

Abstract
To investigate whether TLR agonists can replace mycobacteria in adjuvant to induce EAE in Lewis rats, we immunized rats with MBP peptide (MBP(68-86)) in IFA, supplemented with TLR agonists. Rats immunized with MBP(68-86) plus CpG-ODN or LPS in IFA did not develop EAE. In contrast, rats immunized with MBP(68-86) plus CpG-ODN and LPS in IFA developed clinical EAE. Spleen cells proliferated and secreted IFN-γ in response to MBP(68-86), and secreted IL-6 and IL-12p40 in response to CpG-ODN and LPS. However, rats immunized with MBP(68-86) plus CpG-ODN and PolyI:C, a TLR3 agonist, did not develop EAE. We conclude that selected combinations of TLR agonists can facilitate the induction of EAE by MBP peptide via the innate immune system.
Keywords: Autoimmunity, EAE, innate immunity, TLR agonists
1. Introduction
Experimental autoimmune encephalomyelitis (EAE) in the Lewis rat is an acute T cell-mediated inflammatory disease that can be readily induced with a single injection of myelin basic protein (MBP) or MBP-derived peptides administered in complete Freund's adjuvant (CFA) (reviewed in Swanborg, 2001). In contrast, injection of MBP or MBP peptides in incomplete Freund's adjuvant fails to elicit EAE in Lewis rats. Rather, this regimen induces tolerance with respect to EAE, which is due, at least in part, to the generation of T suppressor/regulatory cells that prevent the pathogenic T cells from eliciting paralysis (Swierkosz and Swanborg, 1975). These regulatory cells secrete TGF-β, which downregulates the production of proinflammatory cytokines (e.g., IFN-γ) by the encephalitogenic T cells (Karpus and Swanborg, 1991; Miller et al., 1992).
Until recently, the use of CFA and, in some models, other ancillary adjuvants were a requirement for the induction of EAE in rodents (reviewed in Swanborg, 1995). CFA was developed in the 1940's and led to pioneering studies of EAE (Kabat et al., 1947; Morgan, 1947), and other autoimmune diseases. However, the role of adjuvants in EAE was unclear until recently, when Segal et al. (1997) discovered that microbial products including bacterial lipopolysaccharide (LPS) and cytosine-guanine dinucleotide-containing oligodeoxynucleotides (CpG-ODN) could facilitate the induction of EAE by promoting IL-12 production. CpG-ODN is a Toll-like receptor (TLR)-9 agonist that activates APCs through innate immunity to secrete IL-12 and IL-6, which in turn activates the adaptive immune system (Hemmi et al., 2000; Janeway, 2001). LPS is a TLR-4 agonist that activates NF-κB in the nucleus (Medzhitov et al., 1997).
Recently, Ichikawa et al. (2002) reported that unresponsive T cells from SJL mice tolerized to an encephalitogenic peptide fragment of proteolipid protein are converted into pathogenic effector cells that proliferate and transfer EAE when activated in the presence of CpG-ODN. We confirmed their in vitro findings and showed that Lewis rats tolerized against the major dominant self MBP epitope from rat MBP (MBP(68-86)) harbor potentially encephalitogenic T cells that can be activated in vitro to proliferate to the tolerizing peptide by exposure to CpG-ODN (Conant and Swanborg, 2004). In the present report, we employ the peptide-IFA tolerance protocol to demonstrate that selected TLR agonists can replace the mycobacterial component of CFA and induce EAE in Lewis rats.
2. Materials and Methods
2.1. Peptide synthesis
The major encephalitogenic epitope from rat (self) MBP was synthesized on an Applied Biosystems Synergy model 432A peptide synthesizer (PerkinElmer, Foster City, CA) as previously described (Conant and Swanborg, 2004). The sequence of the peptide was YGSLPQKSQRTQDENPV. This peptide was designated MBP(68-86).
2.2. Animals and immunization
Female Lewis rats were immunized s.c. with MBP(68-86) in either incomplete Freund's adjuvant (IFA) or complete Freund's adjuvant (CFA), as previously described (Swanborg and Stepaniak, 1996; Conant and Swanborg, 2004). In some experiments, the MBP(68-86)-IFA emulsion was supplemented with CpG oligonucleotide (CpG-ODN, ATAATCGACGTCAAGCAAG, synthesized by Qiagen, Santa Clarita, CA), E. coli 0111:B4 lipopolysaccharide (LPS, Sigma, St. Louis, MO), or Polyinosinic-Polycytidylic acid (PolyI:C, Amersham, Piscataway, NJ). A control oligonucleotide for CpG-ODN (CO, sequence ATAATAGAGCTTACAAGCAAG, Qiagen) was used in in vitro experiments.
The rats were observed for clinical signs of EAE, which were graded as 0 (no disease), 1 (loss of tail tonicity), 2 (hind limb weakness), or 3 (hind limb paralysis). The study had IACUC approval: Protocol # A-08-06-06.
2.3. Proliferation assays
Splenic T cells were purified by adherence and passage through T cell columns, and cultured with irradiated (2000 rad) syngeneic thymocytes as APCs and peptide with or without CpG-ODN, LPS or CO in triplicate for 96 hr, and pulsed with 0.5 μCi 3H-thymidine 18 hr before harvesting, as previously reported (Swanborg and Stepaniak, 1996). Stimulation indices (SI) were calculated as cpm with peptide/background cpm of T cells and APCs without peptide.
