

Abstract
We show that supercoiling of a DNA trefoil, the simplest knotted ring, perturbs differently the spatial writhe of its two chiral forms. As a consequence, the negative-noded and positive-noded DNA trefoils can be resolved by gel electrophoresis. Analysis of the chirality of trefoils produced by cyclization of two linear DNAs demonstrates that the two chiral trefoils are produced in equal amounts, suggesting that these DNAs do not prefer intrinsic writhe of one chirality or the other. In contrast, knotting of nicked DNA rings by a molar excess of Saccharomyces cerevisiae DNA topoisomerase II produces more negative-noded than positive-noded trefoils, indicating an asymmetry in the interaction between the enzyme and DNA crossovers of different signs. These results suggest that asymmetry in DNA crossovers and intrinsic or ligand-induced writhe in a DNA might be detectable from an analysis of trefoil chirality.
Keywords: DNA topology, knotted DNA rings, DNA crossovers, spatial conformation of DNA
Crossovers or nodes between two duplex DNA segments are a common motif in higher order structures of DNA and DNA–protein complexes. When two segments along a DNA are brought into proximity, they may form a positive or negative node, as illustrated in Fig. 1 a and b. In topological transformations of DNA rings catalyzed by DNA topoisomerases and site-specific recombinases, the topology of the final products are often critically dependent on the nodes present in the DNA and their signs.
Figure 1.

(a and b) Sign convention for nodes (crossovers) formed by segments along a DNA. Arrows specify an arbitrarily chosen direction along the DNA helical axis. The node is negative if alignment of the two arrows, which specify the directions of the crossing DNA segments, can be achieved by a clockwise rotation of the upper arrow by an angle less than 180° (b); otherwise the node is positive (a). (c) The structure of a crossover in a crystal of the self-complementary decameric DNA 5′-CCAACITTGG-3′. The structure can be viewed as a pair of quasi-continuous DNA helices in close contact, with the backbone of one helix fitting snugly into the grooves of the other (taken from figure 6 of ref. 6). [Reproduced with permission from the American Chemical Society (Copyright 1995)]. (d) Idealized drawings of a negative-noded trefoil (Left) and positive-noded trefoil (Right).
A question that often arises in the study of reactions involving intramolecular synapsis of two remote DNA segments is whether the structure of DNA might impose a bias for nodes of a particular sign. In general, two factors may affect the nodal sign. First, a DNA of a particular sequence might have a preferred writhe in solution and thus favor the formation of a node of a particular sign: positive writhe or preferential formation of a right-handed loop would favor the formation of a positive node, and negative writhe or preferential formation of a left-handed loop would favor the formation of a negative node. Second, if the crossing segments are in close contact, then interactions between the pair of DNA double helices might favor one geometry over another. A number of studies have revealed the structures of DNA helices in close contact. In several crystals, a pair of contacting DNA segments were found to form crossovers with unique structures (1–6). In the crossover illustrated in Fig. 1c, for example, the pair of helices form an acute angle of 60°, with the backbone of one helix fitting snugly into the grooves of the other; this type of structure was seen in several crystals, with the acute angle ranging from 43° to 77° (6). A pair of DNA helices with a backbone-groove fit was also deduced earlier through a variety of measurements in solution for the Holliday junction, a four-way DNA junction involved in DNA recombination (7–10).
The crossover exemplified by the structure shown in Fig. 1c is chiral, and inverting its chirality by pushing the upper helix through the lower one would destroy the backbone-groove fit. The formation of DNA crossovers with well-defined structures is by itself insufficient, however, to impose a nodal sign in a DNA ring. The structure shown in Fig. 1c, for example, may specify a negative node if the ends A and A′ are joined, and a positive node if the ends A and B′ are joined. To test whether a DNA might possess an intrinsic writhe or whether the local structures of DNA crossovers might influence nodal signs between distant segments in a DNA, we have analyzed the chirality of DNA trefoils formed under a variety of conditions. A trefoil is the simplest knot with irreducible negative or positive nodes (Fig. 1d), and therefore any bias favoring the formation of nodes of a particular sign may in turn favor the formation of one chiral trefoil over the other. Our results are presented here.
