

Summary
Intrinsic resistance to multiple drugs in many gramnegative bacterial pathogens is conferred by resistance nodulation cell division efflux pumps, which are composed of three essential components as typified by the extensively characterized Escherichia coli AcrA-AcrB-TolC system. The inner membrane drug: proton antiporter AcrB and the outer membrane channel TolC export chemically diverse compounds out of the bacterial cell, and require the activity of the third component, the periplasmic protein AcrA. The crystal structures of AcrB and TolC have previously been determined, and we complete the molecular picture of the efflux system by presenting the structure of a stable fragment of AcrA. The AcrA fragment resembles the elongated sickle shape of its homolog Pseudomonas aeruginosa MexA, being composed of three domains: β-barrel, lipoyl, and α-helical hairpin. Notably, unsuspected conformational flexibility in the α-helical hairpin domain of AcrA is observed, which has potential mechanistic significance in coupling between AcrA conformations and TolC channel opening.
Introduction
The rising incidence of multidrug-resistant bacterial pathogens poses a serious threat to human health. Intrinsic multidrug resistance is conferred in a number of gram-negative bacterial pathogens, including Pseudomonas aeruginosa and Haemophilus influenzae, by transporters belonging to the resistance nodulation cell division (RND) family of proteins (Putman et al., 2000; Walsh, 2000). RND-type transporters utilize the proton electrochemical gradient to energize efflux of antibiotics and other compounds out of the bacterial cell. The major mechanistic feature of these transporters is the ability to transfer multiple substrates across both the inner and the outer membranes of gram-negative bacteria directly into external media without periplasmic intermediates (Thanassi et al., 1995). To achieve drug efflux across two membranes, RND-type transporters assemble into multiprotein efflux systems. Two of the most extensively characterized RND multidrug efflux systems are Escherichia coli AcrA-AcrB-TolC and P. aeruginosa MexA-MexB-OprM (Hirakata et al., 2002; Ma et al., 1995).
RND systems consist of large complexes of three essential components. The first is an RND inner membrane protein, which is energized by the proton-motive force. The RND inner membrane protein assembles into a trimer, as shown by the X-ray crystal structure of the proton antiporter AcrB (Murakami et al., 2002; Yu et al., 2003). Each protomer of the trimer has 12 transmembrane α-helical segments and two large ∼300 residue periplasmic domains that extend ∼70 Å above the plane of the inner membrane. Chemically diverse substrates, such as rhodamine 6G, ethidium, dequalinium, and ciprofloxacin, have been seen to bind through hydrophobic interactions to a central cavity in the periplasmic domain (Murakami et al., 2002; Yu et al., 2003). How these compounds are pumped outward from their binding sites is not yet known.
The second essential component of the RND system is an outer membrane protein, also known as outer membrane factor (OMF), that like the RND inner membrane protein is trimeric. The structures of the OMFs E. coli TolC, P. aeruginosa OprM, and Vibrio cholerae VceC have been determined (Akama et al., 2004a; Federici et al., 2005; Koronakis et al., 2000), revealing similarly shaped cylindrical channels. The trimeric channel is embedded in the outer membrane as a 12 stranded β-barrel that continues ∼100 Å into the periplasmic space as an α-helical barrel. TolC, OprM, and VceC have been visualized in their closed states, and hypothesized to open through an iris-like mechanism (Andersen et al., 2002; Koronakis et al., 2000). TolC has been shown to interact physically with AcrB by chemical crosslinking and intermolecular disulfide bond formation experiments (Tamura et al., 2005; Tikhonova and Zgurskaya, 2004; Touze et al., 2004). This interaction suggests that substrates are transported directly from the RND inner membrane protein through the open OMF channel and out into the extracellular space.
The third essential component is a periplasmic protein that belongs to the membrane fusion protein (MFP) family (Dinh et al., 1994; Saier et al., 1994), named for sequence similarity in these proteins to the membrane fusion protein (F protein) of paramyxovirus 5 (Dinh et al., 1994; Saier et al., 1994). Interestingly, MFPs are not only essential components of RND systems but also of other energy-dependent transport systems, such as the ATP binding cassette (ABC) system and the major facilitator system (MFS) (Putman et al., 2000). MFPs are attached to the inner membrane via lipid acylation of a cysteine residue or through an N-terminal transmembrane segment. However, membrane attachment is not essential for drug efflux activity, as seen by the functionality of soluble, periplasmic mutants of the E. coli MFP AcrA and the P. aeruginosa MFP MexA (Yoneyama et al., 2000; Zgurskaya and Nikaido, 1999a). AcrA has been shown to interact physically with both AcrB and TolC (Husain et al., 2004; Tikhonova and Zgurskaya, 2004; Touze et al., 2004; Zgurskaya and Nikaido, 2000). Recent experimental evidence indicates that interaction of AcrA with these components is likely to play an active role in the efflux process (Aires and Nikaido, 2005; Zgurskaya and Nikaido, 1999b).
In this study, we have determined the 2.7 Å resolution X-ray crystal structure of the stable core of AcrA, thereby helping to complete the atomic resolution model of the AcrA-AcrB-TolC drug extrusion system and providing a point of comparison to recently determined structures of the P. aeruginosa MFP MexA (Akama et al., 2004b; Higgins et al., 2004). The structure of AcrA is found to provide unsuspected evidence for conformational flexibility in MFPs. Intriguingly, this flexibility coincides with conformational changes predicted to occur during opening of OMF channels by an iris-like mechanism.
Results
Domain Mapping of AcrA
AcrA was subjected to limited proteolytic digestion to map its domain architecture. For this purpose, a soluble form of mature AcrA (residues 26-397, 40 kDa), which lacks the cleaved signal sequence (residues 1-24) and the lipid acylation site at residue Cys-25, was expressed cytoplasmically in E. coli and purified using an introduced C-terminal histidine tag (Figure 1). This cytoplasmically expressed, soluble form of AcrA has been shown to be functional in restoring in vivo drug efflux activity to a ΔacrA strain of E. coli (Zgurskaya and Nikaido, 1999a). After 1 hr of digestion with the relatively nonspecific protease thermolysin at a 50:1 substrate:protease (mass) ratio, AcrA is found to be trimmed slightly at its N- and C-terminal ends. The resulting 38 kDa products begin heterogeneously at either residue 26 or 30 and end homogeneously at residue 386, as determined by N-terminal sequencing and mass spectrometry. These products are further digested over the next two to four hours to yield the stable, protease-resistant core of AcrA (Figure 1). The 28 kDa stable core begins at residue 45 and ends heterogeneously at residue 305 or 312. This thermolytic fragment is very similar to a recently reported tryptic fragment of AcrA that encompasses residues 45-315 (Touze et al., 2004).
