

Abstract
Although much is known about environmental factors that predispose individuals to hypertension and cardiovascular disease, little information is available regarding the genetic and signaling events involved1-3. Indeed, few genes associated with the progression of these pathologies have been discovered despite intensive research in animal models and human populations1-3. Here we identify Vav3, a GDP-GTP exchange factor that stimulates Rho and Rac GTPases4, as an essential factor regulating the homeostasis of the cardiovascular system.Vav3-deficient mice exhibited tachycardia, systemic arterial hypertension and extensive cardiovascular remodeling. These mice also showed hyperactivity of sympathetic neurons from the time of birth. The high catecholamine levels associated with this condition led to the activation of the renin-angiotensin system, increased levels of kidney-related hormones and the progressive loss of cardiovascular and renal homeostasis. Pharmacological studies with drugs targeting sympathetic and renin-angiotensin responses confirmed the causative role and hierarchy of these events in the development of theVav3-null mouse phenotype. These observations uncover the crucial role of Vav3 in the regulation of the sympathetic nervous system (SNS) and cardiovascular physiology, and reveal a signaling pathway that could be involved in the pathophysiology of human disease states involving tachycardia and sympathetic hyperactivity with unknown etiologies2,5,6.
Vav proteins are phosphorylation-dependent GDP-GTP exchange factors that activate Rho and Rac GTPases7. This activity is important for connecting stimulated membrane receptors to downstream biological effects such as cytoskeletal reorganization, mitogenesis and transcriptional regulation7. There are three Vav family members in vertebrates, Vav1, Vav2 and Vav3, which have a common structure as well as similar regulatory and catalytic properties7. Recent genetic experiments using knockout mice have shown that Vav proteins are important for the development and/or function of hematopoietic system–derived cells such as lymphocytes, natural killer cells and osteoclasts8-10. Despite the widespread expression of Vav2 and Vav3 proteins in mouse tissues7, no defects have been observed in nonhematopoietic cells8,10.
To look for additional Vav3 functions, we generated mice harboring a null Vav3 gene using homologous recombination techniques (Supplementary Fig. 1 online). Vav3−/− mice were born at the expected mendelian ratios, had normal growth curves and were fertile independent of their gender (Supplementary Fig. 2, Supplementary Table 1 online and data not shown). Analysis of their hematopoietic system revealed no major defects in the total number of megakaryocytes, erythroblasts, eosinophils, neutrophils, monocytes and, as previously described11, T and B lymphocytes (data not shown). We observed, however, that these mice were hypertensive, showing high systolic and diastolic blood pressure. Additionally, and unlike most hypertension models2, Vav3−/− mice had tachycardia. We observed these cardiovascular defects in mice in both anaesthetized and conscious states (Supplementary Table 1). Vav3−/− mice also had cardiovascular defects, which typically occur in humans with essential hypertension2,12,13, including left ventricular hypertrophy, left ventricular fibrosis, and severe thickening and remodeling of the aorta media wall (Fig. 1a, Supplementary Fig. 3 and Supplementary Note online). We first detected cardiovascular defects in 4-month-old mice; these defects became accentuated in older specimens (Fig. 1a). We observed no defects in the right ventricle or in pulmonary arteries (Fig. 1a, Supplementary Fig. 3 and data not shown), indicating that hypertension is restricted to the systemic circulation system. Vav1−/−mice had normal cardiovascular parameters (Supplementary Table 1 and data not shown).
Figure 1.