MATERIALS AND METHODS
Preparation of Trefoils by Cyclization of Linear DNAs.
A 5.6-kb plasmid pCC56cos (11) was first linearized at its unique EcoRI site. Similarly, YEplac181, a 5.7-kb plasmid containing the origin of replication of the yeast 2-μ plasmid and the LEU2 gene of yeast was linearized at a unique HindIII site. Both plasmids were separately treated with phage T4 DNA ligase (Boehringer Mannheim) at 16°C overnight at a final DNA concentration of 5 μg/ml and in reaction mixtures containing 10 mM Tris·HCl (pH 7.5), 0.1 mg/ml bovine serum albumin, 10 mM 2-mercaptoethanol, 0.5 mM ATP, 4% (vol/vol) glycerol, and 7.5 mM or 75 mM magnesium glutamate (Fluka). The reaction mixtures were phenol extracted twice and concentrated 40-fold by vacuum dialysis against 10 mM Tris·HCl (pH 8.0)/0.1 mM EDTA. The cyclized DNA in each mixture was then nicked with DNase I in the presence of a saturating amount of ethidium bromide (11, 12), phenol extracted three times, and ethanol precipitated. Following resuspension, the DNase-treated sample was subject to three successive rounds of fractionation by electrophoresis in 1% agarose gels at 4°C (approximately 40–45 hr for each round at a voltage gradient of 2.6 V/cm; the electrophoresis buffer, which contained a 36 mM Tris base, 30 mM NaH2PO4, and 0.6 μg/ml ethidium bromide, was recirculated and changed every 16–20 hr). At the end of each round of electrophoresis, gel slices containing knotted DNA rings were electroeluted using tRNA as a carrier. The recovered DNA samples were phenol extracted and ethanol precipitated.
Knotted DNA Rings Produced by Treatment with Type II DNA Topoisomerases.
Trefoil and figure-eight knots were also obtained by incubating nicked pCC56cos with Saccharomyces cerevisiae DNA topoisomerase II according to ref. 13. The desired DNAs were then isolated as described above. Phage T4 DNA topoisomerase was used to generate a series of supercoiled knots from negatively supercoiled pCC56cos, as described in ref. 14; supercoiled knots were also generated from positively supercoiled pCC56cos, as described later. DNA samples were then nicked by HindIII in the presence of excess ethidium bromide (15, 16), and knotted rings were purified from the nicked samples as described above.
Relaxation by Vaccinia Virus Topoisomerase.
Positively supercoiled topoisomers were converted into their negatively supercoiled counterparts by treatment with vaccinia virus topoisomerase in the presence of calculated amounts of ethidium bromide. Reaction conditions were as described in ref. 17, except that they contained 50 μg/ml unlabeled carrier DNA, 0.52 μg/ml ethidium bromide, and 50 ng/ml vaccinia virus topoisomerase. After incubation for 1 hr at 37°C, the reaction mixtures were phenol extracted twice, and ethanol precipitated.
Analysis of Knotted Topoisomer Distributions.
The gel-purified nicked DNA knots were radiolabeled by nick-translation in reaction mixtures containing 50 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 50 μg/ml bovine serum albumin, 0.8 mM each dTTP, dCTP, and dGTP, 25 μCi [α-32P]dATP (3000 Ci/mmol; 1 Ci = 37 GBq), and 0.02 unit per ml of Escherichia coli DNA polymerase I (Boehringer Mannheim). Following a 20-min incubation at 20°C, NAD and E. coli DNA ligase were added to 26 μM and 0.3 unit/ml, respectively, and incubation was continued for 30 min at the same temperature. Reactions were terminated by the addition of 0.2 M Na3EDTA (pH 8.0) to 50 mM, phenol extracted, and dialyzed first against 2 M NaCl/10 mM Tris·HCl (pH 8), to remove unincorporated nucleotides, and then against 10 mM Tris·HCl (pH 8)/0.1 mM EDTA. The topoisomer distribution in each reaction mixture was analyzed by electrophoresis at room temperature and 2.4 V/cm for 44 hr. Agarose gel slabs (0.9% agarose in electrophoresis buffer containing 90 mM Tris base, 90 mM boric acid, and 1.8 mM Na3EDTA) were used in all experiments. The electrophoresis buffer was replaced every 16–20 hr. The gels were dried for one hour at room temperature and an additional hour at 60°C prior to autoradiography.
