

Abstract
The identification of substrates of protein tyrosine phosphatases (PTPs) is an essential step toward a complete understanding of the physiological function of members of this enzyme family. PTPs are defined by a conserved catalytic domain harboring 27 invariant residues. From a mutagenesis study of these invariant residues that was guided by our knowledge of the crystal structure of PTP1B, we have discovered a mutation of the invariant catalytic acid (Asp-181 in PTP1B) that converts an extremely active enzyme into a “substrate trap.” Expression of this D181A mutant of PTP1B in COS and 293 cells results in an enzyme that competes with endogenous PTP1B for substrates and promotes the accumulation of phosphotyrosine primarily on the epidermal growth factor (EGF) receptor as well as on proteins of 120, 80, and 70 kDa. The association between the D181A mutant of PTP1B and these substrates was sufficiently stable to allow isolation of the complex by immunoprecipitation. As predicted for an interaction between the substrate-binding site of PTP1B and its substrates, the complex is disrupted by vanadate and, for the EGF receptor, the interaction absolutely requires receptor autophosphorylation. Furthermore, from immunofluorescence studies, the D181A mutant of PTP1B appeared to retain the endogenous EGF receptor in an intracellular complex. These results suggest that the EGF receptor is a bona fide substrate for PTP1B in vivo and that one important function of PTP1B is to prevent the inappropriate, ligand-independent, activation of newly synthesized EGF receptor in the endoplasmic reticulum. This essential catalytic aspartate residue is present in all PTPs and has structurally equivalent counterparts in the dual-specificity phosphatases and the low molecular weight PTPs. Therefore we anticipate that this method may be widely applicable to facilitate the identification of substrates of other members of this enzyme family.
The protein tyrosine phosphatases (PTPs) are a structurally diverse family of receptor-like and nontransmembrane enzymes that have been implicated in the control of numerous physiological processes, including growth and differentiation (1, 2). Approximately 75 PTPs have been identified to date. These enzymes are characterized by the presence of a conserved catalytic domain of ≈240 residues which contains the unique signature motif, 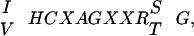 that defines this enzyme family (3). The structural diversity is manifested by the variability in noncatalytic sequences fused to the N or C terminus of the catalytic domain. These noncatalytic segments frequently serve a regulatory function, including ligand binding for receptor PTPs and targeting of cytoplasmic PTPs to defined subcellular locations (1, 2).
that defines this enzyme family (3). The structural diversity is manifested by the variability in noncatalytic sequences fused to the N or C terminus of the catalytic domain. These noncatalytic segments frequently serve a regulatory function, including ligand binding for receptor PTPs and targeting of cytoplasmic PTPs to defined subcellular locations (1, 2).
Although great progress has been made in illustrating the, somewhat unexpected, structural diversity within this large enzyme family, relatively little is known of the physiological function of individual PTPs. Presumably this structural diversity reflects a broad range of functions for the PTP family in vivo. To understand fully the function of PTPs, it will be necessary to identify the physiological substrates of the individual members of the family. However, progress in this area to date has been limited. In this study, we have developed an approach by which this may be achieved.
Our procedure was based on PTP1B, the prototypical PTP. PTP1B was first purified from human placenta as a 37-kDa protein of 321 amino acids constituting predominantly the catalytic domain (4, 5). Isolation of cDNA illustrated that this purified protein was derived from a full-length molecule of 435 residues, containing a C-terminal regulatory segment fused to the catalytic domain (6–8). The extreme C-terminal 35 residues constitute a hydrophobic segment that is both necessary and sufficient for targeting the enzyme to the cytoplasmic face of membranes of the endoplasmic reticulum (9). This motif is preceded by a stretch of hydrophilic residues that contains sites of serine phosphorylation; PTP1B is phosphorylated in vivo in a manner that varies during the cell cycle (10). We determined the crystal structure of the 37-kDa form of PTP1B, alone (11) and in a complex with a peptide substrate (12). When the sequences of PTP catalytic domains were aligned, a number of invariant residues were identified. The structure of PTP1B illustrated that, for the most part, these residues are clustered in regions of the protein surrounding the active site, where they presumably facilitate enzyme–substrate recognition and catalysis.
The signature motif functions as a phosphate-binding cradle in which the catalytically essential cysteine (Cys-215 in PTP1B) is positioned to act as a nucleophile to attack the phosphorus atom of the phosphotyrosyl residue (pTyr) of the substrate (11, 13). In PTP1B this motif lies at the base of a cleft on the surface of the protein, defined at one end by Tyr-46 and the other by Phe-182. The depth of this cleft is a primary determinant of the specificity of the enzyme for pTyr. Upon substrate binding a localized conformational change is induced in which the loop containing Phe-182 moves 5.5 Å into the catalytic site, contributing to a hydrophobic pocket that buries the pTyr of the substrate (12). This conformational change positions the side chain of Asp-181 to act as a general acid to facilitate protonation of the phenolic oxygen atom of the tyrosyl leaving group (12, 14). Cleavage of the scissile bond yields a Cys-215 thiol-phosphate intermediate and releases the free substrate protein product (15). Asp-181 then also may serve to activate a water molecule responsible for hydrolysis of the enzyme-phosphate intermediate (16).
