural killer cell immune synapse. Proc. Natl. Acad. Sci. USA. 96:15062–15067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedl, P., A.T. den Boer, and M. Gunzer. 2005. Tuning immune responses: diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 5:532–545. [DOI] [PubMed] [Google Scholar]
- 4.Grakoui, A., S.K. Bromley, C. Sumen, M.M. Davis, A.S. Shaw, P.M. Allen, and M.L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science. 285:221–227. [DOI] [PubMed] [Google Scholar]
- 5.Huppa, J.B., and M.M. Davis. 2003. T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 3:973–983. [DOI] [PubMed] [Google Scholar]
- 6.Monks, C.R., B.A. Freiberg, H. Kupfer, N. Sciaky, and A. Kupfer. 1998. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 395:82–86. [DOI] [PubMed] [Google Scholar]
- 7.Dustin, M.L., and J.A. Cooper. 2000. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat. Immunol. 1:23–29. [DOI] [PubMed] [Google Scholar]
- 8.Lee, K.H., A.D. Holdorf, M.L. Dustin, A.C. Chan, P.M. Allen, and A.S. Shaw. 2002. T cell receptor signaling precedes immunological synapse formation. Science. 295:1539–1542. [DOI] [PubMed] [Google Scholar]
- 9.Kupfer, A., and S.J. Singer. 1989. The specific interaction of helper T cells and antigen-presenting B cells. IV. Membrane and cytoskeletal reorganizations in the bound T cell as a function of antigen dose. J. Exp. Med. 170:1697–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanchard, N., V. Di Bartolo, and C. Hivroz. 2002. In the immune synapse, ZAP-70 controls T cell polarization and recruitment of signaling proteins but not formation of the synaptic pattern. Immunity. 17:389–399. [DOI] [PubMed] [Google Scholar]
- 11.Holdorf, A.D., K.H. Lee, W.R. Burack, P.M. Allen, and A.S. Shaw. 2002. Regulation of Lck activity by CD4 and CD28 in the immunological synapse. Nat. Immunol. 3:259–264. [DOI] [PubMed] [Google Scholar]
- 12.Yachi, P.P., J. Ampudia, N.R. Gascoigne, and T. Zal. 2005. Nonstimulatory peptides contribute to antigen-induced CD8-T cell receptor interaction at the immunological synapse. Nat. Immunol. 6:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mossman, K.D., G. Campi, J.T. Groves, and M.L. Dustin. 2005. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 310:1191–1193. [DOI] [PubMed] [Google Scholar]
- 14.Yokosuka, T., K. Sakata-Sogawa, W. Kobayashi, M. Hiroshima, A. Hashimoto-Tane, M. Tokunaga, M.L. Dustin, and T. Saito. 2005. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6:1253–1262. [DOI] [PubMed] [Google Scholar]
- 15.DeFalco, J., M. Tomishima, H. Liu, C. Zhao, X. Cai, J.D. Marth, L. Enquist, and J.M. Friedman. 2001. Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 291:2608–2613. [DOI] [PubMed] [Google Scholar]
- 16.O'Keefe, J.P., and T.F. Gajewski. 2005. Cutting edge: cytotoxic granule polarization and cytolysis can occur without central supramolecular activation cluster formation in CD8+ effector T cells. J. Immunol. 175:5581–5585. [DOI] [PubMed] [Google Scholar]
- 17.Purbhoo, M.A., D.J. Irvine, J.B. Huppa, and M.M. Davis. 2004. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5:524–530. [DOI] [PubMed] [Google Scholar]
- 18.Huse, M., B.F. Lillemeier, M.S. Kuhns, D.S. Chen, and M.M. Davis. 2006. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 7:247–255. [DOI] [PubMed] [Google Scholar]
- 19.Davis, D.M., and M.L. Dustin. 2004. What is the importance of the immunological synapse? Trends Immunol. 25:323–327. [DOI] [PubMed] [Google Scholar]
- 20.Thomas, C.E., G. Schiedner, S. Kochanek, M.G. Castro, and P.R. Lowenstein. 2000. Peripheral infection with adenovirus causes unexpected long-term brain inflammation in animals injected intracranially with first-generation, but not with high-capacity, adenovirus vectors: toward realistic long-term neurological gene therapy for chronic diseases. Proc. Natl. Acad. Sci. USA. 97:7482–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stevenson, P.G., S. Hawke, D.J. Sloan, and C.R. Bangham. 1997. The immunogenicity of intracerebral virus infection depends on anatomical site. J. Virol. 71:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry, V.H. 1998. A revised view of the central nervous system microenvironment and major histocompatibility complex class II antigen presentation. J. Neuroimmunol. 90:113–121. [DOI] [PubMed] [Google Scholar]