Preparation of Positively Supercoiled DNA.
Positively supercoiled pCC56cos was prepared using a chimeric protein in which the DNA-binding domain of yeast GAL4 protein (residues 1–147) is fused to the amino end of phage T7 RNA polymerase (18). This fusion protein generates simultaneously positive and negative supercoils as it transcribes the DNA template, and positive supercoils accumulate in the DNA template in the presence of E. coli DNA topoisomerase I, which preferentially relaxes the negative supercoils (18).
RESULTS
Resolution of Supercoiled Trefoils of Opposite Chirality by Agarose Gel Electrophoresis.
It has been shown previously that cyclization of a linear DNA yields a small percentage of trefoils, which, in the nicked form, migrate electrophoretically in agarose gel as a sharp band (11, 19). Upon conversion of the nicked trefoils to the supercoiled form, however, two distinct ladders of topoisomers were observed upon agarose gel electrophoresis (Fig. 2). In this experiment, nicked trefoil knots of a 5.6-kb plasmid pCC56cos were purified from two sources: following cyclization of a linear DNA or following the addition of a nonhydrolyzable ATP analog, 5′-adenylyl imidodiphosphate (AMPPNP), to a mixture of unknotted nicked DNA rings and a molar excess of yeast DNA topoisomerase II. In the former reaction, the joining of the ends of linear molecules of different spatial configurations traps the various knotted forms, of which the trefoils are the major species. In the latter reaction, binding of the nonhydrolyzable ATP analog triggers the type II topoisomerase-mediated transport of one DNA segment through another to give various topological isomers, including the trefoils. The nicked trefoils from both reactions were purified as described and then treated with DNA ligase at 20°C and analyzed by agarose gel electrophoresis. Because the temperature and ionic medium employed for agarose gel electrophoresis differed from those employed for covalent closure of the DNA rings with DNA ligase, the various forms of covalently closed DNA rings were positively supercoiled during electrophoresis (20).
Figure 2.

Conversion of a nicked DNA trefoil to the positively supercoiled form yields doublets of topoisomers of different linking numbers upon agarose gel electrophoresis. Lanes 1 and 2, trefoil produced by cyclization of a 5.6-kb linear pCC56cos DNA in the presence of 7.5 mM (lane 1) or 75 mM (lane 2) magnesium glutamate. Lane 3, trefoil produced by the addition of AMPPNP to mixtures containing nicked pCC56cos DNA and a 10-fold molar excess of S. cerevisiae DNA topoisomerase II; the reaction medium contained 7.5 mM magnesium glutamate. nc, nicked circle; n3, nicked trefoil; lin, linear DNA.
It is well-known that there are two topologically distinct chiral forms of trefoils, one with negative nodes and the other with positive nodes (Fig. 1d). Therefore, a plausible interpretation of the double ladders shown in Fig. 2 is that each represents one particular chiral form, and that bands of each ladder represent topoisomers of different linking numbers. To test this interpretation, DNA trefoil of a unique chirality was prepared and examined. Treatment of a negatively supercoiled DNA with a molar excess of phage T4 topoisomerase in the absence of ATP is known to give predominantly negative-noded twist knots, including the trefoil, in the initial reaction products (21). As depicted in Fig. 3 (left lane), positively supercoiled topoisomers of a negative-noded trefoil so prepared migrate in agarose gel as a single ladder. Furthermore, this ladder of topoisomers comigrates with the trailing ladder of the samples shown in Fig. 2, one of which was run again in the gel shown in Fig. 3 (right lane) for comparison. This experiment provides strong evidence that the trailing band of each doublet shown in Fig. 2 and the right lane of Fig. 3 is a negative-noded trefoil of a particular linking number. By implication then, the leading band of each doublet in the gel patterns depicted is a positive-noded trefoil of the same linking number.