2.4. Cytokine analysis
Spleen cell (SpC) culture supernatants (48hr) were evaluated for IL-6, IL-12, and IFN-γ using rat-specific ELISA kits (Biosource, Camarillo, CA), according to the manufacturer's instructions.
Western blots were run as previously described (Trivedi et al. 2005), except that SpC culture supernatants rather than cell lysates were analyzed. Briefly, 40 μl of culture supernatant in native sample buffer (Bio-Rad) was electrophoresed in 10% SDS-PAGE gel for 1 hr at 200 V then transferred to PVDF membranes (Bio-Rad). Non-specific binding sites were blocked with 5% nonfat dry milk in Tris-buffered saline, pH 7.5 overnight at 4 C. After washing, biotinylated anti-IL-12p40 + p70 (Biosource) in blocking buffer was added at 1 μg/ml for 1 hr at room temperature. The membrane was washed and streptavidin-alkaline phosphatase (1 μg/ml) was added for 1 hr at room temperature. After washing, BCIP/NBT alkaline phosphatase substrate (Sigma) was added for 30 minutes at room temperature in the dark. The reaction was stopped with distilled water and the blot was photographed.
2.5. RNA isolation
SpC from rats with EAE induced by MBP(68-86) + CpG + LPS in IFA were cultured for 48 hr with MBP(68-86), CpG and/or LPS. RNA was isolated using RNeasy Mini Kits (Qiagen) and treated with RNase-free DNase (Promega, Madison, WI), according to the manufacturers' directions. Purity was determined by comparing OD260/OD280 using an Ultraspec III spectrophotometer (Amersham). Single-stranded cDNA was synthesized using the iScript™ cDNA Synthesis Kit (Bio-Rad, Hercules, CA), according to the manufacturer's instructions.
2.6. Real-Time PCR
The relative amount of mRNA was determined using the SYBR Green method using the Bio-Rad MyiQ Single Color PCR Detection System. All primer sets had an optimal annealing temperature of 57°C. Quantitative RT-PCR was performed in duplicate in a 25 μl reaction volume consisting of 2X SYBR Green Supermix (Bio-Rad), 500 nM of each primer and 2 μl of cDNA. Amplification conditions were denaturation for 3.5 min at 95°C, 35 cycles of denaturation for 10 seconds at 95°C and annealing/extension for 50 seconds at 57°C. A melt curve was performed for 1 min at 95°C, 1 min at 55°C, and 80 cycles for 10 sec at 55°C. The relative fold change (2−ΔΔCT) of the target transcript was determined by comparing it to the reference gene transcript, β-actin using the formula:
We analyzed IL-12p40, IL-12p19 and IL-12p35 by Real-Time PCR, using primers described by Wefer et al. (2004)(synthesized by Qiagen).
2.7. Statistical analysis
Results are presented as mean ± SD. Statistical significance was calculated using Student's t test.
3. Results
3.1. Studies of rats immunized with MBP(68-86) in IFA
Lewis rats do not develop EAE when immunized with MBP or MBP peptides in IFA. Rather, this treatment protects them against EAE, due in part to the generation of suppressor/regulatory T cells (Swierkosz and Swanborg, 1975). Recently we reported that MBP(68-86) + IFA-immunized Lewis rats harbor encephalitogenic T cells. These could be activated in vitro by culturing T cells in the presence of MBP(68-86) and CpG-ODN; T cells cultured only with MBP(68-86) were not activated (Conant and Swanborg, 2004). Thus, unresponsive Lewis rats harbor potentially encephalitogenic T cells without developing overt signs of EAE. To determine whether CpG-ODN could elicit EAE, we conducted a pilot experiment in which four Lewis rats were immunized with MBP(68-86) in IFA supplemented with 100 μg CpG-ODN. A second group received peptide plus 300 μg CpG-ODN. As shown in Table 1, these rats did not develop EAE. In confirmation of our earlier finding (Conant and Swanborg, 2004), T cells from these rats proliferated poorly when cultured with MBP(68-86) alone (SI = 5.2), but responded vigorously when cultured with MBP(68-86) + CpG-ODN (SI = 43, Fig. 1). Cells cultured with CpG-ODN alone also proliferated, but to a lesser extent than those also cultured with MBP(68-86), which presumably reflects polyclonal stimulation of T cells of irrelevant specificity in the bulk T cell cultures by cytokines produced by APCs after stimulation with CpG-ODN. Supernatants of SpC cultures activated in the presence of MBP(68-86) contained detectable levels of IFN-γ, whereas CpG-ODN alone did not elicit secretion of this cytokine (Fig. 2). In contrast, secretion of significant quantities of IL-6 and IL-12 was associated with the presence of CpG-ODN, not MBP(68-86) (Fig. 2), indicating that the APCs secrete IL-6 and IL-12, whereas the T cells produce IFN-γ. Responses to other TLR agonists were not evaluated in this preliminary experiment.
Table I.
Induction of EAE in Lewis rats with rat68-86 plus TLR agonists
| Immunization with 100 μg rat68-86 in IFA plus | EAE incidence | EAE severity |
|---|---|---|
| 100 μg CpG-ODN | 0/4 | 0.0 |
| 300 μg CpG-ODN | 0/4 | 0.0 |
| 50 μg LPS | 0/4 | 0.0 |
| 100 μg CpG-ODN + 50 μg LPS | 11/14 | 2.0a |
| 100 μg CpG-ODN + 50 μg PolyI:C | 0/4 | 0.0 |
| 50 μg rat68-86 + CFAb | 6/6 | 2.3 |
| IFA | 0/4 | 0.0 |
Mean severity for sick rats (max = 3.0).