Figure 1.
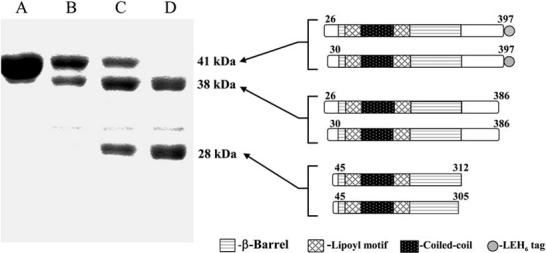
Domain Mapping of AcrA(26-397) by Thermolytic Digestion
AcrA(26-397) was digested with thermolysin for 0, 1, 2, and 4 hr (lanes A, B, C, D, respectively) at a 50:1 (mass) substrate:protease ratio and analyzed by 12% SDS-PAGE. Molecular weights and schematics of proteolytic products are shown to the right of the corresponding fragments. Fragments were identified by N-terminal sequencing and mass spectrometry.
These results demonstrate that the ∼90 C-terminal residues of AcrA are likely to be flexible and hence proteolytically sensitive. The C-terminal residues (313-397) removed from AcrA by proteolysis are digested into small peptides and do not yield a resistant fragment. This proteolytically sensitive C-terminal region of AcrA is required for association with the inner membrane RND protein AcrB, as shown by chimera studies using AcrA residues 290-357 and direct binding studies using an AcrA fragment composed of residues 172-397 (Elkins and Nikaido, 2003; Touze et al., 2004). The C-terminal region also appears to be required for interaction with TolC (Touze et al., 2004). The flexibility of both N- and C-terminal regions of AcrA is also consistent with X-ray crystal structures of the homolog MexA, the periplasmic component of the P. aeruginosa MexA-MexB-OprM multidrug efflux system (Akama et al., 2004b; Higgins et al., 2004). Although intact mature MexA, with a molecular weight of 38 kDa, had been crystallized, electron density was observed only for a region of the protein equivalent to the 28 kDa stable core of AcrA.
Structure Determination of the Stable Core of AcrA
To gain functional insight, we set out to crystallize the 28 kDa stable core of AcrA (residues 45-312). Although AcrA(45-312) was found to readily crystallize (e.g., under one fifth of Hampton crystal screen I and II conditions), almost all crystals were found to diffract anisotropically to a maximum resolution of only 5-10 Å. Only one condition was identified to yield crystals that diffracted X-rays to a reasonable resolution limit (∼3.5 Å). These crystals appeared to belong to space group P42212, but were discovered, after several unsuccessful attempts at obtaining phase information, to have arisen through a rare case of perfect pseudomerohedral twinning of P212121 crystals, in which the a and b axes are fortuitously identical in length (169 Å). Analysis of intensity distributions in local areas of reciprocal space proved instrumental in detecting pseudomerohedral twinning in these crystals (Padilla and Yeates, 2003).
Due to great difficulties in determining structures from perfectly twinned crystals, an alternate crystal form was sought. Untwinned crystals that diffracted X-rays isotropically to 2.7 Å resolution were obtained from a quadruple methionine substitution mutant (F223M, L224M, L287M, and L288M) which had been constructed for the purpose of multiwavelength anomalous dispersion (MAD) phase determination. Wild-type AcrA(45-312) also crystallized under essentially the same conditions as the quadruple methionine substitution mutant, called AcrA(45-312)-4M, but diffracted X-rays anisotropically to a maximum resolution of only 3.3 Å resolution. Crystals of AcrA(45-312)-4M contain four molecules per asymmetric unit and a solvent content of 60%. The structure was determined by MAD techniques using selenomethionine-labeled protein. Continuous electron density for the main chain from residue 53 to 299 is observed in one of the monomers (Figure 2A, labeled C), whereas small breaks (at residues 230-241) are observed in the other three monomers (Figure 2A, labeled A, B, and D). The model has been refined to 2.71 Å resolution with an Rwork of 23.7% and an Rfree of 27.5% (Table 1).
Figure 2.
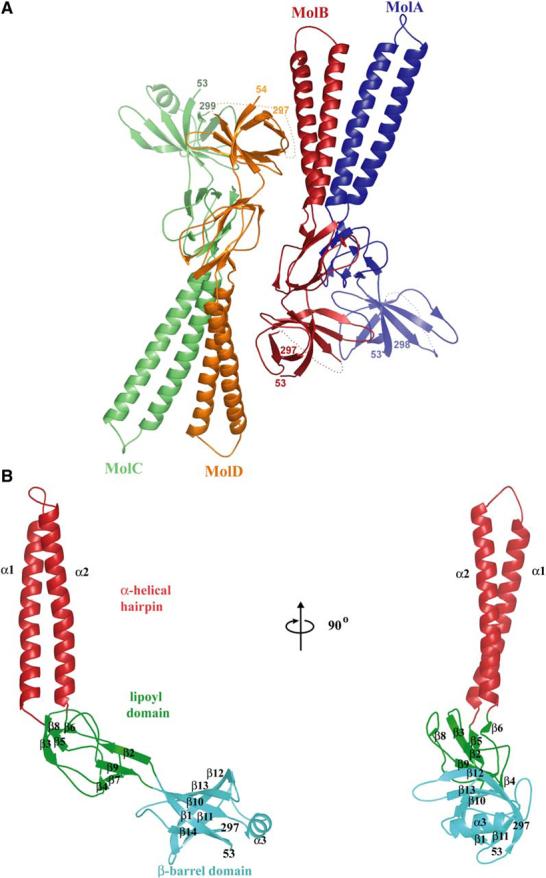
Structure of AcrA(45-312)-4M (A) Ribbon representation of AcrA(45-312)-4M, showing two apparent dimers per asymmetric unit in the crystal.
(B) Ribbon representation of a monomer of AcrA(45-312)-4M (molecule C), with the α-helical hairpin domain in red, lipoyl domain in green, and β-barrel domain in cyan. Molecular graphics were made with PyMOL (DeLano, 2002).
Table 1.