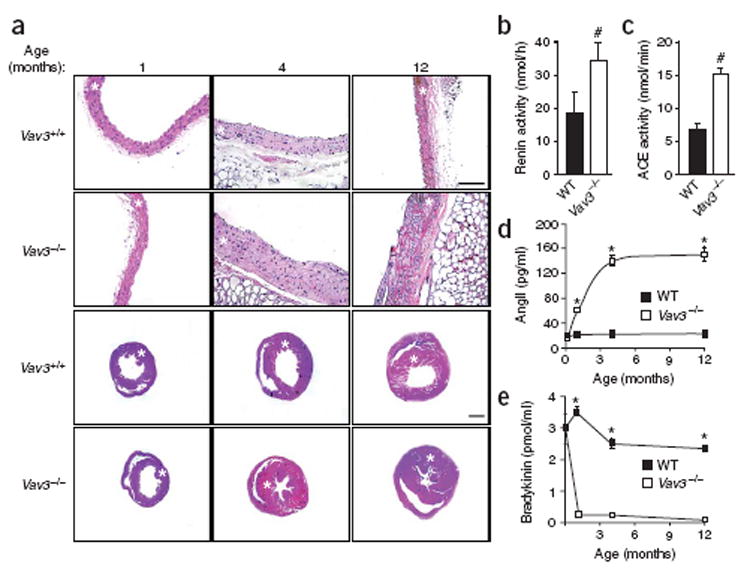
The cardiovascular system in Vav3−/−mice. (a) Histological sections of aortas (first and second row) and hearts (third and fourth row) from mice of the indicated genotypes and ages. Asterisks indicate aortic media layers and left ventricles. Scale bars, 100 μm. Sections are representative of 6–14 mice of each genotype. (b,c) Levels of renin (n =9;b) and ACE (n = 5; c) activity in wild-type (WT) and Vav3−/− mice. (d,e) Plasma levels of AngII (n ≥ 5; d) and bradykinin (n ≥ 5; e) in wild-type (WT) and Vav3−/− mice of the indicated ages. Error bars represent s.e.m. #P < 0.05, *P < 0.01.
To identify the cause of the cardiovascular defects of Vav3−/− mice, we investigated the status of two essential physiological circuits involved in hypertension, the renin-angiotensin system (RAS) and endothelin system1, 14, 15. In the case of the RAS, the protease renin cleaves angiotensinogen to yield the inactive angiotensin I (AngI) peptide. Angiotensin-converting enzyme (ACE) then catalyzes the cleavage of AngI to AngII. This peptide modulates blood pressure by promoting vasoconstriction and reabsorption of renal sodium through interaction with AT1, a G-protein-coupled receptor14. ACE also cleaves the vasodilator peptide bradykinin into inactive peptides, further tilting the cardiovascular balance toward vasoconstriction14. In the case of the endothelin system, endothelins are also generated through a proteolytic cascade in the cytoplasm and at the plasma membrane15. They mediate potent vasoconstrictor responses by activating a specific G-protein-coupled receptor (ETA)15. Analysis of Vav3−/− mice showed high levels of renin and ACE in the plasma and the heart, respectively (Fig. 1b,c). These mice also showed an agedependent increase of AngII and a concomitant reduction of bradykinin in the plasma (Fig. 1d,e). The increased AngII levels were accompanied by a parallel upregulation of Agtr1a (also known as At1) mRNA expression in aortic smooth muscle cells and heart, but not in pulmonary arteries (Supplementary Fig. 4 online ). In contrast, expression of a second AngII receptor involved in vasodilatation, AT2 (ref. 14), showed no variation (Supplementary Fig. 4). We also did not detect changes in levels of mRNAs coding for endothelin receptors (Supplementary Fig. 4), suggesting that the hypertension of Vav3−/−mice is RAS dependent. Consistent with these results, injection of the AT1 antagonist losartan promoted a decline in the blood pressure of Vav3−/− mice that, within minutes, reached levels similar to those found in wild-type mice (Fig. 2a,b). Bosentan, an inhibitor of endothelin receptors, had no significant effect on the blood pressure of Vav3−/− mice (Fig. 2c). Oral administration of captopril, an ACE inhibitor, halted the increase of AngII levels (Fig. 2d), the reduction of bradykinin levels (Fig. 2e) and the cardiovascular remodeling observed in Vav3−/− mice (Fig. 2g). These results indicate that the RAS system, and not the endothelin system, contributes to the development of the cardiovascular dysfunction of Vav3−/− mice.
Figure 2.