Complete Freund's adjuvant.
Fig. 1.
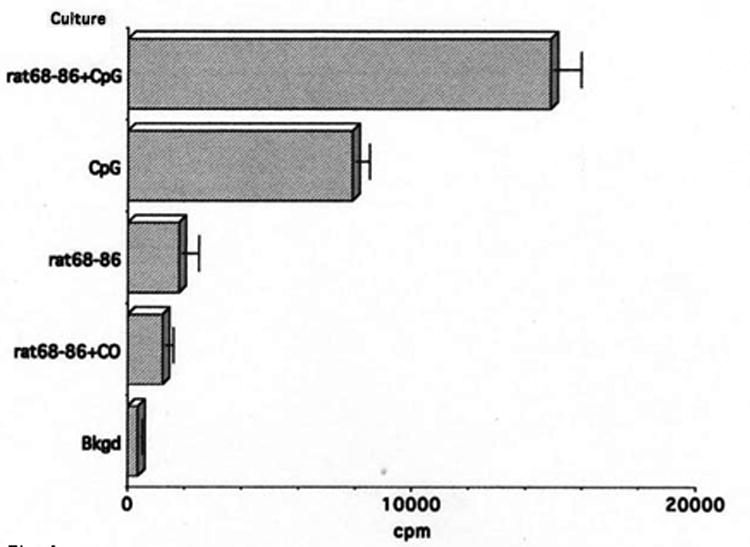
Proliferative response of T cells from MBP(68-86) + IFA tolerized Lewis rats to MBP(68-86) + CpG-ODN, MBP(68-86) + control oligonucleotide (CO), and to CpG-ODN or MBP(68-86) alone. The T cells respond weakly to MBP(68-86) (SI = 5.2), but proliferate vigorously if CpG-ODN is added to wells with the peptide (SI = 43). The results represent mean cpm ± SD of triplicate wells.
Fig. 2.
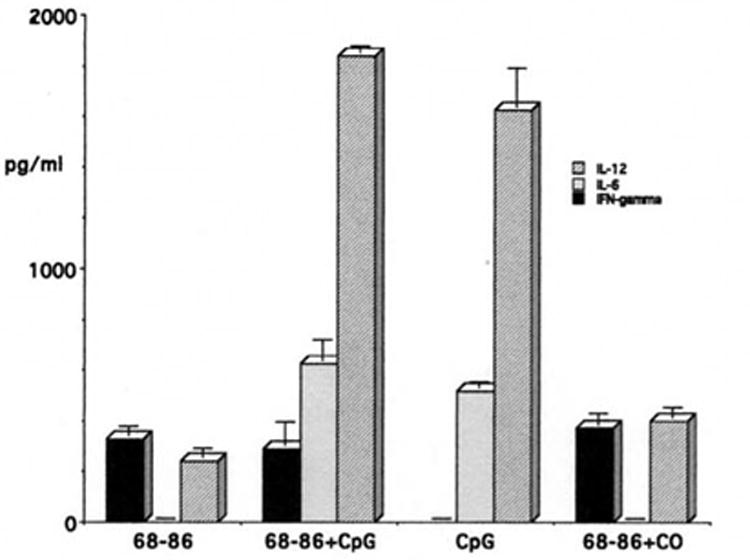
Cytokine production by SpC from MBP(68-86) tolerized Lewis rats after culture for 48 hr. IFN-γ was secreted in cultures containing MBP(68-86), (327 ± 36 pg/ml), MBP(68-86) + CpG-ODN or control oligonucleotide (CO) (290 ± 90 and 370 ± 45 pg/ml, respectively), but not in cultures containing only CpG-ODN. In contrast high levels of IL-12 were secreted in response to MBP(68-86) + CpG-ODN and CpG-ODN alone (1840 ± 25 and 1625 ± 150 pg/ml, respectively), but not to MBP(68-86) + CO or MBP(68-86) alone. IL-6 was not secreted in response to MBP(68-86) alone or MBP(68-86) + CO, but was produced in response to MBP(68-86) + CpG-ODN or CpG-ODN alone (625 ± 78 and 515 ± 22 pg/ml respectively). Results represent mean pg/ml ± SD of duplicate ELISA samples.
3.2. Single TLR agonists do not replace CFA to induce EAE
To determine whether TLR agonists can replace the mycobacterial component of Freund's adjuvant and elicit clinical manifestations of EAE in Lewis rats, we conducted a series of experiments in which either CpG-ODN, or LPS was added to emulsions of MBP(68-86) and incomplete Freund's adjuvant, which were then injected into Lewis rats. As shown in Table 1, neither CpG-ODN at 100 or 300 μg, nor LPS at 50 μg was effective at inducing EAE when added to MBP(68-86) + IFA emulsions. Similar to results obtained with T cells from rats immunized with MBP(68-86) in IFA (Fig. 1), T cells from rats that received an emulsion supplemented with CpG-ODN or LPS proliferated in vitro when stimulated with MBP(68-86) plus CpG-ODN or LPS, but not MBP(68-86) alone (data not shown).
3.3. Induction of EAE with a selected combination of TLR agonists
Based on a recent report that selected TLR agonist combinations function synergistically to trigger Th1 T cell responses (Napolitani et al., 2005), we investigated whether a similar approach would be effective with respect to inducing EAE in Lewis rats. Accordingly, we immunized Lewis rats with MBP(68-86)-IFA emulsions supplemented with 100 μg CpG-ODN plus 50 μg LPS. The results of three experiments are summarized in Table 1. Eleven of 14 rats (79%) immunized with this emulsion developed clinical EAE, compared with 6 of 6 rats that received MBP(68-86) in CFA. The mean severity for symptomatic rats in these two groups was comparable (Table 1). Thus, a combination of TLR4 and TLR9 agonists have adjuvant activity necessary for EAE induction.