Figure 3.

A single ladder of positively supercoiled topoisomers prepared from the negative-noded trefoil formed by treatment of negatively supercoiled pCC56cos with phage T4 DNA topoisomerase (left lane, T4). For comparison, the sample analyzed in lane 3 of Fig. 2, which was prepared by converting nicked trefoil formed by S. cerevisiae DNA topoisomerase II to the positively supercoiled form, was run again in the right lane (Sc). The drawings to the left and right schematically depict the sign of the knot nodes and that of crossings in the plectonemically wound supercoiled DNA. The single ladder of topoisomers in the left lane comigrate with the trailing ladder of topoisomers in the right lane.
Effect of Supercoiling on the Electrophoretic Mobilities of Chiral DNA Knots.
The above results suggest that positive supercoiling makes positive-noded trefoil topoisomers migrate faster than their negative-noded counterparts. By symmetry, negative supercoiling is expected to do the opposite. To test this prediction, a nicked DNA trefoil preparation containing both chiral forms was first treated with DNA ligase and then relaxed by the vaccinia virus topoisomerase in the presence of ethidium. Ethidium was removed to introduce negative supercoils into the trefoils, and the sample was then analyzed by agarose gel electrophoresis (Fig. 4, right lane). For comparison, a sample of a negative-noded trefoil of the same DNA was similarly treated and analyzed in the same gel (Fig. 4, left lane). Clearly, negative supercoiling makes the negative-noded trefoil topoisomers migrate faster than their positive-noded counterparts.
Figure 4.

Negatively supercoiled topoisomers of the negative-noded trefoil prepared by phage T4 DNA topoisomerase (left lane, T4), and from knotting of nicked DNA by yeast DNA topoisomerase II (right lane, Sc). Samples identical to those analyzed in Fig. 3 were first relaxed by vaccinia virus topoisomerase in the presence of calculated amounts of ethidium bromide, and ethidium was removed prior to electrophoresis to give the negatively supercoiled topoisomers. The single ladder of topoisomers in the left lane sample now comigrate with the leading ladder in the right lane sample.
Ligation of an Achiral Knot Results in a Single Topoisomer Distribution.
The above results provide strong evidence that the presence of two distinct distributions of a supercoiled trefoil is due to the presence of two enantiomeric trefoil knots that interact differently with the writhe introduced by supercoiling. Each ladder represents topoisomers of a particular chiral trefoil that differ in their linking numbers. Therefore, two topoisomer ladders are observed with a mixture of positive- and negative-noded trefoils, but not with negative-noded trefoil molecules alone, or with unknotted rings. As a further confirmation of this requirement, we examined the figure-eight knot, an achiral knot with two positive and two negative nodes (see the line portion of Fig. 5). Because of its lack of chirality, the interpretation discussed above would predict that ligation of a nicked figure-eight knot should give rise to a single ladder of topoisomers of different linking numbers. A nicked figure-eight knot was obtained by treatment of the nicked 5.6-kb unknotted plasmid DNA with yeast DNA topoisomerase II and AMPPNP, and the nicked form was converted to the positively supercoiled form as before. Electrophoresis of the positively supercoiled figure-eight knot in agarose gel yields a single ladder of topoisomers, as predicted (Fig. 5).
Figure 5.