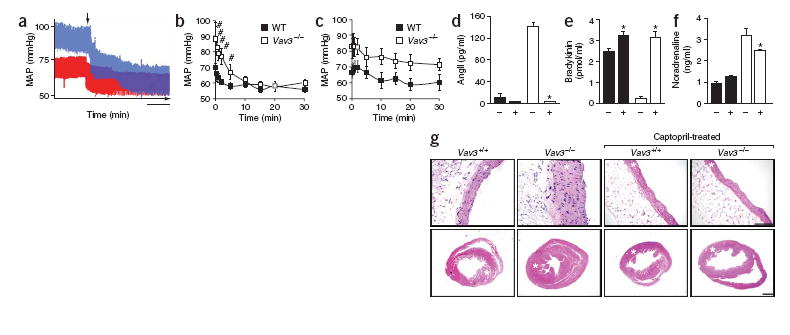
The RAS and endothelin systems in Vav3−/− mice. (a) Real-time recording of the blood pressure of either a wild-type (red) or a Vav3−/− (blue) mouse upon administration of losartan. MAP, mean arterial pressure. Scale bar, 1 min. (b,c) Quantification of the changes in the MAP in wild-type (WT) and Vav3−/− mice after administration of either losartan (b) or bosentan (c) during the indicated periods of time (n = 6). (d–f) Effect of captopril on the plasma levels of AngII (n = 5–7;d), bradykinin (n = 5–6;e) and noradrenaline (n = 5–6;f) in wild-type (black) and Vav3 null (white) mice. +, captopril-treated mice. (g) Histological sections of aortas (top) and hearts (bottom) of 4.25-month-old mice of the indicated genotypes, which were previously left untreated or treated with captopril for 5 weeks as indicated. Asterisks indicate the aortic media walls and the left ventricles. Sections are representative samples of five mice of each genotype. Scale bars, 100 μm. Error bars represent s.e.m. #P < 0.05, *P < 0.01.
The SNS mediates cardiovascular activity by modulating heart rate, cardiac contraction, vascular tone and release of renin1, 16. The action of the SNS is promoted by three catecholamines: adrenaline, noradrenaline and dopamine17. The concentration of these molecules was elevated from birth to adulthood in the plasma of Vav3−/− mice (Fig. 3a–c), indicating hyperactivity of the SNS. Several observations allowed us to infer that this phenotype was not derived from an indirect stimulation of the SNS by the elevated levels of AngII. The increase in catecholamine levels, which was readily detected in newborn Vav3−/− mice (Fig. 3a–c), preceded that observed for AngII by at least 1 month (Fig. 1d). Additionally, captopril blocked the production of AngII but had only marginal effects on the levels of noradrenaline (Fig. 2d,f). We verified the upstream action of the SNS in relation to the RAS system by treating Vav3−/− mice with propranolol, a nonselective β-adrenergic receptor inhibitor. This drug blocked development of hypertension (Fig. 3d), tachycardia (Fig. 3e), production of AngII (Fig. 3f), cardiovascular remodeling (Fig. 3g) and heart fibrosis (Supplementary Fig. 3). These results strongly suggest that the cardiovascular defects of Vav3−/− mice are due to a SNS-dependent stimulation of RAS.
Figure 3.
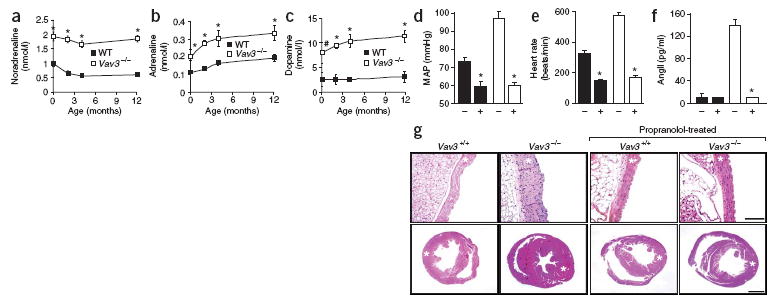
SNS activity is deregulated in Vav3−/− mice. (a–c) Catecholamine levels in wild-type (WT) and Vav3−/− mice of the indicated ages (n ≥ 5). (d–f) Effects of propranolol on mean arterial pressure (MAP;n = 7; d), heart rate (n = 8;e) and AngII levels (n = 8; f) of wild-type (black bars) and Vav3−/− (white bars) mice. +, propranolol-treated mice. (g) Histological sections of aortas (top) and hearts (bottom) of 4.25-month-old mice of the indicated genotypes that were previously left untreated or treated with propranolol for 5 weeks as indicated. Asterisks indicate aortic media and left ventricles. Sections are representative samples of eight mice of each genotype. Scale bars, 100 μm. Error bars represent s.e.m. #P < 0.05, *P < 0.01.