3.4. In vitro responses of lymphoid cells from immunized rats
In vitro proliferative responses were performed to determine whether T cells responded to MBP(68-86) and/or TLR agonists. Results of one representative experiment out of five are presented in Fig. 3. As shown, T cells from rats that developed EAE following immunization with MBP(68-86)-IFA emulsions supplemented with 100 μg CpG-ODN plus 50 μg LPS proliferated in vitro in response to MBP(68-86) (SI = 28), although responses to MBP(68-86) plus the TLR agonists CpG-ODN and LPS were more vigorous (SI = 45, Fig. 3). Stimulation indices were comparable in cultures stimulated with MBP(68-86) plus one or both TLR agonists (Fig. 3) despite the fact that both CpG-ODN and LPS were required to elicit clinical EAE (Table 1). T cells from the rats that did not develop EAE proliferated in similar fashion to MBP(68-86) indicating that they had been immunized to the peptide. It is not clear why they did not exhibit clinical signs of disease.
Fig. 3.
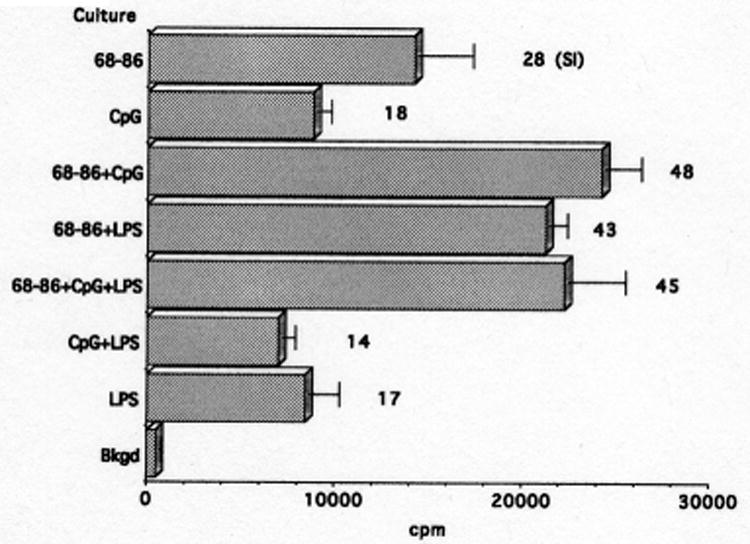
Proliferation of T cells from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS. A significant proliferative response is induced by MBP(68-86) alone (SI = 28).
IFN-γ was secreted by SpC following activation with MBP(68-86) regardless whether TLR agonist was also added to the cultures (Fig. 4 top panel). As expected, TLR agonists alone did not elicit the production of IFN-γ, since these act primarily on APCs. In support of this, IL-12 and IL-6 were secreted in SpC cultures containing CpG-ODN, and/or LPS, but not in cultures containing only MBP(68-86) (Fig. 4 middle and bottom panels, respectively). Each experiment was repeated 3–4 times with similar results.
Fig. 4.
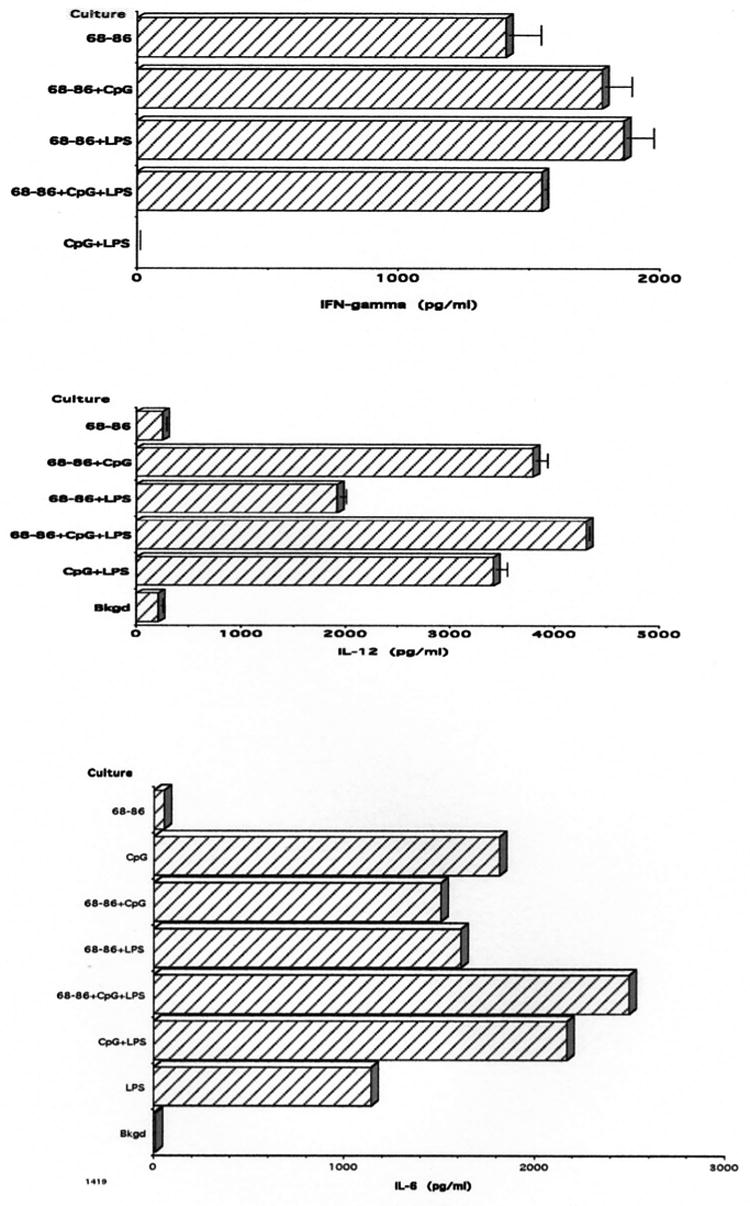
Top panel: IFN-γ production by SpC from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, after culture for 48 hr, as measured by ELISA. IFN-γ is secreted only in cultures containing MBP(68-86) in the presence or absence of TLR agonists. No IFN-γ is secreted in response to CpG-ODN and LPS without MBP(68-86). No IFN- was detected in SpC cultured in medium alone (background, not shown).