We and others have shown that alteration of the nucleophilic Cys to Ser or Ala allows some PTPs to be isolated in a complex with their target substrates (12, 17–20). However, for other PTPs this interaction is too weak to permit isolation of the complex. Thus it appears that the Cys → Ser/Ala mutants may be limited in their applicability and may not be utilized to isolate all combinations of PTPs and substrates. In an attempt to generate alternative “substrate-trapping” PTP mutants, we undertook a systematic mutational analysis of the invariant residues that were predicted from the crystal structure to be important in catalysis. In this paper we report the identification and characterization of such a mutant form of PTP1B, in which Asp-181, the residue that functions as a general acid to facilitate cleavage of the scissile P—O bond in the substrate, has been altered to Ala.
MATERIALS AND METHODS
Preparation and Assay of Mutants of PTP1B.
Site-directed mutagenesis of either the complete coding sequence of human PTP1B or a clone encoding amino acids 1–321 (37-kDa PTP1B) in pSK was accomplished by using the U-labeled template method of Kunkel (21) and the Bio-Rad Muta-gene kit. For expression of mutants of 37-kDa PTP1B in Escherichia coli, DNA fragments containing the mutation were exchanged with the corresponding wild-type sequences in the T7-based expression vector pET19b (Novagen) using the unique XhoI site at Arg-169 and either a 5′ NcoI site or a 3′ BamHI site. Similarly, an XhoI/EcoRI fragment containing mutations in Cys-215 (M1 in figures) or Asp-181 (M2 in figures) replaced the wild-type sequence in full-length PTP1B or the glutathione S-transferase (GST)-PTP1B fusion protein in the pMT2 eukaryotic expression vector (10). All subcloned fragments were sequenced completely to confirm that no other mutations had been introduced. Mutants of 37-kDa PTP1B were expressed in E. coli strain BL21 and purified by chromatography on Fast Flow S-Sepharose and Mono Q (22).
Tyrosine phosphatase activity was measured using [32P]pTyr reduced, carboxamidomethylated, and maleylated lysozyme (RCML) as described previously (10). Kinetic parameters were determined using a direct nonlinear curve-fitting routine. Protein concentrations were determined by Bradford with a bovine serum albumin standard (23).
Transfection, Immunoprecipitation, and Immunofluorescence.
Calcium phosphate-mediated transfection of COS1 or 293 cells was performed with 20 μg of CsCl-purified DNA per 10-cm dish of cells or 8 μg per 6-cm dish. The efficiency of transfection, as assessed by 5-bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) staining of pSV-β-gal-transfected COS and 293 cells, was 16% and 20%, respectively. Cells were lysed 44–48 h after transfection in 50 mM Tris·HCl, pH 7.5/5 mM EDTA/150 mM NaCl/1% Triton X-100/5 mM iodoacetic acid/10 mM sodium phosphate/10 mM NaF/5 μg/ml leupeptin/5 μg/ml aprotinin/1 mM benzamidine (1 ml per 10-cm dish, 0.5 ml per 6-cm dish). PTP1B was immunoprecipitated from lysates with mAb FG6 and GST-PTP1B fusion proteins were precipitated with glutathione-Sepharose, 4 μg of mAb or ≈10 μl of beads respectively per mg of cell lysate. Precipitates were collected by centrifugation (15 s, 5000 × g) at 4°C, washed four times with 0.7 ml of ice-cold lysis buffer, and finally heated at 95°C for 5 min in 60 μl of 2× Laemmli sample buffer. mAbs to pTyr were used for immunoblotting at the following concentrations: 4G10 (Upstate Biotechnology) 1 μg/ml, RC20b (Transduction Laboratories) 0.5 μg/ml, G98 (Cold Spring Harbor Laboratory) 1:1000 dilution of ascites fluid. mAb to epidermal growth factor (EGF) receptor (EGFR) (Transduction Laboratories) was used at 1 μg/ml and rabbit polyclonal antisera to EGFR, #1964 and KSM (kindly provided by G. Gill, University of California at San Diego), were used at 1:5000 and 1:1000 dilution, respectively. The levels of expression of wild-type, C215A, and mutant PTP1B were always equivalent. For immunofluorescence experiments, cells were seeded onto glass coverslips at 2.5–3 × 105 per 6-cm dish, transfected with 10 μg of PTP1B cDNA, wild-type or mutant, and processed for immunofluorescence staining at 36 h after transfection as previously described (24).