As hypertension in humans is usually associated with renal dysfunction1-3, 14, 17, we evaluated the functional status of kidneys in Vav3−/−mice. Kidneys had normal weight and morphology in haematoxylin and eosin–stained tissue sections (data not shown). Closer examination, however, showed functional and histological alterations. We observed that the urinary flow of Vav3−/− mice was approximately 85% lower than that in control littermates (Fig. 4a). Additionally, kidneys of Vav3−/− mice had lower rates of creatinine clearance (Fig. 4b), Na+ excretion (Fig. 4c) and, as a compensatory mechanism to keep the electrolytic balance, Cl− excretion (Fig. 4c). Instead, K+ excretion rates of Vav3−/− kidneys were lower than wild-type rates (Fig. 4c). Finally, Vav3−/− mice developed renal fibrosis is an agedependent manner (Fig. 4d,e). The alterations in urine production and sodium excretion are probably due to elevated vasopressin and aldosterone levels. Vasopressin is an SNS-activated hormone that regulates water reabsorption in the kidney17. Aldosterone is secreted by the adrenal cortex upon stimulation with AngII and is responsible for regulating sodium reabsorption by nephrons1, 2. Consistent with this hypothesis, we observed high levels of vasopressin in newborn Vav3−/− mice and observed a further increase as they aged (Supplementary Fig. 5, online). Aldosterone levels became elevated from 4 months of age onward (Supplementary Fig. 5). These kinetics correlate well with the pattern of catecholamine and AngII production observed in Vav3−/− mice, respectively (Figs. 1d and 3a–c).
Figure 4.
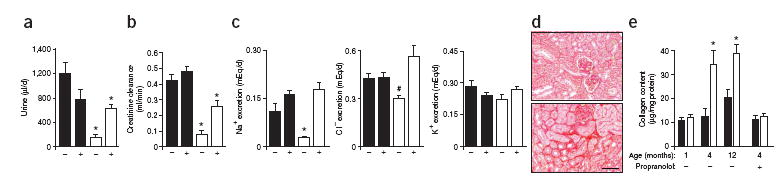
Functional status of the kidneys of Vav3−/− mice. (a–c) Rates of urine production (a), creatinine clearance (b), Na+ excretion (c, left), Cl– excretion (c, middle) and K+ excretion (c, right) in wild-type (black bars) and Vav3−/− (white bars) mice either untreated (–) or treated (+) with propranolol for 5 d (n = 5). (d) Sirius red staining of kidney sections from wild-type (top) and Vav3−/− (bottom) mice to detect interstitial collagen (n = 5). Scale bar, 50 μm. (e) Levels of renal fibrosis estimated by total collagen content of wild-type (black bars) and Vav3−/− (white bars) mice at indicated ages. Where indicated (+), 3-month-old mice were treated with propranolol for 5 weeks before kidney collection (n = 5). Error bars represent s.e.m. #P < 0.05, *P < 0.01.
To test whether the SNS hyperactivity of these mice caused these defects, we evaluated the effect of propranolol on the above parameters. A 5-d treatment of Vav3−/− mice with this β-adrenergic inhibitor restored urine production (Fig. 4a), sodium excretion (Fig. 4c), normal plasma levels of both vasopressin and aldosterone (Supplementary Fig. 5), and partially rescued the defective rates of creatinine clearance by kidneys (Fig. 4b). As expected, we observed that normal levels of aldosterone were recovered upon a 5-d treatment with captopril (Supplementary Fig. 5), a result that further links the SNS and the RAS to these physiological dysfunctions. As with cardiovascular remodeling (Supplementary Fig. 3), a 5-week administration of propranolol prevented the development of kidney fibrosis in Vav3−/− mice (Fig. 4e). Together, these results indicate that the renal defects are the consequence, rather than the cause, of the hypertensive state found in Vav3−/− mice. Furthermore, they rule out the possibility that the SNS overstimulation observed in these mice could be due to defects in kidney function.