Center panel: IL-12 production by SpC from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, after culture for 48 hr, as measured by ELISA. IL-12 is secreted in response to CpG-ODN and LPS, but not to MBP(68-86) alone.
Bottom panel: IL-6 production by SpC from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, after culture for 48 hr, as measured by ELISA. IL-6 is secreted in response to CpG-ODN and LPS, but not to MBP(68-86) alone.
3.5. CpG-ODN plus PolyI:C does not facilitate EAE
To evaluate whether another combination of TLR agonists could elicit EAE, a group of four rats was immunized with MBP(68-86) in IFA supplemented with 100 μg CpG-ODN plus 50 μg PolyI:C. As shown in Table 1, these rats did not develop EAE. T cells from these rats proliferated to MBP(68-86) and the response was enhanced in the presence of one or both TLR agonists (Fig. 5). However, in contrast with the finding that CpG-ODN stimulated IL-12 production, only minimal levels of this cytokine were produced by PolyI:C (Fig. 6). PolyI:C did not induce IL-6 or IFN-γ (data not shown)
Fig. 5.
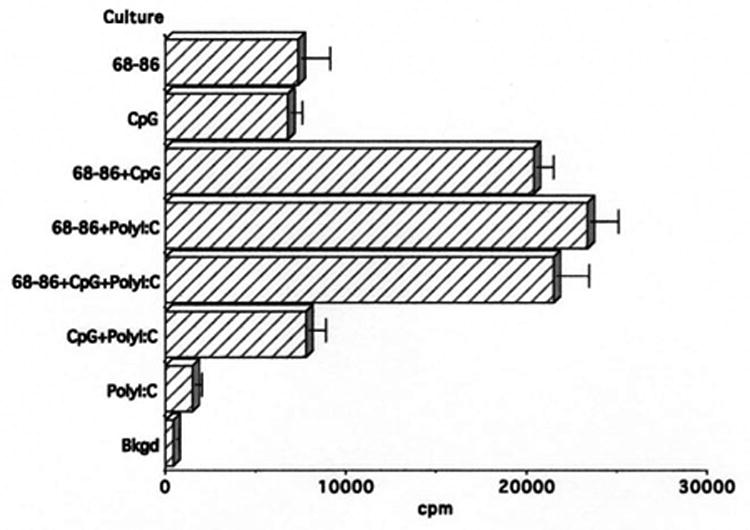
Proliferation of T cells from Lewis rats immunized with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg PolyI:C. PolyI:C enhances the proliferative response to MBP(68-86) despite failure to induce EAE.
Fig. 6.
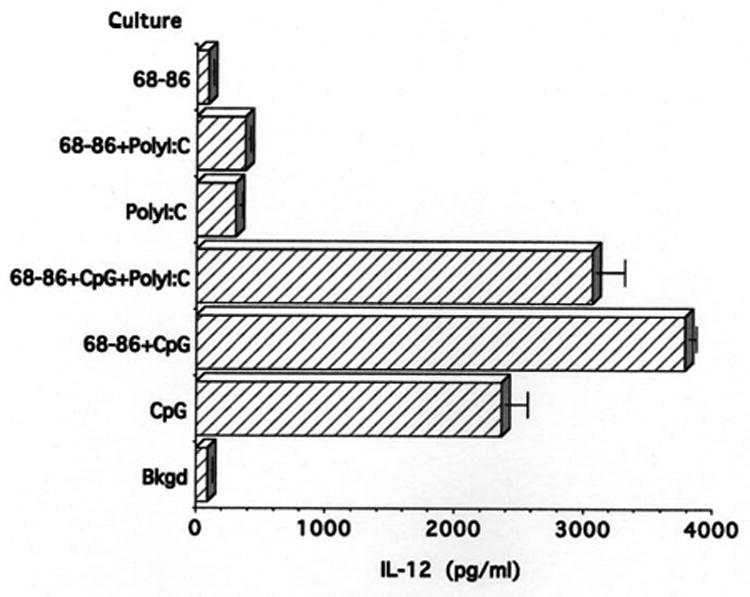
IL-12 production by SpC from Lewis rats immunized with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg PolyI:C, after culture for 48 hr, as measured by ELISA. Neither PolyI:C alone or in the presence of MBP(68-86) induces significant amounts of IL-12.