X-Ray Data Collection and Refinement, AcrA(45-312)-4M
| Data Collection | ||||
|---|---|---|---|---|
| Beamline | APS-BM19 | ALS-8.2.2 | ||
| Data set | λ1 | λ2 | λ3 | λ1 |
| Wavelength (Å) | 0.97938 | 0.97955 | 0.96415 | 1.0000 |
| Resolution (Å)a | 50-3.00 (3.11-3.00) | 50-3.39 (3.59-3.39) | 50-3.50 (3.63-3.50) | 50-2.70 (2.77-2.70) |
| Completeness (%) | 99.3 (99.3) | 99.8 (99.9) | 99.8 (99.9) | 97.3 (91.1) |
| Redundancy | 4.2 (3.4) | 4.4 (4.5) | 4.2 (3.8) | 5.0 (3.9) |
| I/σ | 13.5 (2.3) | 16.6 (4.6) | 12.0 (4.0) | 14.6 (2.3) |
| Rmergeb | 13.9 (76.1) | 9.8 (38.9) | 18.5 (53.4) | 9.0 (42.9) |
| Phasing | |||
|---|---|---|---|
| Se λ1 | Se λ2 | Se λ3 | |
| Anomalous and dispersive differences (%)c | |||
| Se λ1 | 4.2 | ||
| Se λ2 | 1.8 | 2.4 | |
| Se λ3 | 2.0 | 3.8 | 3.1 |
| Anomalous phasing powerd | 1.5 | 0.8 | 0.7 |
| Number of sites | 16 | ||
| Figure of merit (40-3.0 Å) | 0.59 |
| Refinement | |||
|---|---|---|---|
| Resolution | 50.0-2.71 (2.77-2.71) | ||
| Rcryst (%)e | 23.7 (40.8) | Rms deviation | |
| Rfree (%)e | 27.5 (47.4) | Bonded B factor (Å2) | 3.30 |
| Number of reflections | From ideal geometry | ||
| Working set | 35,426 | Lengths (Å) | 0.010 |
| Test set | 1,873 | Angles (°) | 1.608 |
| Number of atoms | Average B factors (Å2) | ||
| Protein | 6936 | Protein | 89.2 |
| Solvent | 36 | Solvent | 64.0 |
Highest resolution shell is in parentheses.
Anomalous and dispersive differences = 100 × rms ΔF/rms F, where ΔF for anomalous differences is (F+h - F-h)/2 (diagonal element) and for dispersive differences is Fλi - Fλj (off-diagonal).
FH″/E, with FH″ being the anomalous component of the heavy atom structure factor and E the rms lack-of-closure error.
Overview of the Structure
The four molecules of AcrA(45-312)-4M in the asymmetric unit of the crystal pack as an apparent dimer of dimers. The molecules labeled A and B are related to one another by approximate dyad symmetry, as are the molecules labeled C and D; each set of dimers is in turn related to one another by yet another approximate 2-fold axis (Figure 2A). Close structural relationship is seen (and detailed below) between AcrA(45-312)-4M and MexA, which is not surprising given their 62% and 73% sequence identities and similarities, respectively. Despite the close sequence and structural similarity, the two proteins do not functionally complement each other (Tikhonova et al., 2002). Both AcrA(45-312)-4M and MexA are elongated, sickle-shaped molecules comprised of three domains: a β-barrel domain at one end, a lipoyl domain centrally located, and a coiled-coil α-helical hairpin at the other end of the molecule (Figure 2B). The existence in MFPs of the lipoyl and α-helical hairpin domains had been predicted from primary sequence considerations alone (Johnson and Church, 1999). The α-helical hairpin in AcrA is seven residues longer than in MexA, resulting in a total length of 105 Å for AcrA as compared to the 89 Å length of MexA.
β-Barrel Domain
The β-barrel domain consists of six antiparallel β strands and a short α helix (Figure 2B). The N and C termini of the AcrA fragment form two of the apposing strands (β1, residues 54-61 and β14, residues 292-297, respectively). This domain is predicted to be proximal in vivo to the inner membrane due to lipid acylation of Cys-25. The 28 flexible residues connecting Cys-25 to the β-barrel domain are more than sufficient to reach the periplasmic top of AcrB, which extends ∼70 Å above the inner membrane surface (Murakami et al., 2002; Yu et al., 2003). The short α helix (α3, residues 222-230) is located between β strands 10 and 11, and closes off the end of the β-barrel near the C terminus of the AcrA fragment. This helix in intact AcrA would, therefore, lie close to the flexible, proteolytically sensitive C-terminal 100 residues that are required for interaction with AcrB and TolC (Elkins and Nikaido, 2003; Touze et al., 2004).
The β-barrel domain is the site of all four methionine substitutions introduced for crystallization and phasing purposes. Two of the methionine substitutions, at positions 223 and 224, are located on the α3 helix, and the other two, at positions 287 and 288, on a loop connecting β strands 13 and 14 (Figure 3). Among these four substitutions, the only methionine that makes intermolecular contacts (within 4 Å) is Met-288 (molecules A and C), which abuts residues Val-265 and Val-267. The adjacent methionine substitution Met-287 makes intramolecular contacts, and is positioned against a hydrophobic pocket formed by residues Phe-254, Leu-246, and Met-291 near the center of the molecule’s β-barrel domain. Overall, the β-barrel domains of all four AcrA molecules are found to be structurally similar (rmsd 1.02 Å, 63 Cα atoms).
Figure 3.

Intermolecular Associations
(A) Sequence alignment of the α-helical hairpins of AcrA and MexA. The corresponding heptad position is shown above the sequence with residues in the c and f positions highlighted in gray (including Ala-99, which is situated at a c position but is a b position residue according to sequence) and hydrogen bonding residues in cyan.
(B) Parallel interactions of AcrA(45-312)-4M. Molecules A and B make similar dimeric contacts as molecules C and D, with the latter pair being shown here in Cα trace. Side chains are shown for residues in the c and f heptad positions as well as for residues that were substituted with methionine. The boxed region is enlarged at right, with hydrogen bonds colored in cyan. Below is a cross-section of the α-helical hairpin showing the intra- and intermolecular packing between helices. The positions of the heptad repeat are labeled a-g with side chains for the a and d heptad positions shown in yellow, and the e and f heptad positions shown in gray.
(C) Parallel interactions of MexA. Molecules A and B of the 13 total molecules in the asymmetric unit of MexA are shown, with representation as in (B).
(D) Antiparallel interactions of AcrA(45-312)-4M, with molecules related by crystallographic symmetry. Representation as in (B).