In summary, this study shows that ablation of the Vav3 gene promotes the gradual development of an SNS-dependent cardiovascular disease in mice. Although we do not know the cellular basis for the sympathetic hyperactivity found in Vav3−/− mice, these results are appealing from a clinical point of view because SNS-dependent tachycardia is now recognized as a major risk factor for developing cardiovascular-related diseases in humans5, 6, 18, 19. It has been also postulated that sympathetic-dependent cardiac overactivity is responsible for the elevation of blood pressure in the early stages of essential hypertension2, 5. These alterations have an unknown etiology2, 6. Our results also show that renal dysfunction can be a downstream consequence, rather than the trigger1-3, of hypertensive conditions. Based on our results, it will be intriguing to investigate the involvement of Vav3 in sympathetic-dependent cases of human cardiovascular disease. Arguably, our Vav3-deficient mice will serve as a promising experimental model in which to discover genes, biological processes and drugs that could influence the development of these complex disorders.
METHODS
Generation of knockout mice
We generated knockout mice by homologous recombination techniques, which replaced the 5´ end of the Vav3 locus with a DNA fragment containing the neomycin resistance gene, the Pgk1 promoter, the polyadenylation signal of mRNA encoding bovine growth hormone, and two loxP sequences (Supplementary Fig. 1). We verified the ablation of Vav3 expression using PCR and immunohistochemistry in different tissues of knockout mice. Vav1 knockout mice20 were provided by V. Tybulewicz (National Institute for Medical Research, London). Animal care and work protocols were approved and carried out following the regulations set forth by the Committee of Animal Research and Bioethics of Salamanca University. Unless otherwise stated, we used 4-month-old mice in the experiments.
Genotyping of mice
Mice were initially characterized by Southern blot using genomic DNA extracted from tail biopsies. DNA was digested with either EcoRV or BamHI, electrophoresed in agarose gels, transferred to nitrocellulose membranes (Schleicher & Schuell), screened with [32P]dCTP-labeled 5´ and 3´ diagnostic cDNA probes (Supplementary Fig. 1), and exposed to X-ray film. We then performed routine genotyping by PCR from tail biopsies using the following primers: bGH2 (5´-GCATCGCATTGTCTGAGTAGG-3´), Vav3−/− 3´ (5´-TGAGCAGCTGGCAGAGCAGG-3´) and Vav3−/− 5´ (5´-GGACATGGA GCCGTGGAAGC-3´).
Hematopoietic cell populations
We obtained cells by dissociations of bone marrow, spleen and thymus, and evaluated the percentage of each hematopoietic lineage by flow cytometry using specific antibodies to surface markers.
Hemodynamic studies
We anesthetized mice with sodium pentobarbital (40 mg/kg body weight; Sigma). We recorded arterial pressures by catheterization of the right carotid artery with a pressure probe connected to a digital data recorder (MacLab/4e; AD Instruments), as previously described21. We analyzed the recordings with Chart v 3.4 software (AD Instruments). We recorded blood pressure and heart rates after a 20-min stabilization period after catheterization. Alternatively, we recorded blood pressure and heart rate in mice that were awake with an automated multichanel system using the tail-cuff method with a photoelectric sensor (Niprem 546; Cibertec SA).
Histology and pathological assessment of mice
We fixed selected tissues in 4% paraformaldehyde in PBS and paraffin-embedded them. We stained 2–3 μm sections with hematoxylin and eosin (Sigma). We quantified stained sections in a blinded manner using Metamorph-Metaview software (Universal Imaging).
Tissue fibrosis analysis
For the in situ visualization of fibrosis, we stained paraffin-embedded sections from hearts and kidneys with Sirius red (Fluka) to visualize collagen. For quantification of total collagen in tissues, we determined the hydroxyproline content using a spectrophotometric method22. We calculated total collagen assuming that collagen contains 12.7% hydroxyproline.