IL-12 is a p70 heterodimer consisting of a p35 and a p40 chain. IL-12p40 is shared with IL-23, which is a p40-p19 heterodimer that has been implicated in the activation of pathogenic T cells in EAE (Gran et al., 2002; Langrish et al., 2005). The IL-12 ELISA we employed detects IL-12p40 and IL-12p70. To gain insight into which IL-12 components were secreted by the SpC obtained from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, we analyzed the SpC supernatants by immunoblot using the antibodies specific for p40 and p70. As shown in Fig. 7, IL-12p40 was the only protein identified in the supernatants that contained the TLR agonists CpG-ODN and/or LPS, with or without MBP(68-86). IL-12p70 was not detected. No IL-12 was detected in the supernatant cultured with MBP(68-86) alone.
Fig. 7.
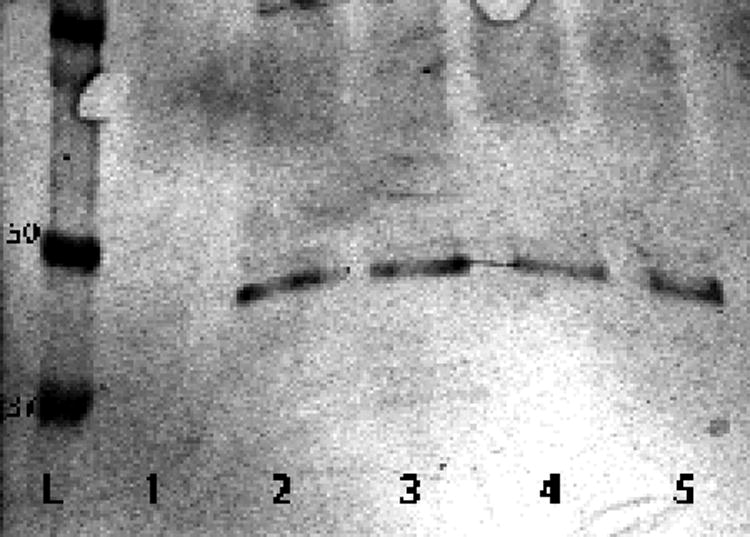
Immunoblot analysis of SpC supernatants from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, after culture for 48 hr. L, ladder showing 37, 50 and 100 kDa bands. Lanes 1-5, SpC supernatants cultured for 48 hr with: 1, MBP(68-86); 2, MBP(68-86) + CpG-ODN + LPS; 3, MBP(68-86) + CpG-ODN; 4, MBP(68-86) + LPS; 5, CpG-ODN + LPS. IL-12p40 is present only in bands containing supernatants incubated with CpG-ODN and/or LPS (bands 2-5).
3.6. IL-12 Real-Time PCR
Since antibodies specific for rat IL-12p19 are not available, we employed Real-Time PCR to ascertain whether IL-23 was implicated in the activation of encephalitogenic effector cells. We synthesized primers specific for IL-12p40, which is common to both cytokines, for IL-12p19, which is specific for IL-23, and for IL-12p35, which is specific for IL-12, as described by Wefer et al. (2004). We prepared cDNA from SpC obtained from Lewis rats that developed EAE following immunization with MBP(68-86)-IFA emulsions supplemented with 100 μg CpG-ODN plus 50 μg LPS, and activated in vitro with MBP(68-86) with/without CpG-ODN and/or LPS. These were analyzed by Real-Time PCR.
As shown in Fig. 8, only IL-12p40 mRNA was detected in significant amounts (fold change relative to β-actin), and this was from the cDNA of SpC activated with MBP(68-86) plus LPS and MBP(68-86) plus CpG-ODN and LPS. Neither IL-12p19 nor IL-12p35 transcripts were detected in significant amounts.
Fig. 8.
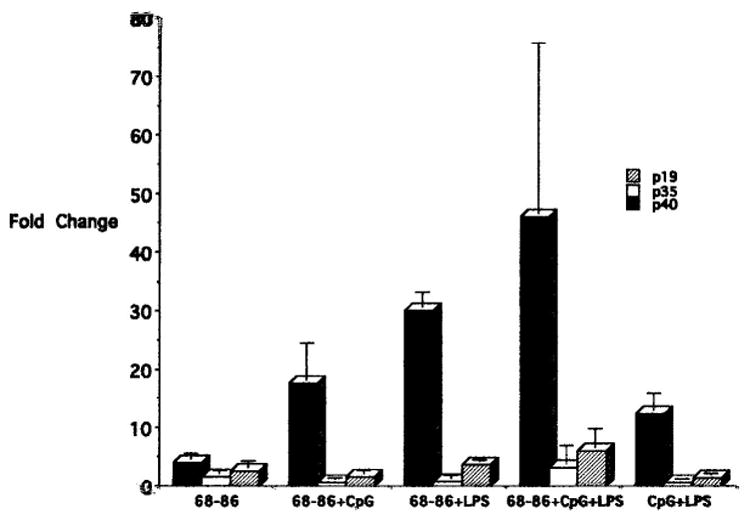
Real time PCR analysis of cDNA from SpC from Lewis rats with EAE induced by immunization with 100 μg MBP(68-86) + 100 μg CpG-ODN + 50 μg LPS, after culture for 48 hr. The results are presented as mean fold change relative to β-actin ± SD, after 35 cycles of PCR. A significant increase in IL-12p40 mRNA is present in SpC cultured with (MBP)68-86 plus LPS and MBP(68-86) plus CpG-ODN and/or LPS. Neither IL-12p19 nor IL-12p35 transcripts were produced in abundance.