(E) Antiparallel interactions of MexA, with molecules G and H shown and representation as in (B).
Lipoyl and Coiled-Coil Domains
The central portion of AcrA consists of a lipoyl domain, best thought of as two lipoyl half-motifs interrupted by an α-helical hairpin formed by helices α1 and α2(Figure 2B). The N- and C-terminal lipoyl half-motifs are homologous to each other (Johnson and Church, 1999) and consist of four β strands (β2-β5 and β6-β9, respectively)that intertwine with one another to form a β sandwich. Lipoyl domains generally serve as carriers of lipoyl or biotinyl cofactors, which are attached to a conserved lysine on a loop connecting the two half-motifs. In AcrA and other MFPs, a coiled-coil α-helical hairpin intervenes between the lipoyl half-motifs instead of a lysine-containing loop.
The coiled-coil α-helical hairpin is 58 Å long and has five heptad repeats per helix. This is longer than the 47 Å long α-helical hairpin of MexA, which has four rather than five heptad repeats. The additional heptad in AcrA is located at residues 120-126 and 142-148, and entirely accounts for the difference in length between AcrA and MexA (Figure 3A). In general, MFPs are found to have coiled-coil lengths varying between four and six heptad repeats per helix. As seen with MexA, packing between the two helices (α1, residues 99-130; α2, residues 140-172) resembles canonical knobs-into-holes packing of hydrophobic side chains in the a and d positions of the heptad repeat (Akama et al., 2004b; Higgins et al., 2004; Johnson and Church, 1999). Also as seen in MexA, the left-handed superhelical coiling in the α1 helix is more pronounced than in the α2 helix, so that α2 appears to be relatively straighter than α1.
Functional Properties of Methionine Substitution Mutants
While the methionine mutations introduced for phasing have no apparent effect on protein stability in vitro, they have surprising effects on AcrA-AcrB-TolC efflux activity in vivo. To understand in vivo activity, we first asked whether AcrA truncated at residue 312 confers drug resistance. AcrA(1-312) was placed under control of its native promoter in an operon that also contains acrB. This operon was expressed in a strain of E. coli that lacks AcrAB and its close homolog AcrEF (ΔacrABΔacrEF). AcrA(1-312) contains its endogenous signal sequence, which directs it to the periplasm and promotes its attachment to the inner membrane via lipid acylation of Cys-25. The strain expressing AcrA(1-312) was found to be highly susceptible to a number of AcrA-AcrB-TolC substrates, including erythromycin, novobiocin, ethidium bromide, sodium dodecyl sulfate, and puromycin (Table 2). Interestingly, the truncation mutant was seen to have a drastically lower steady-state level of protein expression than wild-type AcrA (Figure 4).
Table 2.
Antimicrobial Susceptibility of E. coli Cells Expressing Wild-Type and Mutant AcrA
| Minimum Inhibitory Concentration (μg/ml) |
||||||
|---|---|---|---|---|---|---|
| pUC18 | AcrA | AcrA F223M/L224Ma | AcrA L287M/L288Mb | AcrA-4Mc | AcrA(1-312) | |
| Erythromycin | 2 | 64 | 128 | 128 | 2 | 2 |
| Novobiocin | 4 | 128 | 256 | 256 | 2 | 1 |
| Ethidium bromide | 6.25 | 100 | 100 | 100 | 6.25 | 6.25 |
| SDS | 62.5 | >2000 | >2000 | >2000 | 62.5 | 40 |
| Puromycin | 2 | 256 | 256 | 256 | 2 | ND |
ND, not determined.
AcrA(1-397) F223M/L224M.
AcrA(1-397) L287M/L288M.
AcrA(1-397) F223M/L224M/L287M/L288M.
Figure 4.

Steady-State Expression of AcrA and Mutants
Western blotting analysis with anti-AcrA antibodies of total cell protein (1 μg per lane) separated on an SDS-polyacrylamide (12%) gel. AcrA was expressed along with AcrB under control of the native acrAB promoter from various derivatives of pAHisB plasmid in E. coli AG100AX (ΔacrAB::kan ΔacrEF::spe). Steady-state expression of intact AcrA(1-397) (WT), AcrA L287M/L288M, AcrA F223M/L224M, AcrA F223M/L224M/L287M/L288M, and AcrA(1-312) is shown.
Because truncation at residue 312 abrogates steady-state protein expression and function, the methionine substitutions were introduced as double or quadruple substitutions into intact AcrA for functional assays. Both AcrA F223M/L224M and AcrA L287M/L288M, when directed to the periplasm for attachment to the inner membrane through Cys-25, were found to confer resistance to various compounds at levels nearly or fully equivalent to wild-type AcrA (Table 2). Surprisingly, the quadruple substitution mutant, AcrA F223M/L224M/L287M/L288M, did not confer resistance (Table 2). Steady-state protein expression levels of the two double methionine substitutions are at levels approximately equivalent to that of wild-type AcrA. In contrast, the steady-state expression level of the quadruple substitution mutant was undetectable, as was also the case for the truncation mutant (Figure 4).
It is not readily apparent why such large differences exist between the double and the quadruple substitution mutants. Association among RND components has been observed to promote stability (Tikhonova et al., 2002), and so it is possible that the low steady-state levels of some of the AcrA mutants above reflect compromised affinity for AcrB, TolC, or both. However, other explanations are possible and remain to be tested.
Packing of AcrA Monomers
Unlike trimeric AcrB and TolC, the association state of AcrA is uncertain. While soluble forms of AcrA and MexA have been found to be monomeric in vitro, crosslinking of AcrA in vivo suggests a trimeric form (Akama et al., 2004b; Higgins et al., 2004; Zgurskaya and Nikaido, 1999a, 2000). In crystals, both AcrA and MexA are observed to form oligomers involving similar sets of contacts. For AcrA, two molecules in parallel orientation associate as a dimer through extensive contacts involving the α-helical hairpins as well as adjoining lipoyl and β-barrel domains (Figure 3B). Approximately 1650 Å2 of surface area per molecule is buried at this interface. The same interactions are seen in MexA between adjacent molecules (Figure 3C), but in MexA, they are extended beyond dimers to form unusual six- and seven-membered superhelical rings that associate head to head (Akama et al., 2004b; Higgins et al., 2004).