Analysis of cardiovascular-, renal- and blood-related molecules
We evaluated renin activity in plasma using a radioimmunoassay kit (Kit Ren-CT2; Schering España). We determined ACE activity in heart extracts using a luminescence method, as previously described23. We used enzyme-linked immunosorbent assay (ELISA) kits to measure plasma levels of AngII (Angio-tensin II ELISA kit; SPI Bio), bradykinin (Markit-M Bradykinin; Dainippon), noradrenaline (CatCombi ELISA; IBL), adrenaline (CatCombi ELISA; IBL), aldosterone (Aldosterone ELISA; IBL) and vasopressin (Arg-Vasopressin EIA kit; Assay Designs). We measured dopamine levels by radioimmunoassay (Dopamine RIA; IBL). Agtr1a, Agtr2, Ednra and Ednrb mRNA levels were determined by quantitative; reverse-transcription PCR in the indicated tissues. To this end, we extracted total RNAs using Trizol (Life Technologies), reverse transcribed them (using the Quantitec SYBER Green RT-PCR kit; Qiagen), and PCR-amplified them (using iCycler iQ; BioRad) using Sybr Green as the fluorescent probe. PCR controls included RpLp0 mRNA. Information about primers is available upon request.
Pharmacological studies
We adminstered losartan (Du Pont) and bosentan (Sigma) in physiological solution as single-dose boluses in the catheterized left jugular veins of mice at a concentration of 10 mg/kg body weight. In the case of captopril and propranolol, 3-month-old mice were given drinking water alone or supplemented with captopril (100 μg/ml; Sigma) or propranolol (500 μg/ml; Sigma). Mice were killed 5 weeks later and tissues samples collected. In renalfunction studies, we treated 4-month-old mice with either captopril or propranolol for 5 d as indicated above.
Kidney function analysis
We collected urine samples from single mice in metabolic cages during 24 h. Urine was collected into graduated cylinders containing 100 μl 0.1% sodium azide (to minimize bacterial contamination) and 1 ml mineral oil (to avoid evaporation). We also collected blood samples (150 μl) from the caudal vein of the mice under study to evaluate the concentration of creatinine in plasma. We determined urine and plasma creatinine concentrations by a modification of the Jaffé reaction, as previously described24. We measured urinary electrolyte concentration using a Hitachi autoanalyzer.
Heart performance assays
Details are available in the Supplementary Note online.
Supplementary Material
Note: Supplementary information is available on the Nature Medicine website.
Acknowledgments
We thank J. Tamame, M. Blázquez, T. Iglesias, M. Jerkic and F. Núñez for technical help. We also thank V. Tybulewicz for making available to us his Vav1 knockout mice and M. Dosil for helpful comments on the manuscript. This work was supported by grants from the US National Institutes of Health to X.R.B. and the Spanish Ministry of Education and Science to X.R.B. and J.M.L.-N. V.S. is supported by a European Molecular Biology Organization (EMBO) long-term postdoctoral fellowship.
Footnotes
V.S. contributed to study design and performed all experiments. M.A.S. and M.J.M. collaborated on experiments shown in Figure 1c and Figure 3d,f. J.V.R.-E. collaborated on experiments shown in Figure 2a–c. E.d.Á. collaborated on histological analysis of mice. J.M.L.-N. analyzed data and collaborated on experiments shown in Figure 4a–c. X.R.B. directed and designed the study, analyzed the data and wrote the manuscript.