Thus, our findings suggest that the TLR4 and 9 agonists, but not the TLR3 agonist, induce APCs to produce IL-12p40.
4. Discussion
The major finding in the present report is that the addition of CpG-ODN and LPS to emulsions consisting of MBP(68-86) in incomplete Freund's adjuvant renders the emulsions encephalitogenic for Lewis rats. Thus, the two TLR agonists can replace mycobacteria, suggesting that M. tuberculosis in CFA functions to activate innate immunity, which in turn, initiates an adaptive immune response to the antigen in the adjuvant emulsion. Neither CpG-ODN nor LPS alone initiated EAE when included in the MBP(68-86) + IFA emulsion, suggesting that the combination of two TLR agonists probably functioned through the activation of different cytokines required for the activation of encephalitogenic T cells. Napolitani et al. (2005) reported that the combination of CpG-ODN and LPS or PolyI:C strongly enhanced the production of IL-12 and IL-23 by mouse dendritic cells, and resulted in Th1 cell polarization. CpG-ODN, LPS and PolyI:C are agonists for TLR-9 (Hemmi et al., 2000), TLR-4 (Medzhitov et al., 1997) and TLR-3 (Alexopoulou et al., 2001), respectively. Our present data differ from those of Napolitani et al. (2005) in that synergistic interactions between TLR-4 and TLR-9, but not between TLR-3 and TLR-9, promote the development of EAE in Lewis rats (Table 1).
In mice it has been shown that CpG-ODN reverses tolerance of PLP-tolerant lymph node cells (Ichikawa et al., 2002; Waldner et al., 2004). It has also been reported that LPS induces relapses of EAE in MBP peptide 85–99-imunized mice, which usually develop monophasic disease (Nogai et al., 2006). Our attempts to induce relapses with LPS in Lewis rats that had recovered from EAE induced with MBP(68-86) in CFA were unsuccessful (results not shown).
Hansen et al. recently reported that multiple TLR agonists could serve as adjuvants to induce EAE in C57BL/6 mice (Hansen et al., 2006). They found that LPS or mycobacteria (which activates TLR-1, 2 and 4) promoted induction of moderate to severe EAE when added to emulsions of MOG peptide in IFA. Zymosan (which is a TLR-2 agonist) induced mild disease, whereas PolyI:C was inefficient as an adjuvant. In their study, the mice also received pertussis toxin as an ancillary adjuvant. In the present report, no additional adjuvants were required for induction of EAE in Lewis rats.
It has recently been reported that PolyI:C suppresses relapses of EAE in SJL/J mice (Touil et al. 2006). This TLR3 agonist induced IFN-β, which regulates EAE and has therapeutic efficacy in some MS patients. This might provide an explanation for the failure of PolyI:C to promote EAE in the present study and in the report of Hansen et al. (2006). Thus, it is conceivable that not all TLR agonists promote innate immune responses.
We also observed that CpG-ODN and LPS induced the secretion of IL-6 and IL-12p40 from SpC of tolerized rats (Fig. 2) and rats with EAE induced by MBP(68-86) emulsions containing these TLR agonists (Figs. 5 and 6), whereas SpC from rats with EAE secreted IFN-γ when cultured with MBP(68-86) (Fig. 4). This is consistent with the findings of Segal et al., who first reported that CpG-ODN induced the production of IL-12p40 by MBP-reactive mouse lymph node cells (Segal et al., 1997). The IL-12p40 subunit is common to both IL-12 and IL-23, and it was subsequently determined that IL-23-dependent T cells are essential for the induction of EAE (Gran et al., 2002), (Langrish et al., 2005). These IL-23-dependent cells have been distinguished from IL-12-producing cells by the production of IL-6, IL-17, and several other proinflammatory cytokines (Langrish et al., 2005). Our IL-12 ELISA was specific for both IL-12p40 and IL-12p70, so it was necessary to resort to immunoblots and Real-Time PCR to ascertain which subunits the TLR agonist-stimulated SpC produced. We were only able to detect IL-12p40 mRNA by Real-time PCR, and only IL-12p40 protein by Western blot analysis. It is unclear at present why we only could detect the p40 subunit because it is well established that IL-12p40 tends to form homodimers which function as IL-12 antagonists and inhibit IL-12-induced T cell responses (Ling et al., 1995). However, a recent report implicates the induction of IL-12p40 by dendritic cells in the maturation of dendritic cells to a T cell-activating phenotype in mice exposed to Mycobacterium tuberculosis (Khader et al., 2006). This response is associated with the induction of chemokines by the dendritic cells, and does not require either IL-23 or IL-12p70, suggesting that IL-12p40 may play a unique role in the initiation of the adaptive immune response. Moreover, it was recently reported that Kilham rat virus infection of BBDR/Wor rats induces autoimmune diabetes and IL-12p40 production by spleen cells, presumably by signaling through TLR9 (Zipris et al., 2007). Thus, although IL-12p40 homodimers antagonize T cell proliferation (Ling et al., 1995), the possibility that IL-12p40 may also be implicated in the induction of immune responses remains to be established.
It is of interest that PolyI:C failed to induce the proinflammatory cytokines IL-12p40 or IL-6 in SpC, which may account in part for the failure of this TLR agonist to facilitate EAE.
In conclusion, we have shown that a combination of CpG-ODN and LPS, but not of CpG-ODN and PolyI:C, can replace the requirement for mycobacteria in the adjuvant to facilitate induction of EAE in Lewis rats. These TLR agonists activate SpCs to secrete IL-6 and IL-12, and Real-Time PCR analyses suggest that only the IL-12p40 chain is produced. These findings imply a role for selected TLR agonists which activate the innate immune system to facilitate the initiation of autoimmune disease. This gives credence to the hypothesis that infectious agents, which are known to stimulate innate responses, may be involved in triggering human autoimmune diseases.