The parallel intermolecular association observed in both AcrA and MexA crystals involves antiparallel interaction of the α1 helix of one monomer with the α2 helix of a second monomer (Figures 3B and 3C). The association involves knobs-into-holes packing of residues at c and f positions in AcrA, as assessed using the Socket algorithm (Figure 3B) (Walshaw and Woolfson, 2001). The association in MexA is similar but a less ideal form of knobs-into-holes packing (Figure 3C). As noted previously (Higgins et al., 2004), residues at c and f positions are generally conserved between AcrA and MexA (Figure 3A) as well as in other MFPs.
In addition to parallel associations, extensive antiparallel associations are observed. This occurs in the crystallographic packing of adjacent AcrA molecules and noncrystallographic packing of six- and seven-membered rings of MexA (Figures 3D and 3E). In the case of antiparallel association, adjacent equivalent helices, α1 for MexA and α2 for AcrA, pack in antiparallel fashion and contribute residues predominantly at the c and f positions to the interface. Therefore, the observation that MFPs are monomeric in solution but oligomeric in the high-concentration environment of crystals may be reconciled by the propensity of their α-helical hairpins to interact homotypically.
Conformational Flexibility of the α-Helical Hairpin Domain
Four conformations of AcrA(45-312)-4M have been captured in the crystal and provide evidence for flexibility of the hinge between the α-helical hairpin and lipoyl domain (Figure 5). The greatest variation is between AcrA molecules B and C (Figure 5A). Using the lipoyl domain as a reference, the α-helical hairpins of molecules B and C are found to differ by ∼15° overall and by 21 Å at the loop located at the tip of the hairpin. The lipoyl domain, which is connected to the base of the hairpin, superposes with high structural similarity in all four AcrA molecules (rmsd 0.12 Å, 66 Cα atoms). Superposition of the α-helical domains alone also shows the hairpins to be similar (rmsd 0.46 Å, 51 Cα atoms). The conformation flexibility evident in AcrA contrasts with MexA, which has essentially the same conformation in all 13 views captured in its crystal (maximum rmsd 1.17 Å, 230 Cα atoms) (Figure 5B). Notably, molecule D of AcrA has nearly the same α-helical hairpin orientation as the one seen in MexA (rmsd 0.85 Å, 183 Cα atoms) (Figure 5C). Thus, the AcrA crystal captures a range of α-helical hairpin orientations including the one observed for MexA.
Figure 5.

Conformational Flexibility of α-Helical Hairpin
(A) Comparison of four conformations of AcrA(45-312)-4M observed in the crystal, with molecules A, B, C, and D superposed on the lipoyl domain and displayed as Cα traces. The greatest difference, ∼15°, is between molecules B and C.
(B) Comparison of the 13 conformations of MexA observed in its crystal, with molecules superposed on the lipoyl domain and displayed as Cα traces.
(C) Superposition of molecule B of AcrA(45-312)-4M (orange) and molecule M of MexA (purple), with molecules superposed on all domains and displayed as Cα traces.
The difference in orientation of the α-helical hairpin of AcrA stems from an effective hinge located at the base of the hairpin. The hinge is composed of residues 99-106 in α1 and 169-173 in α2 (Figure 3A). This part of the coiled-coil in AcrA is underwound in molecules A and C, having a ∼200 Å pitch that is greater than the 150 Å pitch of a canonical α-helical coiled-coil (Seo and Cohen, 1993). This part also has imperfect knobs-into-holes packing. A canonical coiled-coil pitch (∼150 Å) and knobs-into-holes packing is evident in the rest of the α-helical coiled-coil. In contrast to molecules A and C, the hinge regions of molecules B and D are observed to have a nearly canonical coiled-coil pitch. Thus, the four conformations of AcrA taken together indicate that the base of the α-helical hairpin domain accommodates variation in helical winding and thereby permits hinge-like flexion of the rigid body of this domain.
Discussion
We have carried out biochemical and structural studies on the membrane fusion protein AcrA. Thermolytic domain mapping of AcrA revealed a stable central core to the protein similar to the one observed crystallographically in MexA (Akama et al., 2004b; Higgins et al., 2004) and recently reported from tryptic digestion of AcrA (Touze et al., 2004). We also found that AcrA strongly resembles MexA in three-dimensional structure, which is not surprising given their high sequence identity. However, an unsuspected and potentially significant aspect of AcrA was revealed in the four conformations of the molecule seen in the crystal. In particular, these conformations provide evidence for hinge-like conformational flexibility at the base of the α-helical hairpin domain of AcrA. The magnitude of this conformational flexibility is remarkably congruent with conformational changes implicated in opening of the OMF channel (Figure 6)(Andersen et al., 2002; Koronakis et al., 2000).
Figure 6.
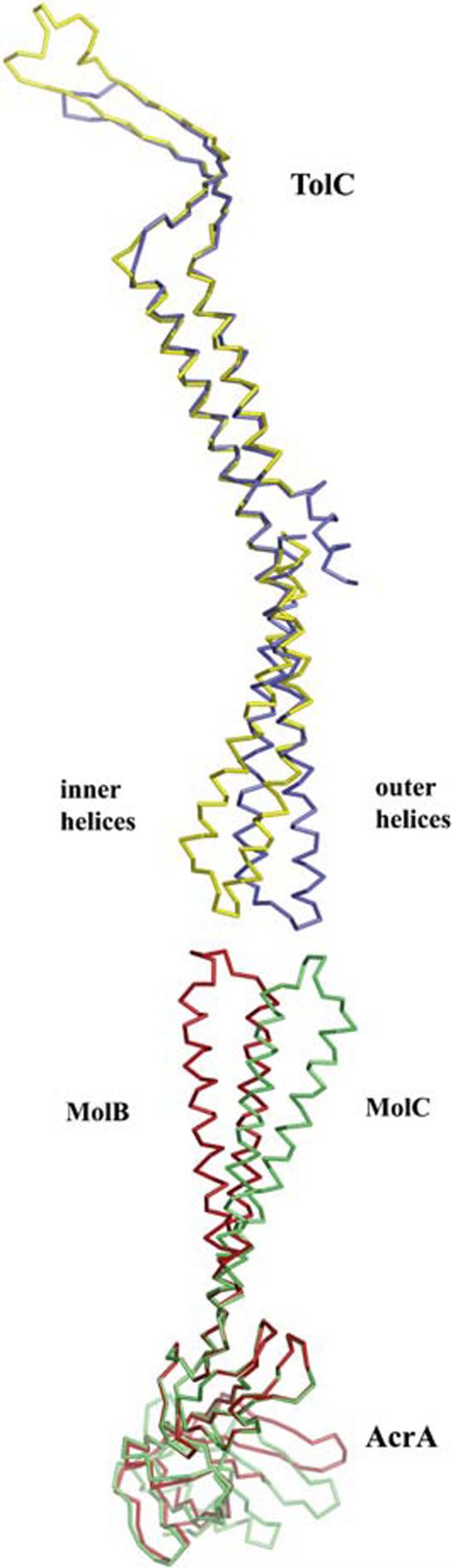
Comparison of the TolC and AcrA(45-312)-4M Coiled-Coils
The magnitude of conformational change predicted for the opening of the TolC channel coincides with the flexibility observed in the AcrA α-helical hairpin domain. The inner (yellow) and outer helices (blue) of TolC are shown superposed based on the internal structural repeat (residues 16-98 and 222-316). Below these are molecules B (red) and C (green) of AcrA(45-312)-4M, as superposed on the lipoyl domain. Direct engagement of AcrA and TolC is not modeled.