The authors declare that they have no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 2.Beevers G, Lip GY, O’Brien E. ABC of hypertension: the pathophysiology of hypertension. Br Med J. 2001;322:912–916. doi: 10.1136/bmj.322.7291.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Staessen JA, Wang J, Bianchi G, Birkenhager WH. Essential hypertension. Lancet. 2003;361:1629–1641. doi: 10.1016/S0140-6736(03)13302-8. [DOI] [PubMed] [Google Scholar]
- 4.Movilla N, Bustelo XR. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol Cell Biol. 1999;19:7870–7885. doi: 10.1128/mcb.19.11.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palatini P, Julius S. Relevance of heart rate as a risk factor in hypertension. Curr Hypertens Rep. 1999;1:219–224. doi: 10.1007/s11906-999-0024-7. [DOI] [PubMed] [Google Scholar]
- 6.Palatini P, Julius S. The physiological determinants and risk correlations of elevated heart rate. Am J Hypertens. 1999;12:3S–8S. [PubMed] [Google Scholar]
- 7.Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner M, Billadeau DD. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat Rev Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 9.Faccio R, et al. Vav3 regulates osteoclast function and bone mass. Nat Med. 2005;11:284–290. doi: 10.1038/nm1194. [DOI] [PubMed] [Google Scholar]
- 10.Tybulewicz VL, Ardouin L, Prisco A, Reynolds LF. Vav1: a key signal transducer downstream of the TCR. Immunol Rev. 2003;192:42–52. doi: 10.1034/j.1600-065x.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 11.Fujikawa K, et al. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J Exp Med. 2003;198:1595–1608. doi: 10.1084/jem.20030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmieder RE, Messerli FH. Hypertension and the heart. J Hum Hypertens. 2000;14:597–604. doi: 10.1038/sj.jhh.1001044. [DOI] [PubMed] [Google Scholar]
- 13.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38:581–587. doi: 10.1161/hy09t1.096249. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi N, Smithies O. Gene targeting approaches to analyzing hypertension. J Am Soc Nephrol. 1999;10:1598–1605. doi: 10.1681/ASN.V1071598. [DOI] [PubMed] [Google Scholar]
- 15.Remuzzi G, Perico N, Benigni A. New therapeutics that antagonize endothelin: promises and frustrations. Nat Rev Drug Discov. 2002;1:986–1001. doi: 10.1038/nrd962. [DOI] [PubMed] [Google Scholar]
- 16.DiBona GF. The sympathetic nervous system and hypertension: recent developments. Hypertension. 2004;43:147–150. doi: 10.1161/01.HYP.0000113047.47711.fa. [DOI] [PubMed] [Google Scholar]
- 17.Carrasco GA, Van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Pharmacol. 2003;463:235–272. doi: 10.1016/s0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- 18.Palatini P, Julius S. Elevated heart rate: a major risk factor for cardiovascular disease. Clin Exp Hypertens. 2004;26:637–644. doi: 10.1081/ceh-200031959. [DOI] [PubMed] [Google Scholar]
- 19.Palatini P, Benetos A, Julius S. Impact of increased heart rate on clinical outcomes in hypertension: implications for antihypertensive drug therapy. Drugs. 2006;66:133–144. doi: 10.2165/00003495-200666020-00001. [DOI] [PubMed] [Google Scholar]
- 20.Turner M. A requirement for the Rho-family GTP exchange factor Vav in positive and negative selection of thymocytes. Immunity. 1997;7:451–460. doi: 10.1016/s1074-7613(00)80367-2. [DOI] [PubMed] [Google Scholar]
- 21.Jerkic M, et al. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J. 2004;18:609–611. doi: 10.1096/fj.03-0197fje. [DOI] [PubMed] [Google Scholar]
- 22.Flores O, et al. Beneficial effect of the long-term treatment with the combination of an ACE inhibitor and a calcium channel blocker on renal injury in rats with 5/6 nephrectomy. Exp Nephrol. 1998;6:39–49. doi: 10.1159/000020503. [DOI] [PubMed] [Google Scholar]
- 23.Chevillard C, Brown NL, Mathieu MN, Laliberte F, Worcel M. Differential effects of oral trandolapril and enalapril on rat tissue angiotensin-converting enzyme. Eur J Pharmacol. 1988;147:23–28. doi: 10.1016/0014-2999(88)90629-2. [DOI] [PubMed] [Google Scholar]
- 24.Valdivielso JM, et al. Renal ischemia in the rat stimulates glomerular nitric oxide synthesis. Am J Physiol Regul Integr Comp Physiol. 2001;280:R771–R779. doi: 10.1152/ajpregu.2001.280.3.R771. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Medicine website.