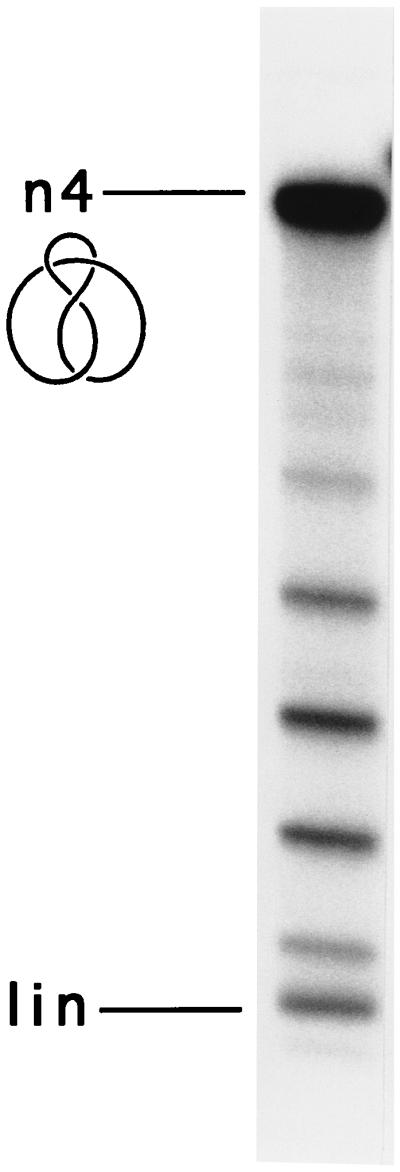
Singlet topoisomer distribution resulting from ligation of the nicked figure-eight knot. The figure-eight knot was prepared by the action of S. cerevisiae DNA topoisomerase II on nicked DNA when AMPPNP was added. n4, nicked figure-eight knot; an idealized drawing of the knot is shown on the left. lin, linear DNA.
Chirality of Knots Produced by Treatment of a Supercoiled DNA with a Type II DNA Topoisomerase Reflects Retention of Supercoil Nodes.
The preferential formation of negative-noded twist knots in the phage T4 topoisomerase-catalyzed reaction has been attributed to the retention of negative nodes in the DNA substrate: a subset of crossovers in the plectonemically wound and negatively supercoiled DNA are converted to negative knot nodes in the products (21). In addition to catalyzing the passage of one duplex DNA through another, a type II DNA topoisomerase may also effect the retention of positive or negative nodes by binding to crossovers of either sign (22). In the case of yeast DNA topoisomerase II, retention of nodes in supercoiled DNA substrates is readily detected in low salt media containing magnesium ions (23). These findings predict that predominantly positive- and negative-noded twist knots, including the trefoils, can be obtained from positively and negatively supercoiled DNA, respectively, by treatment with a molar excess of a eukaryotic type II DNA topoisomerase.
Fig. 6 depicts the formation of trefoil knots under several sets of conditions. As expected, trefoils that were derived from positively supercoiled DNA, either by treatment with yeast DNA topoisomerase II and AMPPNP, or by treatment with phage T4 DNA topoisomerase in the absence of ATP, are predominantly positive-noded: positively supercoiled trefoil topoisomers migrate with the leading ladder of the positively supercoiled topoisomer doublets (compare the patterns shown in lanes 1 and 3 with that shown in lane 2). Control experiments suggest that the minor negative-noded trefoil bands in the samples analyzed in lanes 1 and 3 of Fig. 6 were formed from a small amount of DNA that became relaxed during the topoisomerase knotting reactions (data not shown).
Figure 6.

The sign of crossovers in a plectonemically wound supercoiled DNA determines the sign of the trefoil produced by DNA topoisomerases. All trefoil topoisomers were made to be positively supercoiled under electrophoresis conditions. Samples run in lanes 2 and 6 were identical to that run in the lane designated Sc in Fig. 3 and contained positively supercoiled trefoils prepared from nicked trefoils formed by the action of S. cerevisiae DNA topoisomerase II on nicked unknotted DNA rings. These samples serve as markers for the positions of negative- and positive-noded trefoil topoisomers. Lanes: 1, trefoil formed by treatment of positively supercoiled pCC56cos with S. cerevisiae DNA topoisomerase II and AMPPNP; 3, trefoil formed by treatment of positively supercoiled pCC56cos with T4 DNA topoisomerase; 4, topoisomers of unknotted pCC56cos; 5, trefoil formed by treatment of negatively supercoiled pCC56cos with S. cerevisiae DNA topoisomerase II and AMPPNP. nc, nicked circle; n3, nicked trefoil; lin, linear DNA.