In the OMFs TolC, OprM, and VceA, the periplasmic entrance to the channel is blocked by coiled-coil helices, termed inner helices (H7 and H8) (Akama et al., 2004b; Federici et al., 2005; Higgins et al., 2004). The inner helices along with a second pair of coiled-coil helices, termed outer helices (H3 and H4), constitute the periplasmic α-helical barrel of OMFs. Opening of the channel requires movement of the inner helices from the center to the periphery of the channel. This movement is suggested to involve an iris-like untwisting of the inner helices, which have a canonical coiled-coil conformation, to resemble the noncanonical coiled-coil conformation of the outer helices (H3 and H4) (Koronakis et al., 2000). This iris-like opening model is supported by the fact that the inner and outer pairs of helices are similar in sequence, suggesting that both canonical and noncanonical coiled-coil conformations are available to them. The model is further supported by mutational data (Andersen et al., 2002).
AcrA, which has been shown to associate with both TolC and AcrB (Husain et al., 2004; Tikhonova and Zgurskaya, 2004; Touze et al., 2004; Zgurskaya and Nikaido, 2000), may therefore not only promote the formation and maintenance of the tripartite AcrA-AcrB-TolC complex, but also may function in the opening of the TolC channel. An appealing model is for the coiled-coil hairpin of AcrA to engage the coiled-coils of TolC. Interestingly, modeling of just this interaction by docking and energy minimization required a hinge-like rearrangement of the α-helical hairpin domain of AcrA (Fernandez-Recio et al., 2004). Further studies are required to determine whether conformational changes in the AcrA α-helical hairpin play an active role in the opening of the TolC channel in response to drug efflux, or a passive role in maintaining contact between AcrA and the open channel state of the TolC channel. Additional evidence for conformational flexibility in AcrA has been obtained from electron paramagnetic resonance studies (Ip et al., 2003). Despite the similarities between the conformational flexibility of AcrA seen in the crystal structure and the predicted iris-like opening of TolC, it is currently unclear how AcrA and TolC specifically associate.
While conformational flexibility is evident in AcrA, the 13 copies of MexA in the crystal are seen to have essentially the same conformation (Figure 5B) (Akama et al., 2004b; Higgins et al., 2004). However, it must be noted that MexA molecules are closely packed in the crystal into six- and seven-membered rings, and this may constrain the flexibility of the hairpin to one orientation. The unusual oligomeric state seen in the MexA crystals is not believed to be biologically relevant because the stacking of the six- and seven-membered rings is incompatible with the anchoring of MexA to the inner membrane. Furthermore, the diameter of each ring is likely to be insufficient to accommodate the inner membrane RND protein MexB.
Recent results suggest a more complicated mode of interaction between MFPs and OMFs than simple engagement of opposing coiled-coils. Most significantly, the tryptic fragment of AcrA, composed of residues 45-315, has been found to be insufficient to bind TolC (Touze et al., 2004). Similarly, the thermolytic fragment of AcrA studied here, residues 45-312, was not detected by chemical crosslinking to associate with TolC (data not shown). These results suggest a requirement for either or both the N- and C-terminal portions lacking in truncated AcrA for interaction with TolC. Because the proteolytically sensitive ∼20 residue N-terminal region of AcrA tethers the molecule to the inner membrane and is predicted to be distal from the periplasmic end of the molecule, it seems unlikely that it confers TolC association. The region more likely required for association with TolC is the proteolytically sensitive C-terminal ∼90 residues of AcrA, which are also implicated in conferring association with the inner membrane protein AcrB (Elkins and Nikaido, 2003; Touze et al., 2004). In addition to the C-terminal region, the β-barrel domain of AcrA has also been implicated in conferring TolC association. This implication comes from mutations in AcrA that suppress the drug hypersensitivity phenotype of a TolC mutant being found in the β-barrel domain (Gerken and Misra, 2004).
Whether the β-barrel and C-terminal regions of AcrA directly affect TolC association is not clear. One possibility is that these regions mediate AcrA oligomerization and therefore indirectly influence TolC association. The stoichiometry of AcrA remains uncertain, with a trimeric form found in vivo but a monomeric form in vitro (Zgurskaya and Nikaido, 1999a, 2000). Similarly, MexA is found to be monomeric in vitro (Akama et al., 2004b; Higgins et al., 2004). The monomeric forms of both AcrA and MexA lack membrane anchors and are observed to form oligomers at high concentrations used for crystallization. In both crystal structures of AcrA and MexA, conserved residues at the c and f positions of the α-helical hairpin heptad repeat along with residues in the β-barrel domain are seen to mediate similar homotypic interactions. It is possible that oligomeric contacts, which appear to be weak in the absence of a membrane anchor, may be strengthened in vivo by confinement to the two-dimensional surface of the inner membrane, association with other RND components, or both. Clearly more structural and biochemical studies are need to clear up such ambiguities and determine whether hinge-like flexibility in the helical hairpin domain of AcrA has mechanistic significance. The structure of AcrA(45-312) presented here completes the picture of the AcrA-AcrB-TolC efflux system, and serves as a guide for future studies in the elucidation of the function of MFPs and assembly of multidrug efflux pumps.
Experimental Procedures
Cloning, Expression, and Purification
Coding sequences for AcrA(26-397) and (45-312) were amplified using PCR from E. coli JM101 chromosomal DNA and ligated into the NcoI and XhoI restriction sites of pET28b (Novagen). These constructs contain a C-terminal His tag (LEHHHHHH) for purposes of protein purification. The methionine substitution mutations F223M/L224M and L287M/L288M were introduced by site-directed mutagenesis using strand overlap extension PCR (Higuchi et al., 1988).