RESULTS
The goal of this study was to create mutant forms of PTP1B in which the affinity for substrate remained similar to that of the wild-type enzyme but in which catalytic activity had been reduced, to such an extent that an enzyme–substrate complex, once formed, would be stable enough to withstand isolation. To implement this we generated a series of point mutants in PTP1B, expressed the proteins in E. coli, and purified them to homogeneity. The effects of these mutations on the ability of the enzyme to dephosphorylate pTyr-RCML in vitro were then tested (Table 1).
Table 1.
Kinetic parameters of purified point mutants of 37-kDa PTP1B assessed with pTyr-RCML as substrate
| Enzyme | Vmax, nmol/min/mg | Km, nM | kcat, min−1 |
|---|---|---|---|
| Wild type | 60,200 | 102 | 2244 |
| Tyr-46 → Ser | 4,120 | 1700 | 154 |
| → Leu | 4,160 | 1700 | 155 |
| Glu-115 → Ala | 5,700 | 45 | 212 |
| → Asp | 5,900 | 20 | 220 |
| Lys-116 → Ala | 68,600 | 150 | 2557 |
| Lys-120 → Ala | 19,000 | 80 | 708 |
| Asp-181 → Ala | 0.61 | ≤126 | 0.023 |
| → Glu | 97 | 10 | 3.6 |
| His-214 → Ala | 700 | 20 | 26 |
| Cys-215 → Ser | No detectable activity | ||
| Arg-221 → Lys | 11 | 80 | 0.41 |
| → Met | 3.3 | 1060 | 0.12 |
| Gln-262 → Ala | 720 | 9 | 27 |
In agreement with our predictions based on the x-ray structure of PTP1B in complex with a substrate peptide, affinity for substrate was significantly weakened by replacement of the aromatic ring of Tyr-46 by Ser or Leu (Km increased by 20-fold) or by loss of the electrostatic interaction between Arg-221 and the phosphate oxygens in Arg-221 → Met. Whereas the Arg-221 → Lys mutant bound substrate normally, it has severely impaired activity, suggesting that this invariant Arg in the signature motif also has a critical catalytic role, most likely to stabilize the transition state as proposed for YopH (25). As noted previously by many groups, mutation of the active site Cys to Ala or Ser inactivated the enzyme. In the context of our goal, mutation of Asp-181 also dramatically reduced activity. Conversion of Asp-181 to Glu reduced kcat ≈600-fold; however, the residual activity, kcat = 3.6 min−1, was sufficient to preclude the use of this mutant as a substrate trap. In contrast, the kcat was reduced by a factor of ≈105 in the D181A mutant, equivalent to ≈1 catalytic cycle per hour. In this mutant one might anticipate that an enzyme–substrate complex, further stabilized by chilling to 4°C, would be sufficiently long-lived to allow isolation.
To test the effects of PTP1B, and mutant forms thereof, on the extent of tyrosine phosphorylation of proteins in vivo, we expressed wild-type and C215A and D181A mutants of PTP1B (either untagged or GST-tagged) in COS cells and immunoblotted cell lysates with an anti-pTyr antibody (Fig. 1). In cells expressing mutant forms of PTP1B, we detected accumulation of pTyr in several proteins, predominantly p180, and to lesser extents p120 and p70. We also noted that the enhancement of phosphorylation was more pronounced in cells expressing D181A than C215A PTP1B. As expected, the level of tyrosine phosphorylation in cells expressing the wild-type enzyme was comparable to that in the vector control. These data suggest that the mutant enzymes bind to and protect these proteins from dephosphorylation by endogenous PTP1B.
Figure 1.

Induction of tyrosine phosphorylation resulting from expression of PTP1B mutants in COS cells. Samples (50 μg) of protein from lysates of COS cells transfected with pMT2 alone (vector) or with pMT2 plasmids expressing wild-type PTP1B, PTP1B with a C215A mutation (M1), PTP1B with a D181A mutation (M2), or GST fusions of each of these proteins were immunoblotted with anti-phosphotyrosine mAb 4G10. Proteins showing substantial increases in their phosphotyrosine content are marked with the dark arrows on the left. The positions of the molecular mass markers (Sigma) myosin, β-galactosidase, phosphorylase, bovine serum albumin, ovalbumin, and carbonic anhydrase are marked on the right with thin arrows.