In contrast, trefoils prepared from negatively supercoiled DNA are predominantly negative-noded, and yielded positively supercoiled topoisomers that migrate with the trailing ladder of doublets (compare the pattern shown in lane 5 with those shown in lanes 2 and 6). In lane 4, positively supercoiled topoisomers of the 5.6-kb unknotted DNA are shown. The spacing between adjacent topoisomers is not significantly different for trefoils of either chirality and the unknotted DNA ring.
Chiral Knot Formation by Yeast DNA Topoisomerase II.
Inspection of the double-ladder pattern of topoisomers derived from a randomly cyclized trefoil (Fig. 2, lanes 1 and 2) suggests that the intensities of each pair of bands in a doublet are about the same; this equality is confirmed by the densitometric tracing of the distribution (Fig. 7a). This demonstrates that the probabilities of forming positive- and negative-noded trefoils are about the same when the ends of a linear DNA are joined. Identical results were also obtained with a 5.7-kb plasmid containing the LEU2 gene of yeast and the replication origin of the yeast 2-μ plasmid (results not shown).
Figure 7.

Densitometric tracing of positively supercoiled trefoil topoisomers. (a) Trefoil was produced by cyclization of linear pCC56cos in the presence of 7.5 mM Mg(II). The tracing shows the electrophoretic resolution of positive-noded trefoils (+) and negative-noded trefoils (−), and that the two forms were produced in equal amounts. (b) Trefoil was produced by treatment of nicked pCC56cos with S. cerevisiae DNA topoisomerase II in the presence of 7.5 mM magnesium ions. The production of negative-noded trefoils (−) was favored by approximately a factor of 2 over that of positive-noded trefoils (+). N3, nicked trefoil; L, linear DNA. Gel slabs were dried and analyzed in a PhosphorImager (Molecular Dynamics); quantitation of radioactivity in the bands was done with Molecular Dynamics imagequant software, version 3.15.
In contrast to the production of equal amounts of both chiral forms of trefoils when a linear DNA is circularized, unequal amounts of the two chiral forms were formed by yeast DNA topoisomerase II acting on a nicked DNA ring. In the experiment depicted in Fig. 2 (lane 3), AMPPNP was added to a mixture of nicked pCC56cos and a 10-fold molar excess of yeast DNA topoisomerase II, and the resulting nicked trefoils were gel purified and converted to the positively supercoiled form for the separation of the two chiral forms by agarose gel electrophoresis. A densitometric tracing of the topoisomer distribution of the sample analyzed in lane 3 of Fig. 2 is shown in Fig. 7b. Clearly, the formation of the negative-noded trefoil is favored by approximately a factor of 2 over the positive-noded one.
DISCUSSION
Several conclusions can be drawn from the results presented above. First, whereas nicked trefoil of either chirality has the same electrophoretic mobility in agarose gel, this equivalence breaks down when the positive- and negative-noded trefoils are supercoiled. Positive supercoiling increases the mobility of a positive-noded trefoil more than it does that of a negative-noded one; similarly, negative supercoiling results in a preferential increase in the electrophoretic mobility of the negative-noded trefoil. These preferential increments can be attributed to the writhe in each trefoil lobe imposed by the topology of the knot. An idealized positive-noded trefoil, for example, can be viewed as one with three equivalent lobes, each with a positive writhe (see Fig. 1d). Positive writhe in such a molecule would be favored by both its knotted configuration as well as by positive supercoiling. In contrast, in a negative-noded trefoil the knot topology would oppose the contribution of positive supercoiling to the overall writhe of the molecule.
Second, we found that equal amounts of the two chiral trefoils are formed upon cyclization of a linear DNA. This suggests a lack of preferential writhe in the DNAs used. In other words, a nicked trefoil with loops of positive writhe and its mirror image with loops of negative writhe appear to have the same unfavorable free energy relative to the nicked DNA ring in its unknotted form. This equality holds over a wide range of magnesium ion concentrations, suggesting that the well-defined crossover structures formed by a pair of closely packed DNA double helices, such as the one illustrated in Fig. 1c, do not significantly influence the chirality of nicked DNA trefoils in solution.