Expression of AcrA(26-397) was induced in E. coli BL21 (DE3) using 1 mM isopropyl-β-D-thiogalactopyranoside (25°C). Bacteria were harvested by centrifugation and lysed by sonication (in 50 mM phosphate buffer [pH 8.0], 150 mM NaCl, 10 mM imidazole), and AcrA(26-397) was purified by Ni2+ chelation chromatography (Poros MC). Pooled fractions were dialyzed (50 mM Tris [pH 8.0], 150 mM NaCl), concentrated using Amicon ultrafiltration (MW cutoff 30 kDa; Millipore), and applied to a size exclusion column (Superdex 200). Purified AcrA(26-397)-His was concentrated to ∼30 mg/ml (calculated ε280 of 17,210 M-1 cm-1), dialyzed in 10 mM Tris (pH 8.0), and flash frozen at -80°C. AcrA(45-397) was expressed and purified similarly.
Selenomethionine was incorporated into a quadruple methionine mutant (F223M, L224M, L287M, and L288M) as described previously (Budisa et al., 1995) and purified as above, except for the addition of 1 mM dithiothreitol to samples following Ni2+ chelation chromatography. Purified protein was concentrated (calculated ε280 of 11,520 M-1 cm-1), dialyzed in 10 mM Tris (pH 8.0), 1 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and flash frozen at -80°C.
Proteolytic Domain Mapping
Proteolysis experiments were carried out at 37°C at a 50:1 AcrA:thermolysin mass ratio in 10 mM Tris (pH 8.0), 0.15 mM ZnSO4, 2 mM CaCl2. The final concentration of AcrA(26-397)-His was 1 mg/ml in a 100 μl reaction. Ten microliter samples were taken at varying times points, mixed with 2× SDS-PAGE sample buffer, boiled, and analyzed by 12% SDS-PAGE. Proteolysis fragments were identified using N-terminal sequencing of bands isolated by SDS-PAGE, and liquid chromatography followed by electrospray ionization mass spectrometry of the proteolysis reaction.
Crystallization and Data Collection
Crystals were grown at 4°C by the vapor diffusion method using a 1:1 mixture of AcrA(45-312)-4M and 30% 2-methyl-2,4-pentadiol, 20 mM MgCl2, 100 mM citrate (pH 5.4), 1 mM TCEP. Crystals were briefly washed in well buffer supplemented with fresh 1 mM TCEP, mounted in fiber loops, and flashed cooled in liquid N2. Inverse beam, three-wavelength oscillation data (0.5°) were collected from cryocooled crystals at beamline 19-BM (Advanced Photon Source, Argonne, IL) to 3.5Å resolution (Table 1). Data were processed using HKL2000 (Otwinowski and Minor, 1997). The crystals belong to space group C2221 with cell dimensions a = 88.7 Å, b = 100.0 Å, and c = 332.6 Å; four protein molecules are contained in the asymmetric unit (60% solvent).
Structure Determination and Refinement
Selenomethionine sites were initially located with SHELXL (Sheldrick and Schneider, 1997), with a total of 16 sites being located after refinement with Sharp (de La Fortelle and Bricogne, 1997). The molecular model was built with O (Jones et al., 1991) after solvent flattening with DM (CCP4, 1994). The initial model was refined against a 2.7 Å resolution data set (beamline 8.3.2, Advanced Light Source, Berkeley, CA) using TLS and noncrystallographic symmetry restrained refinement with REFMAC5 (Table 1) (CCP4, 1994). A random 5% of data were omitted from refinement for Rfree calculations. The model AcrA(45-312)-4M includes residues 54-299 with breaks in the main chain density for residues 230-241 in molecules A, B, and D. All residues are within allowed regions of the Ramachandran plot.
Functional Analysis and Expression of AcrA Mutants
The functional integrity of AcrA truncated at residue 312, and of double (F223M/L224M and L287M/L288M) and quadruple (F223M/L224M/L287M/L288M) methionine substitution mutants were assessed using minimum inhibitory assays, as described previously (Tikhonova and Zgurskaya, 2004). For this purpose, PCR-amplified fragments of acrA mutants were subcloned by replacement of the corresponding EcoNI-XbaI (for AcrA truncated at residue 312) or MscI-XbaI (for double and quadruple methionine substitution mutants) fragments of pAHisB plasmid (Tikhonova and Zgurskaya, 2004). AcrA mutants containing C-terminal His tags were expressed in a single operon with AcrB under control of the native acrAB promoter. Constructs were verified by DNA sequencing. Plasmids were transformed into E. coli AG100AX (ΔacrAB::kan ΔacrEF::spe) and cells were grown to midlogarithmic phase (A600 of 1.0). Luria-Bertani broth media containing 2-fold increasing concentrations of the antibiotic under investigation were inoculated with 2.5 × 104 cells per ml. Cell growth was determined after overnight incubation at 37°C. The steady-state level of expression for these constructs was analyzed by immunoblotting with an anti-AcrA polyclonal antibody, as described previously (Tikhonova and Zgurskaya, 2004).
Acknowledgments
We thank Todd Yeates for his help in the analysis of the twinned data, and the beamline staffs at Advanced Light Source and the Structural Biology Center at Advanced Photon Source for their assistance in data collection. This work was supported by NIH T32 DK007233 (J.M.), NIH R03 AI064312 (J.M. and P.G.), NIH R01 AI052293 (H.I.Z.), and the W.M. Keck Distinguished Young Scholars in Medicine Award (P.G.).
Footnotes
Accession Numbers
Coordinates and structure factors have been deposited in the Protein Data Bank under ID code 2F1M.
References
- Aires JR, Nikaido H. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli. J. Bacteriol. 2005;187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akama H, Kanemaki M, Yoshimura M, Tsukihara T, Kashiwagi T, Yoneyama H, Narita S, Nakagawa A, Nakae T. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: dual modes of membrane anchoring and occluded cavity end. J. Biol. Chem. 2004;279(a):52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- Akama H, Matsuura T, Kashiwagi S, Yoneyama H, Narita S, Tsukihara T, Nakagawa A, Nakae T. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J. Biol. Chem. 2004;279(b):25939–25942. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- Andersen C, Koronakis E, Bokma E, Eswaran J, Humphreys D, Hughes C, Koronakis V. Transition to the open state of the TolC periplasmic tunnel entrance. Proc. Natl. Acad. Sci. USA. 2002;99:11103–11108. doi: 10.1073/pnas.162039399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur. J. Biochem. 1995;230:788–796. doi: 10.1111/j.1432-1033.1995.tb20622.x. [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- de La Fortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.