Abbreviations
- EAE
experimental autoimmune encephalomyelitis
- TLR
Toll-like receptor
- MBP
myelin basic protein
- MBP(68-86)
peptide 68-86 from rat (self) MBP
- LPS
lipopolysaccharide
- CpG-ODN
cytosine-guanine dinucleotide
- CO
control oligonucleotide
- PolyI;C
Polyinosinic-Polycytidylic acid
Footnotes
Grant support: Supported by NIH grants 5-RO1-NS06985-37 and 5-RO1-NS048070-02
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Conant SB, Swanborg RH. Autoreactive T cells persist in rats protected against experimental autoimmune encephalomyelitis and can be activated through stimulation of innate immunity. J Immunol. 2004;172:5322–5328. doi: 10.4049/jimmunol.172.9.5322. [DOI] [PubMed] [Google Scholar]
- Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: Evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. 2002;169:7104–7110. doi: 10.4049/jimmunol.169.12.7104. [DOI] [PubMed] [Google Scholar]
- Hansen BS, Hussain RS, Lovett-Racke AE, Thomas JA, Racke MK. Multiple toll-like receptor agonists act as potent adjuvants in the induction of autoimmunity. J Neuroimmunol. 2006;172:94–103. doi: 10.1016/j.jneuroim.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Ichikawa HT, Williams LP, Segal BM. Activation of APCs through CD40 or Toll-Like Receptor 9 overcomes tolerance and precipitates autoimmune disease. J Immunol. 2002;169:2781–2787. doi: 10.4049/jimmunol.169.5.2781. [DOI] [PubMed] [Google Scholar]
- Janeway CA., Jr How the immune system works to protect the host from infection: a personal view. Proc Natl Acad Sci USA. 2001;98:7461–7468. doi: 10.1073/pnas.131202998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat EA, Wolf A, Bezer AE. The rapid production of acute disseminated encephalomyelitis in rhesus monkeys by injection of heterologous and homologous brain tissue with adjuvants. J Exp Med. 1947;85:117–130. doi: 10.1084/jem.85.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpus WJ, Swanborg RH. CD4+ suppressor cells inhibit the function of effector cells of experimental autoimmune encephalomyelitis through a mechanism involving transforming growth factor-beta. J Immunol. 1991;146:1163–1168. [PubMed] [Google Scholar]
- Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, deSauvage FJ, Lund FE, Cooper AM. Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med. 2006;203:1805–1815. doi: 10.1084/jem.20052545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling P, Gately MK, Gubler U, Stern AS, Lin P, Hollfelder K, Su C, Pan YCE, Hakimi J. Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J Immunol. 1995;154:116–127. [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor β after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan IM. Allergic encephalomyelitis in monkeys in response to injection of normal monkey nervous tissue. J Exp Med. 1947;85:131–140. doi: 10.1084/jem.85.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogai A, Siffrin V, Bonhagen, Pfueller CF, Hohnstein T, Volkmer-Engert R, Bruck W, Stadelmann C, Kamradt T. Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J Immunol. 2006;175:959–966. doi: 10.4049/jimmunol.175.2.959. [DOI] [PubMed] [Google Scholar]
- Segal BM, Klinman DM, Shevach EM. Microbial products induce autoimmune disease by an IL-12-dependent pathway. J Immunol. 1997;158:5087–5090. [PubMed] [Google Scholar]
- Swanborg RH. Experimental autoimmune encephalomyelitis in rodents as a model for human demyelinating disease. Clin Immunol Immunopathol. 1995;77:4–13. doi: 10.1016/0090-1229(95)90130-2. [DOI] [PubMed] [Google Scholar]
- Swanborg RH. Experimental autoimmune encephalomyelitis in the rat: lessons in T cell immunology and autoreactivity. Immunol Reviews. 2001;184:129–135. doi: 10.1034/j.1600-065x.2001.1840112.x. [DOI] [PubMed] [Google Scholar]
- Swanborg RH, Stepaniak JA. Experimental autoimmune encephalomyelitis in the rat. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. Vol. 3. New York: Wiley; 1996. pp. 15.2.1–15.2.14. [Google Scholar]
- Swierkosz JE, Swanborg RH. Suppressor cell control of unresponsiveness to experimental allergic encephalomyelitis. J Immunol. 1975;115:631–633. [PubMed] [Google Scholar]
- Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-β. J Immunol. 2006;177:7505–7509. doi: 10.4049/jimmunol.177.11.7505. [DOI] [PubMed] [Google Scholar]
- Waldner H, Collins M, Kuchroo VK. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J Clin Investig. 2004;113:990–997. doi: 10.1172/JCI19388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefer J, Harris RA, Lobell A. Protective DNA vaccination against experimental autoimmune encephalomyelitis is associated with induction of IFNβ. J Neuroimmunol. 2004;149:66–76. doi: 10.1016/j.jneuroim.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Zipris D, Lien E, Nair A, Xie JX, Greiner DL, Mordes JP, Rossini AA. TLR9-signaling pathways are involved in Kilham rat virus-induced autoimmune diabetes in the Biobreeding Diabetes-Resistant rat. J Immunol. 2007;178:693–701. doi: 10.4049/jimmunol.178.2.693. [DOI] [PubMed] [Google Scholar]