Third, whereas cyclization of a linear DNA yields equal amounts of positive- and negative-noded trefoils, knotting of a nicked DNA ring by a molar excess of yeast DNA topoisomerase II preferentially produces negative-noded trefoils. In principle, a DNA-bound type II topoisomerase could catalyze preferentially the inversion of a positive rather than a negative node to yield a product containing more negative nodes than positive ones. Bacterial gyrase, for example, imposes a right-handed writhe in a DNA segment wrapped around it; as a result, a positive node is preferentially inverted in each reaction cycle, leading to negative supercoiling of a DNA ring (24). Analysis of linking number changes induced in a relaxed DNA by yeast DNA topoisomerase II, however, indicates that the enzyme has no bias in the inversion of nodes of different signs: upon the addition of AMPPNP, the linking number of a relaxed DNA was observed to increase or decrease with about the same probability (25). Therefore, the preponderance of the negative-noded trefoil in the knotting reaction by the yeast enzyme is more likely a result of preferential stabilization of negative nodes by nonproductive binding of the enzyme molecules. Eukaryotic DNA topoisomerase II is known to bind to crossovers, especially in media of relatively low salt concentration (22, 23); this type of binding appears to be nonproductive, however, since the binding of AMPPNP to an enzyme in this state does not trigger a DNA transport event (23).
Our results also raise the possibility of using the relative abundance of the two chiral trefoils of a nicked DNA ring to detect intrinsic or ligand induced writhe in a DNA segment. For instance, in the crystal structure of the complex between the TATA binding protein and DNA containing its recognition sequence, the DNA was found to assume the geometry of a writhing bend, with a compensatory untwisting of the bound DNA (26, 27). Measurements of linking number changes of a DNA ring relaxed in the presence and absence of bound TATA binding protein are incapable of detecting such protein-induced compensatory distortions in DNA (28). The presence of a writhing bend in a linear DNA, however, may favor the formation of one chiral trefoil over the other when the DNA is cyclized, even in the presence of a compensatory change in twist. At present there is no quantitative information on the free energy difference between a positive-noded and a negative-noded nicked trefoil if the DNA contains a segment with a fixed writhe, but experiments with DNA–protein complexes of known structures, and/or Monte Carlo calculations, should provide the required data to assess whether the difference is sufficiently large to be useful in solution measurements of DNA writhe.
Acknowledgments
We thank many of our colleagues for materials and S. Field and M. Greenberg at the Harvard Medical School for the use of their PhosphorImager. This work was supported by U.S. Public Health Services Grant NIH GM24544 and a predoctoral fellowship (to S.Y.S.) from the Howard Hughes Medical Institute.