- Dinh T, Paulsen IT, Saier MH., Jr. A family of extracytoplasmic proteins that allow transport of large molecules across the outer membranes of Gram-negative bacteria. J. Bacteriol. 1994;176:3825–3831. doi: 10.1128/jb.176.13.3825-3831.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins CA, Nikaido H. Chimeric analysis of AcrA function reveals the importance of its C-terminal domain in its interaction with the AcrB multidrug efflux pump. J. Bacteriol. 2003;185:5349–5356. doi: 10.1128/JB.185.18.5349-5356.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federici L, Du D, Walas F, Matsumura H, Fernandez-Recio J, McKeegan KS, Borges-Walmsley MI, Luisi BF, Walmsley AR. The crystal structure of the outer membrane protein VceC from the bacterial pathogen Vibrio cholerae at 1.8Å resolution. J. Biol. Chem. 2005;280:15307–15314. doi: 10.1074/jbc.M500401200. [DOI] [PubMed] [Google Scholar]
- Fernandez-Recio J, Walas F, Federici L, Venkatesh Pratap J, Bavro VN, Miguel RN, Mizuguchi K, Luisi B. A model of a transmembrane drug-efflux pump from Gram-negative bacteria. FEBS Lett. 2004;578:5–9. doi: 10.1016/j.febslet.2004.10.097. [DOI] [PubMed] [Google Scholar]
- Gerken H, Misra R. Genetic evidence for functional interactions between TolC and AcrA proteins of a major antibiotic efflux pump of Escherichia coli. Mol. Microbiol. 2004;54:620–631. doi: 10.1111/j.1365-2958.2004.04301.x. [DOI] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc. Natl. Acad. Sci. USA. 2004;101:9994–9999. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirakata Y, Srikumar R, Poole K, Gotoh N, Suematsu T, Kohno S, Kamihira S, Hancock RE, Speert DP. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J. Exp. Med. 2002;196:109–118. doi: 10.1084/jem.20020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain F, Humbard M, Misra R. Interaction between the TolC and AcrA proteins of a multidrug efflux system of Escherichia coli. J. Bacteriol. 2004;186:8533–8536. doi: 10.1128/JB.186.24.8533-8536.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip H, Stratton K, Zgurskaya H, Liu J. pH-induced conformational changes of AcrA, the membrane fusion protein of Escherichia coli multidrug efflux system. J. Biol. Chem. 2003;278:50474–50482. doi: 10.1074/jbc.M305152200. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Church GM. Alignment and structure prediction of divergent protein families: periplasmic and outer membrane proteins of bacterial efflux pumps. J. Mol. Biol. 1999;287:695–715. doi: 10.1006/jmbi.1999.2630. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta. Crystallogr. A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol. Microbiol. 1995;16:45–55. doi: 10.1111/j.1365-2958.1995.tb02390.x. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–593. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Padilla JE, Yeates TO. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1124–1130. doi: 10.1107/s0907444903007947. [DOI] [PubMed] [Google Scholar]
- Putman M, van Veen HW, Konings WN. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000;64:672–693. doi: 10.1128/mmbr.64.4.672-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier MH, Jr., Tam R, Reizer A, Reizer J. Two novel families of bacterial membrane proteins concerned with nodulation, cell division and transport. Mol. Microbiol. 1994;11:841–847. doi: 10.1111/j.1365-2958.1994.tb00362.x. [DOI] [PubMed] [Google Scholar]
- Seo J, Cohen C. Pitch diversity in α-helical coiled coils. Proteins. 1993;15:223–234. doi: 10.1002/prot.340150302. [DOI] [PubMed] [Google Scholar]
- Sheldrick G, Schneider T. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- Tamura N, Murakami S, Oyama Y, Ishiguro M, Yamaguchi A. Direct interaction of multidrug efflux transporter AcrB and outer membrane channel TolC detected via site-directed disulfide cross-linking. Biochemistry. 2005;44:11115–11121. doi: 10.1021/bi050452u. [DOI] [PubMed] [Google Scholar]
- Thanassi DG, Suh GS, Nikaido H. Role of outer membrane barrier in efflux-mediated tetracycline resistance of Escherichia coli. J. Bacteriol. 1995;177:998–1007. doi: 10.1128/jb.177.4.998-1007.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova EB, Zgurskaya HI. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J. Biol. Chem. 2004;279:32116–32124. doi: 10.1074/jbc.M402230200. [DOI] [PubMed] [Google Scholar]
- Tikhonova EB, Wang Q, Zgurskaya HI. Chimeric analysis of the multicomponent multidrug efflux transporters from Gram-negative bacteria. J. Bacteriol. 2002;184:6499–6507. doi: 10.1128/JB.184.23.6499-6507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touze T, Eswaran J, Bokma E, Koronakis E, Hughes C, Koronakis V. Interactions underlying assembly of the Escherichia coli AcrAB-TolC multidrug efflux system. Mol. Microbiol. 2004;53:697–706. doi: 10.1111/j.1365-2958.2004.04158.x. [DOI] [PubMed] [Google Scholar]
- Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–781. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- Walshaw J, Woolfson DN. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 2001;307:1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- Yoneyama H, Maseda H, Kamiguchi H, Nakae T. Function of the membrane fusion protein, MexA, of the MexA, BOprM efflux pump in Pseudomonas aeruginosa without an anchoring membrane. J. Biol. Chem. 2000;275:4628–4634. doi: 10.1074/jbc.275.7.4628. [DOI] [PubMed] [Google Scholar]
- Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE., Jr. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–980. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. AcrA is a highly asymmetric protein capable of spanning the periplasm. J. Mol. Biol. 1999;285(a):409–420. doi: 10.1006/jmbi.1998.2313. [DOI] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. Bypassing the periplasm: reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proc. Natl. Acad. Sci. USA. 1999;96(b):7190–7195. doi: 10.1073/pnas.96.13.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, Nikaido H. Cross-linked complex between oligomeric periplasmic lipoprotein AcrA and the inner-membrane-associated multidrug efflux pump AcrB from Escherichia coli. J. Bacteriol. 2000;182:4264–4267. doi: 10.1128/jb.182.15.4264-4267.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]