To ascertain whether this interaction was stable to isolation, wild-type and mutant forms of PTP1B were precipitated from COS cell lysates, and associated proteins were visualized by anti-pTyr immunoblotting (Fig. 2). The same proteins that displayed enhanced phosphorylation were observed to coprecipitate with the mutant but not wild-type PTP1B, isolated either by immunoprecipitation with mAb FG6 (untagged) or by precipitation on glutathione-Sepharose (GST-tagged). Expression of GST-fusion proteins and precipitation with GSH-Sepharose provides two advantages as compared with FG6 immunoprecipitations. These precipitates are cleaner to immunoblot because they do not contain any antibody and they are free of any endogenous active PTP1B which exists as a minor component in the FG6 immunoprecipitates. Once again, greater quantities of the pTyr proteins coprecipitated with the D181A mutant than the C215A mutant form of PTP1B. Interestingly, the pattern of tyrosine phosphorylation was the same before and after refeeding of serum-starved cells (Fig. 2). These data suggest that the mutant PTPs are functioning to protect the substrates against dephosphorylation by endogenous phosphatases, thus increasing the basal phosphorylation state. The importance of this observation is that it indicates that the effects of the mutants are manifested primarily within the cell and not after lysis.
Figure 2.

Precipitation of tyrosine-phosphorylated proteins in association with the D181A (M2) mutant of PTP1B. Immunoprecipitates of PTP1B from lysates of COS cells transfected to express PTP1B, C215A (M1), or D181A (M2) and glutathione-Sepharose precipitates from lysates of COS cells transfected to express GST fusions to each of these proteins were immunoblotted with anti-phosphotyrosine mAb 4G10. Pairs of lanes represent samples from duplicate transfections. A − indicates that for 24 h prior to harvesting the cells were maintained in media without serum. Lanes marked with + indicate that these cells were similarly deprived of serum until 10 min before harvesting, during which time they were incubated with 20% fetal bovine serum, a treatment that did not significantly alter the phosphotyrosine content of proteins associated with D181A-PTP1B. The three intense black bands found in all the anti-PTP1B immunoprecipitates, including the vector only control, are derived from the precipitating mAb. (Right) Anti-pTyr (4G10) blot of 50 μg of lysate from untransfected COS cells that were untreated (none) or incubated for 10 min with EGF at 100 ng/ml or platelet-derived growth factor (PDGF) at 5 ng/ml.
To begin the process of identifying the proteins that display enhanced phosphorylation and hence are potential substrates of PTP1B, we starved COS cells of serum, then stimulated them with either EGF or platelet-derived growth factor and examined the patterns of tyrosine phosphorylation that were induced, by immunoblotting lysates with an anti-pTyr antibody. Most notably, in response to EGF the phosphorylation state of a 180-kDa protein, presumably the EGFR, was markedly enhanced. This protein comigrated with pTyr-p180 that coprecipitates with mutant PTP1B (Fig. 2). When this experiment was repeated in 293 cells, which possess much lower levels of endogenous EGFR, only trace quantities of p180 were detected in association with D181A PTP1B. However p120, p70, and an additional pTyr protein of ≈80 kDa were observed to coprecipitate with the mutant PTP (data not shown). We tested further the possibility that p180 was the EGFR by expressing wild-type and mutant forms of PTP1B in COS cells and, after serum starvation, examining the effects of EGF treatment on the patterns of tyrosine phosphorylation (Fig. 3). A comparison of immunoblots of the PTP1B precipitates using either anti-pTyr or anti-EGFR antibodies indicates that p180 is the EGFR. Again, PTP1B-D181A appeared to be more efficient at precipitating the EGFR than C215A, and no association was observed with the wild-type enzyme. Despite the fact that the total cellular content of EGFR was not altered by treatment with EGF, more EGFR was precipitated by PTP1B-D181A after administration of the growth factor. Moreover, more pTyr-p180 protein coprecipitated with the mutant PTP after growth factor treatment, consistent with the concept that EGF-induced autophosphorylation of its receptor produced higher levels of substrate available for trapping.
Figure 3.

Identification of tyrosine-phosphorylated p180 in the D181A precipitates as the EGFR. This experiment was conducted as described in the legend of Fig. 2 except that the resulting immunoprecipitates were split in half, subjected to electrophoresis on duplicate SDS/polyacrylamide gels, and blotted with either anti-EGFR mAb (Transduction Laboratories) (Upper Left) or with anti-pTyr (4G10) (Lower Left). Pairs of lanes represent samples from duplicate transfections. A − indicates that for 24 h prior to harvesting the cells were maintained in media without serum. A + indicates that these cells were similarly deprived of serum until 10 min before harvesting, during which time they were incubated with EGF at 100 ng/ml. The anti-pTyr blot was deliberately overexposed in an attempt to observe pTyr-containing proteins in the C215A (M1) precipitates and to visualize p70 and p80 in the D181A (M2) precipitates. Anti-EGFR staining was weaker than anti-pTyr staining of p180 in D181A (M2) precipitates, presumably either because the anti-EGFR antibody is of lower affinity or because there are multiple pTyr epitopes recognized by 4G10 in the “trapped” EGFR. (Right) Anti-EGFR blot of 50 μg of lysate from untransfected COS cells that were untreated (−) or incubated for 10 min with EGF at 100 ng/ml (+).