- 23.Lowenstein, P.R. 2002. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 23:23–30. [DOI] [PubMed] [Google Scholar]
- 24.Cabarrocas, J., J. Bauer, E. Piaggio, R. Liblau, and H. Lassmann. 2003. Effective and selective immune surveillance of the brain by MHC class I-restricted cytotoxic T lymphocytes. Eur. J. Immunol. 33:1174–1182. [DOI] [PubMed] [Google Scholar]
- 25.McGavern, D.B., U. Christen, and M.B. Oldstone. 2002. Molecular anatomy of antigen-specific CD8(+) T cell engagement and synapse formation in vivo. Nat. Immunol. 3:918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stohlman, S.A., C.C. Bergmann, M.T. Lin, D.J. Cua, and D.R. Hinton. 1998. CTL effector function within the central nervous system requires CD4+ T cells. J. Immunol. 160:2896–2904. [PubMed] [Google Scholar]
- 27.Calzascia, T., F. Masson, W. Di Berardino-Besson, E. Contassot, R. Wilmotte, M. Aurrand-Lions, C. Ruegg, P.Y. Dietrich, and P.R. Walker. 2005. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 22:175–184. [DOI] [PubMed] [Google Scholar]
- 28.Carson, M.J., C.R. Reilly, J.G. Sutcliffe, and D. Lo. 1999. Disproportionate recruitment of CD8+ T cells into the central nervous system by professional antigen-presenting cells. Am. J. Pathol. 154:481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brabb, T., P. von Dassow, N. Ordonez, B. Schnabel, B. Duke, and J. Goverman. 2000. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J. Exp. Med. 192:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ransohoff, R.M., P. Kivisakk, and G. Kidd. 2003. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 3:569–581. [DOI] [PubMed] [Google Scholar]
- 31.Glass, W.G., and T.E. Lane. 2003. Functional expression of chemokine receptor CCR5 on CD4(+) T cells during virus-induced central nervous system disease. J. Virol. 77:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Somersalo, K., N. Anikeeva, T.N. Sims, V.K. Thomas, R.K. Strong, T. Spies, T. Lebedeva, Y. Sykulev, and M.L. Dustin. 2004. Cytotoxic T lymphocytes form an antigen-independent ring junction. J. Clin. Invest. 113:49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawakami, N., U.V. Nagerl, F. Odoardi, T. Bonhoeffer, H. Wekerle, and A. Flugel. 2005. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J. Exp. Med. 201:1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang, A.Y., H. Qi, and R.N. Germain. 2004. Illuminating the landscape of in vivo immunity: insights from dynamic in situ imaging of secondary lymphoid tissues. Immunity. 21:331–339. [DOI] [PubMed] [Google Scholar]
- 35.Lee, K.H., A.R. Dinner, C. Tu, G. Campi, S. Raychaudhuri, R. Varma, T.N. Sims, W.R. Burack, H. Wu, J. Wang, et al. 2003. The immunological synapse balances T cell receptor signaling and degradation. Science. 302:1218–1222. [DOI] [PubMed] [Google Scholar]
- 36.Maldonado, R.A., D.J. Irvine, R. Schreiber, and L.H. Glimcher. 2004. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature. 431:527–532. [DOI] [PubMed] [Google Scholar]
- 37.Dewey, R.A., G. Morrissey, C.M. Cowsill, D. Stone, F. Bolognani, N.J. Dodd, T.D. Southgate, D. Klatzmann, H. Lassmann, M.G. Castro, and P.R. Lowenstein. 1999. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat. Med. 5:1256–1263. [DOI] [PubMed] [Google Scholar]
- 38.Thomas, C.E., E. Abordo-Adesida, T.C. Maleniak, D. Stone, G. Gerdes, and P.R. Lowenstein. 2000. Gene transfer into rat brain using adenoviral vectors. In Current Protocols in Neuroscience. J.N. Gerfen, R. McKay, M.A. Rogawski, D.R. Sibley, and P. Skolnick, editors. John Wiley and Sons, New York, NY. 4.23.21–24.23. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}