ABBREVIATION
- AMPPNP
5′-adenylylimidodiphosphate
References
- 1.Timsit Y, Westhof E, Fuchs R P P, Moras D. Nature (London) 1989;341:459–462. doi: 10.1038/341459a0. [DOI] [PubMed] [Google Scholar]
- 2.Timsit Y, Vilbois E, Moras D. Nature (London) 1991;354:167–170. doi: 10.1038/354167a0. [DOI] [PubMed] [Google Scholar]
- 3.Heinemann U, Alings C, Bansal M. EMBO J. 1992;11:1931–1939. doi: 10.1002/j.1460-2075.1992.tb05246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baikalov I, Grzeskowiak K, Yanagi K, Quintana J, Dickerson R E. J Mol Biol. 1993;231:768–784. doi: 10.1006/jmbi.1993.1325. [DOI] [PubMed] [Google Scholar]
- 5.Lipanov A, Kopka M L, Kaczor-Grzeskowiak M, Quintana J, Dickerson R E. Biochemistry. 1993;32:1373–1389. doi: 10.1021/bi00056a024. [DOI] [PubMed] [Google Scholar]
- 6.Goodsell D S, Grzeskowiak K, Dickerson R E. Biochemistry. 1995;34:1022–1029. doi: 10.1021/bi00003a037. [DOI] [PubMed] [Google Scholar]
- 7.Duckett D R, Murchie A I H, Diekmann S, von Kitzing E, Kemper B, Lilley D M J. Cell. 1988;55:79–89. doi: 10.1016/0092-8674(88)90011-6. [DOI] [PubMed] [Google Scholar]
- 8.Murchie A I H, Clegg R M, von Kitzing E, Duckett D R, Diekmann S, Lilley D M J. Nature (London) 1989;341:763–766. doi: 10.1038/341763a0. [DOI] [PubMed] [Google Scholar]
- 9.Duckett D R, Murchie A I H, Lilley D M J. EMBO J. 1990;9:583–590. doi: 10.1002/j.1460-2075.1990.tb08146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lilley D M J, Clegg R M. Annu Rev Biophys Biomol Struct. 1993;22:299–328. doi: 10.1146/annurev.bb.22.060193.001503. [DOI] [PubMed] [Google Scholar]
- 11.Shaw S Y, Wang J C. Science. 1993;260:533–536. doi: 10.1126/science.8475384. [DOI] [PubMed] [Google Scholar]
- 12.Hsieh T, Wang J C. Biochemistry. 1975;14:527–535. doi: 10.1021/bi00674a011. [DOI] [PubMed] [Google Scholar]
- 13.Hsieh T. J Biol Chem. 1983;258:8413–8420. [PubMed] [Google Scholar]
- 14.Kreuzer K N, Jongeneel C V. Methods Enzymol. 1983;100:144–160. doi: 10.1016/0076-6879(83)00051-8. [DOI] [PubMed] [Google Scholar]
- 15.Parker R C, Watson R M, Vinograd J. Proc Natl Acad Sci USA. 1977;74:851–855. doi: 10.1073/pnas.74.3.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shortle D, Grisafi P, Benkovic S J, Botstein D. Proc Natl Acad Sci USA. 1982;79:1588–1592. doi: 10.1073/pnas.79.5.1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shuman S, Golder M, Moss B. J Biol Chem. 1988;263:16401–16407. [PubMed] [Google Scholar]
- 18.Ostrander E O, Benedetti P, Wang J C. Science. 1990;249:1261–1265. doi: 10.1126/science.2399463. [DOI] [PubMed] [Google Scholar]
- 19.Rybenkov V V, Cozzarelli N R, Vologodskii A V. Proc Natl Acad Sci USA. 1993;90:5307–5311. doi: 10.1073/pnas.90.11.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Depew R E, Wang J C. Proc Natl Acad Sci USA. 1975;72:4275–4279. doi: 10.1073/pnas.72.11.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasserman S A, Cozzarelli N R. J Biol Chem. 1991;266:20567–20573. [PubMed] [Google Scholar]
- 22.Osheroff N, Brutlag D L. In: Mechanism of DNA Replication and Recombination. Cozzarelli N R, editor. New York: Liss; 1983. pp. 55–64. [Google Scholar]
- 23.Roca J, Berger J M, Wang J C. J Biol Chem. 1993;268:14250–14255. [PubMed] [Google Scholar]
- 24.Liu L F, Wang J C. Proc Natl Acad Sci USA. 1978;75:2098–2102. doi: 10.1073/pnas.75.5.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roca J, Wang J C. Genes Cells. 1996;1:17–27. doi: 10.1046/j.1365-2443.1996.01001.x. [DOI] [PubMed] [Google Scholar]
- 26.Kim Y, Geiger J H, Hahn S, Sigler P B. Nature (London) 1993;365:512–520. doi: 10.1038/365512a0. [DOI] [PubMed] [Google Scholar]
- 27.Kim J L, Nikolov D B, Burley S K. Nature (London) 1993;365:520–527. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]
- 28.Lorch Y, Kornberg R. Mol Cell Biol. 1993;13:1872–1875. doi: 10.1128/mcb.13.3.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]