We examined whether the interaction between PTP1B and EGFR involved the catalytic site of the phosphatase by investigating the dependence upon tyrosine phosphorylation of the substrate. Wild-type EGFR or mutant forms, either inactivated as a protein tyrosine kinase or from which the sites of autophosphorylation had been eliminated, were coexpressed in 293 cells with the various forms of PTP1B, and PTP1B immunoprecipitates were blotted with either anti-EGFR or anti-pTyr antibodies (Fig. 4). Although the various forms of the EGFR were expressed to similar levels, association was observed only between the wild-type EGFR and the substrate-trapping mutants of PTP1B. These data established that tyrosine phosphorylation of the EGFR was required for the interaction. Furthermore, treatment with pervanadate, a PTP inhibitor that covalently modifies the nucleophilic Cys (26), disrupted complex formation between PTP1B-D181A and EGFR (data not shown). These observations suggest that this association represents an enzyme–substrate interaction.
Figure 4.

Reconstitution of the interaction between D181A and the EGFR requires EGFR autophosphorylation. 293 cells were cotransfected with plasmids expressing PTP1B, the C215A or D181A PTP1B mutants, and plasmids expressing the human EGFR or a catalytically inactive (K−) mutant, in which the Lys responsible for coordinating ATP, Lys-721, was converted to Arg, or a C-terminally truncated (c′958) form that lacks all the major autophosphorylation sites. Immunoprecipitates of PTP1B were split in half and resolved by SDS/PAGE on duplicate gels. One gel (Upper) was blotted with anti-EGFR antibody KSM and the other (Lower) was blotted for pTyr with G98. Panels on each end represent immunoblots of 50 μg of lysate from the cells cotransfected with D181A-PTP18 (M2) and the various EGFR forms. CMV, cytomegalovirus. Although both the K− and c′958 forms of EGFR are expressed well, neither contains pTyr, and both fail to interact with the D181A mutant of PTP1B.
In addition, we have examined the association between EGFR and the substrate-trapping mutants of PTP1B by immunofluorescence as an independent confirmation that the association occurred in vivo and not after lysis (Fig. 5). Wild-type or C215A PTP1B exhibited a reticular pattern of staining extending from the nucleus throughout the cell, as expected for a protein targeted to the endoplasmic reticulum (Fig. 5A). In comparison, the staining of endogenous EGFR appeared diffuse in cells coexpressing wild-type or C215A PTP1B, which display little or no association with the protein tyrosine kinase(Fig. 5B). In contrast, the staining of the EGFR was altered in cells coexpressing PTP1B-D181A and exhibited a distinct punctate appearance that was not detected in surrounding cells that failed to express the mutant PTP (Fig. 5D). In these PTP1B-D181A-expressing cells, this punctate appearance of anti-EGFR staining precisely matched foci of intense staining with the anti-PTP1B antibody FG6 (Fig. 5C). This coincidence of EGFR staining with PTP1B-D181A, the substrate-trapping mutant that affords greatest protection and ability to coprecipitate the EGFR, reinforces the interpretation that the association occurred in a cellular context rather than in the lysate. It also suggests that in cells expressing PTP1B-D181A mutants, the intracellular location of the EGFR may be determined by colocalization with PTP1B as a result of “substrate trapping.”
Figure 5.


Colocalization of D181A-PTP1B and endogenous EGFRs. COS 1 cells transfected with wild-type PTP1B (data not shown) or C215A (A and B) or D181A expression plasmids (C and D) were fixed with paraformaldehyde at 36 h after transfection and processed for immunofluorescence as previously described (24). Cells were incubated with anti-PTP1B FG6 ascites fluid (10) and anti-EGFR polyclonal serum (1964, kindly provided by G. Gill) at dilutions of 1/4000 and 1/500, respectively. Overexpressed PTP1B (A and C) and endogenous EGFR (B and D) were visualized with fluorescein-conjugated sheep anti-mouse and Texas red-conjugated goat anti-rabbit antibodies (Cappel), respectively.
Although these data illustrate an association between substrate-trapping mutants of PTP1B and EGFR, it was important to test whether such an interaction would be observed after the expression of the equivalent mutant form of another member of the PTP family. Thus we examined the state of basal phosphorylation of tyrosyl residues in proteins from COS cells that had been serum starved after expression of wild-type and mutant PTP1B or the equivalent forms of another cytoplasmic PTP, PTP-PEST (27). Whereas in the presence of PTP1B-D181A the EGFR exhibits enhanced phosphorylation, no such effect was observed after expression of either PTP-PEST C231S or D199A (Fig. 6). Subsequent studies have revealed that PTP-PEST displays a distinct substrate specificity, showing a high degree of selectivity for p130cas as a target substrate (27). This result suggests that individual PTPs display specificity in their interaction with substrates in the cell.
Figure 6.

The analogous substrate-trapping mutant in PTP-PEST does not interact with the EGFR. This figure shows an anti-pTyr blot (RC20b) of precipitates prepared from lysates of COS cells that were transfected to express PTP1B, the C215A-PTP1B mutant (M1), the D181A-PTP1B mutant (M2), the GST fusions of these proteins, or PTP-PEST and the analogous mutants of it: C231S-PTP-PEST (M1) or D199A-PTP-PEST (M2). PTP-PEST was immunoprecipitated with mAb AG25 (27). The expression levels of the PTP-PEST proteins were similar to those for PTP1B.
DISCUSSION
The diversity in structural motifs that have been observed in members of the PTP family is suggestive of important roles in the control of a broad range of fundamental physiological signaling events. Clearly, a definition of the spectrum of phosphotyrosyl proteins dephosphorylated by an individual PTP in vivo will be a prerequisite for the elucidation of the physiological function of that enzyme. In this paper we describe a method by which such substrate identification may be achieved.
Mutants of some members of the PTP family in which the catalytically essential nucleophilic Cys residue from the signature motif has been mutated to Ser or Ala are inactive but retain the ability to bind to substrate in vitro. For example, we used a Cys → Ser mutant form of PTP1B to determine the crystal structure of the enzyme–substrate complex (12), and binding of this mutant to autophosphorylated EGFR in vitro has been examined (19). In addition, there are examples in which such mutants have been shown to bind to substrate in vivo, including MKP-1 (17), YopH (28), and the Drosophila SH2 domain PTP csw (29). However, in some of these cases it is not clear that the interaction is mediated solely by the inactivated catalytic domain (28, 29). In our experience such mutant PTPs do not uniformly bind to substrates in a stable manner in the cell. In this paper we have demonstrated that a mutant of PTP1B in which the invariant aspartate, Asp-181, which functions as a general acid in protonating the tyrosyl leaving group of the substrate, is changed to Ala, retains the ability to bind to substrates in a cellular context but is markedly attenuated in its catalytic function. Thus the enzyme–substrate complex is stabilized sufficiently to permit isolation. In comparison, although the C215A mutant traps the same spectrum of pTyr proteins as PTP1B-D181A, it does so at much lower efficiency. Similar data have been obtained subsequently for PTP-PEST and its substrate, p130cas (27).
There are two features of the PTP1B-D181A mutant that may contribute to its enhanced substrate-trapping properties relative to PTP1B-C215A. First, the dephosphorylation reaction catalyzed by PTPs proceeds via the formation of a cysteinyl phosphate intermediate, generated after nucleophilic attack by the active-site Cys upon the phosphorus atom of the substrate (15). This is accompanied by cleavage of the P—O bond between the phosphorus and the phenolic oxygen of the tyrosyl side chain in the substrate, mediated by the invariant aspartate. Thus, in the absence of this acidic residue one would anticipate that P—O bond cleavage would be suppressed, thereby potentially stabilizing the interaction with the active-site Cys. Stabilization of the complex may be further facilitated by hydrophobic interactions between the residues lining the substrate-binding pocket of the enzyme and the tyrosyl side chain of the substrate that are induced after the loop closure that accompanies substrate binding (12). Second, the mutation of Asp-181 to an uncharged residue, Ala, will result in a decrease in negative charge at this position. Thus the potential for electrostatic repulsion between the negatively charged side chain of Asp-181 and the negatively charged phosphate moiety, which would occur in the PTP1B-C215S/A mutants, would be reduced in PTP1B-D181A. Again this may favor loop closure and stabilization of the enzyme–substrate complex.
Considering that the substrate-trapping mutant PTPs have the ability, through interaction with pTyr residues, to increase the basal phosphorylation of their target substrates, one might expect them to promote ligand-independent signaling. However because these mutants form stable complexes with target substrates, they may also interfere with signaling in vivo in a manner analogous to the wild-type phosphatase. Thus if the site of tyrosine phosphorylation on the substrate is critical for a protein–protein interaction required for signaling or if the substrate is an enzyme and the phosphorylation site is located close to the active site, steric hindrance resulting from binding of the mutant PTP may exert an effect that is functionally equivalent to dephosphorylation. Thus, for many PTPs Cys → Ser/Ala (or Asp → Ala) mutants may not be an appropriate choice as “inactive” controls for effects of expression of wild-type enzymes. Consideration of the data in Table 1 suggests that mutations in the invariant Arg in the signature motif, Arg-221 in PTP1B, which abrogate both catalytic activity and affinity for substrate, might be better suited to this purpose. This Arg residue normally functions in electrostatic and hydrogen-bonding interactions with the phosphate moiety of the substrate and functions to stabilize the transition state (25).
The successful application of this technique to identify additional substrates of PTP1B, and substrates of other PTPs, will require expression of the Asp → Ala mutant PTP in cells and selection of an appropriate stimulus to trigger tyrosine phosphorylation of the substrate and accumulation of a complex with the mutant phosphatase. For PTP1B, EGFR is naturally abundant in COS cells, and its basal rate of autophosphorylation generated enough tyrosine-phosphorylated protein to be trapped by PTP1B-D181A and to be detected by anti-pTyr immunoblotting. Its high molecular weight, characteristic of growth factor receptors, facilitated identification. Of the less-phosphorylated substrates of PTP1B, the finding that phosphorylation of p70 is enhanced upon cotransfection with v-src and D181A may enable enough of this substrate to be isolated to allow its identification by peptide sequencing. In contrast, the substrate of PTP-PEST, p130cas, although relatively abundant in COS cells, does not accumulate in a tyrosine-phosphorylated state that is trapped by PTP-PEST-D199A in unstimulated cells (27). However, treatment of cells with pervanadate, or less potently by cotransfection of v-src, elevates phosphorylation of p130cas to levels that enable it to be trapped specifically by D199A-PTP-PEST and readily detected by anti-pTyr blotting (27). In addition to the use of these trapping mutants to identify substrates, it also may be possible to identify the site within the cell at which a PTP acts on a particular target, through colocalization of the substrate and the Asp → Ala mutant PTP. Thus, whereas we expect that the application of Asp → Ala mutants of other PTPs will be generally useful, the precise experimental approach and choice of cells to study will vary with each individual phosphatase.
There is a considerable body of data in the literature to indicate that PTP1B is somewhat promiscuous in its substrate preference in vitro, dephosphorylating a wide variety of protein and peptide substrates, albeit with widely varying Km values (5, 30, 31). However, the use of the PTP1B-D181A substrate-trapping mutant described in this paper has revealed that within the cell this activity is constrained. PTP1B selected a small number of substrates from the plethora of available pTyr proteins. The major substrate detected was the EGFR, together with no more than three additional proteins, p120, p80, and p70. Our data suggest that PTP1B may exert its effects on newly synthesized EGFRs, preventing inappropriate, ligand-independent, signaling by nascent receptors in transit through the endoplasmic reticulum. The identity of the other substrates remains unclear at present. Using their apparent molecular weights to predict identity, we have tested but failed to detect cross-reactivity between these proteins and antibodies to FAK, p120cas, c-cbl, Src, raf-1, paxillin, p62, or SHP2 in PTP1B-D181A precipitates with antibodies that readily detected these proteins in cell lysates (A.J.F. and N.K.T., unpublished results). In 293 cells expressing v-Src together with PTP1B-D181A, we did not detect phosphorylation of the EGFR but noted that the substrate-trapping mutant interacted selectively with a 70-kDa protein from the greatly increased spectrum of tyrosyl-phosphorylated proteins (data not shown). Interestingly, Dixon’s group reported that dephosphorylation of tyrosyl residues in a 70-kDa protein was the major change in phosphorylation observed in v-Src-transformed cells overexpressing PTP1B (32). However, the identity of this protein remains unclear. Nevertheless, the data further reinforce the importance of targeting PTPs to defined subcellular locations as a mechanism for control of substrate specificity.
In summary, these data highlight the fact that PTP1B can display substrate specificity in vivo and bolster the hypothesis that subcellular targeting is an important mechanism by which such specificity is achieved. The substrate-trapping mutant described in this paper is altered in a residue that is an invariant catalytic acid in all members of the PTP family. Therefore the use of this mutation should be generally applicable to any PTP and may represent a powerful tool with which to delineate the physiological substrate specificity of other members of the family, thereby revealing important insights into their function in vivo.
Acknowledgments
We thank Dr. Andy Garton for PTP-PEST mutants and helpful discussions, Dr. Gordon Gill (University of California at San Diego) for generously providing cDNA and antibodies to the EGFR and Dr. Dave Hill (Calbiochem Oncogene Research Products) for mAb FG6 to PTP1B. We also thank Martha Daddario for technical support, Phil Renna for photography, Regina Whitaker for help with tissue culture, and Carol Marcincuk for typing the manuscript. This work was supported by a grant from the National Institutes of Health (CA53840) and the Mellam Family Foundation. A.J.F. was supported by an Andrew Seligson Memorial Fellowship. T.T. is a National Health and Medical Research Council of Australia C. J. Martin Fellow.
ABBREVIATIONS
- PTP
protein tyrosine phosphatase
- pTyr
phosphotyrosyl residue
- GST
glutathione S-transferase
- RCML
reduced, carboxamidomethylated, and maleylated lysozyme
- EGF
epidermal growth factor
- EGFR
EGF receptor
References
- 1.Tonks N K, Neel B G. Cell. 1996;87:365–368. doi: 10.1016/s0092-8674(00)81357-4. [DOI] [PubMed] [Google Scholar]
- 2.Tonks N K. Adv Pharmacol. 1996;36:91–119. doi: 10.1016/s1054-3589(08)60578-5. [DOI] [PubMed] [Google Scholar]
- 3.Barford D, Jia Z, Tonks N K. Nat Struct Biol. 1995;2:1043–1053. doi: 10.1038/nsb1295-1043. [DOI] [PubMed] [Google Scholar]
- 4.Tonks N K, Diltz C D, Fischer E H. J Biol Chem. 1988;263:6722–6730. [PubMed] [Google Scholar]
- 5.Tonks N K, Diltz C D, Fischer E H. J Biol Chem. 1988;263:6731–6737. [PubMed] [Google Scholar]
- 6.Guan K L, Haun R S, Watson S J, Geahlen R L, Dixon J E. Proc Natl Acad Sci USA. 1990;87:1501–1505. doi: 10.1073/pnas.87.4.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chernoff J, Schievella A R, Jost C A, Erikson R L, Neel B G. Proc Natl Acad Sci USA. 1990;87:2735–2739. doi: 10.1073/pnas.87.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown-Shimer S, Johnson K A, Lawrence J B, Johnson C, Bruskin A. Proc Natl Acad Sci USA. 1990;87:5148–5152. doi: 10.1073/pnas.87.13.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangioni J V, Beahm P H, Shifrin V, Jost C A, Neel B G. Cell. 1992;68:545–560. doi: 10.1016/0092-8674(92)90190-n. [DOI] [PubMed] [Google Scholar]
- 10.Flint A J, Gebbink M F B G, Franza B R, Jr, Hill D E, Tonks N K. EMBO J. 1993;12:1937–1946. doi: 10.1002/j.1460-2075.1993.tb05843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barford D, Flint A J, Tonks N K. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 12.Jia Z, Barford D, Flint A J, Tonks N K. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 13.Stuckey J A, Schubert H L, Fauman E B, Zhang Z-Y, Dixon J E, Saper M A. Nature (London) 1994;370:571–575. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Z-Y, Wang Y, Dixon J E. Proc Natl Acad Sci USA. 1994;91:1624–1627. doi: 10.1073/pnas.91.5.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan K L, Dixon J E. J Biol Chem. 1991;266:17026–17030. [PubMed] [Google Scholar]
- 16.Denu J M, Dixon J E. Proc Natl Acad Sci USA. 1995;92:5910–5914. doi: 10.1073/pnas.92.13.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun H, Charles C H, Lau L F, Tonks N K. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- 18.Shiozaki K, Russell P. Nature (London) 1995;378:739–743. doi: 10.1038/378739a0. [DOI] [PubMed] [Google Scholar]
- 19.Milarski K L, Zhu G, Pearl C G, McNamara D J, Dobrusin E M, MacLean D, Thieme-Sefler A, Zhang Z-Y, Sawyer T, Decker S J, Dixon J E, Saltiel A R. J Biol Chem. 1993;268:23634–23639. [PubMed] [Google Scholar]
- 20.Furukawa T, Itoh M, Krueger N X, Streuli M, Saito H. Proc Natl Acad Sci USA. 1994;91:10928–10932. doi: 10.1073/pnas.91.23.10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunkel T A. Proc Natl Acad Sci USA. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barford D, Keller J C, Flint A J, Tonks N K. J Mol Biol. 1994;239:726–730. doi: 10.1006/jmbi.1994.1409. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Lorenzen J A, Dadabay C Y, Fischer E H. J Cell Biol. 1995;131:631–643. doi: 10.1083/jcb.131.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z-Y, Wang Y, Wu L, Fauman E B, Stuckey J A, Schubert H L, Saper M A, Dixon J E. Biochemistry. 1994;33:15266–15270. doi: 10.1021/bi00255a007. [DOI] [PubMed] [Google Scholar]
- 26.Denu J M, Lohse D L, Vijayalakshmi J, Saper M A, Dixon J E. Proc Natl Acad Sci USA. 1996;93:2493–2498. doi: 10.1073/pnas.93.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garton A J, Flint A J, Tonks N K. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bliska J B, Clemens J C, Dixon J E, Falkow S. J Exp Med. 1992;176:1625–1630. doi: 10.1084/jem.176.6.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herbst R, Carroll P M, Allard J D, Schilling J, Raabe T, Simon M A. Cell. 1996;85:899–909. doi: 10.1016/s0092-8674(00)81273-8. [DOI] [PubMed] [Google Scholar]
- 30.Hippen K L, Jakes S, Richards J, Jena B P, Beck B L, Tabatabai L B, Ingebritsen T S. Biochemistry. 1993;32:12405–12412. doi: 10.1021/bi00097a019. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z-Y, Thieme-Sefler A M, Maclean D, McNamara D J, Dobrusin E M, Sawyer T K, Dixon J E. Proc Natl Acad Sci USA. 1993;90:4446–4450. doi: 10.1073/pnas.90.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woodford-Thomas T A, Rhodes J D, Dixon J E. J Cell Biol. 1992;117:401–414. doi: 10.1083/jcb.117.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]