

Abstract
Mouse genetic resources include inbred strains, recombinant inbred lines, chromosome substitution strains, heterogeneous stocks, and the Collaborative Cross (CC). These resources were generated through various breeding designs that potentially produce different genetic architectures, including the level of diversity represented, the spatial distribution of the variation, and the allele frequencies within the resource. By combining sequencing data for 16 inbred strains and the recorded history of related strains, the architecture of genetic variation in mouse resources was determined. The most commonly used resources harbor only a fraction of the genetic diversity of Mus musculus, which is not uniformly distributed thus resulting in many blind spots. Only resources that include wild-derived inbred strains from subspecies other than M. m. domesticus have no blind spots and a uniform distribution of the variation. Unlike other resources that are primarily suited for gene discovery, the CC is the only resource that can support genome-wide network analysis, which is the foundation of systems genetics. The CC captures significantly more genetic diversity with no blind spots and has a more uniform distribution of the variation than all other resources. Furthermore, the distribution of allele frequencies in the CC resembles that seen in natural populations like humans in which many variants are found at low frequencies and only a minority of variants are common. We conclude that the CC represents a dramatic improvement over existing genetic resources for mammalian systems biology applications.
Keywords: Parental Strain, Inbred Strain, Recombinant Inbred, Collaborative Cross, Chromosome Substitution Strain
Introduction
Since the derivation of the original inbred mouse strains from populations of fancy mice to investigate the genetic basis of cancer (reviewed in Paigen 2003), many additional inbred strains have been derived that harbor a tremendous amount of natural genetic variation (Beck et al. 2000; Ideraabdullah et al. 2004). However, unlike the more recently produced wild-derived strains, the vast majority of commonly used inbred strains trace their ancestry to the original mouse-fancier populations. An analysis of the genomes of extant inbred strains was recently made possible using data from a 15-strain resequencing project (http://www.mouse.perlegen.com/mouse/download.html), which revealed that the most widely used laboratory inbred strains are not random composites of the three main mouse subspecies (Mus musculus domesticus, M. m. musculus, and M. m. castaneus), but have a remarkably high level of shared ancestry largely contributed by the M. m. domesticus subspecies (Yang et al. 2007). Since many of the original inbred strains are also the most widely used in biomedical and laboratory research, the architecture of the genetic variation in derived resources is highly dependent on the interconnected and complex breeding histories of the progenitor inbred strains (Lyon et al. 1996).
Over the last fifty years, numerous genetic resources have been devised and developed for specific purposes using a variety of inbred strains as progenitors (reviewed in Silver 1995). The major genetic resources that are widely used currently include recombinant inbred (RI) lines (Bailey 1971; Broman 2005), recombinant congenic strains (RCS) (Demant and Hart 1986), genome-tagged or congenic (CON) lines (Iakoubova et al. 2001), chromosome substitution strains (CSS) (Hudgins et al. 1985; Nadeau et al. 2000), heterogeneous stocks (HS) (Hitzemann et al. 1994), and, more recently, Laboratory Strain Diversity Panels (LSDP) drawn from the Mouse Phenome Project (Paigen and Eppig 2000) for association studies.
Although the major use conceptualized for RI lines was linkage analysis (Bailey 1971), with the expanded sizes of many RI panels they are now being used to support analysis of more complex polygenic traits (Markel et al. 1996; Williams et al. 2001). Similarly, CSS and LSDP resources are being used for the genetic analysis of polygenic traits. CSS have a simplified genetic structure with only one chromosome differing between a single CSS and the parental recipient strain, a characteristic not shared with the other resources (Nadeau et al. 2000). The HS are significantly different than RI lines or CSS in that they typically contain multiple inbred strain progenitors, which potentially increases the level of genetic diversity represented in the resource (Yalcin et al. 2005). The LSDP were recently envisioned to adapt many of the whole-genome association technologies being developed by the human genetics community (Grupe et al. 2001; Bogue and Grubb 2004; Liao et al. 2004; Pletcher et al. 2004; McClurg et al. 2007; Payseur and Place 2007). In theory, the LSDP should encompass large amounts of variation, but in practice, since analyses of LSDP resources has largely been limited to panels of classical inbred strains, the diversity is most likely restricted to M. m. domesticus. Similar to LSDP resources, a more recently developed resource called the Collaborative Cross (CC) was designed to incorporate large amounts of variation (Threadgill et al. 2002; Churchill et al. 2004; Valdar et al. 2006). The CC is a mammalian genetic reference population that was designed to have controlled randomization of genetic factors, which is essential for causal inference. The CC was designed as a panel of recombinant inbred lines derived from eight parental inbred strains through a mating scheme that minimizes unpredictable genomic interactions between strains and optimizes the contribution from each parental strain. The selection of the parental strains was based upon historical breeding records and suspected relationships drawn from sparse maps of genetic variation.
Herein we sought to reanalyze the structure of genetic variation present in various mouse genetic resources using genome resequencing data (http://www.mouse.perlegen.com/mouse/download.html). We found that the vast majority of resources capture very small amounts of the existing variation and the variation that is captured is not randomly distributed. Unlike other resources, the CC has a high level of variation capture that is normally distributed across the genome. This structure is similar to that found in humans and other randomly breeding mammalian species, showing that the CC is an ideal model for systems biology analyses.
Materials and methods
Genotype data
All genotype data used in this study were obtained from the National Institute of Environmental Health Science’s “Resequencing and SNP Discovery Project” (http://www.niehs.nih.gov/crg/cprc.htm). These data contain over 109 million genotypes that identified 8.3 million SNPs spanning the 19 autosomes, the sex chromosomes, and the mitochondrial genome (http://www.mouse.perlegen.com/mouse/download.html). The 15 resequenced strains include 11 classical inbred strains (129S1/SvImJ, A/J, AKR/J, BALB/cBy, C3H/HeJ, DBA/2J, FVB/NJ, NOD/LtJ, BTBR T + tf/J, KK/HlJ, and NZW/LacJ) and four wild-derived strains (WSB/EiJ, PWD/PhJ, CAST/EiJ, and MOLF/EiJ), representing the M. m. domesticus, M. m. musculus, M. m. castaneus subspecies and M. m. molossinus, a subspecies that arose by natural hybridization between M. m. musculus and M. m. castaneus (http://www.jax.org). In addition, the genotypes of the fully sequenced and annotated C57BL/6J genome were used. Incomplete genotypes were imputed as described previously (Roberts et al. 2007).
Mouse genetic resources
One example was chosen from each of the five major types of resources based on widespread or potential use. In all cases the example represented the maximal amount of diversity captured among similar resources. The BXD, derived from C57BL/6J and DBA/2J by B. Taylor, L. Silver, and R. Williams, was chosen as the prototypical RI line panel because of its past and current popularity (Taylor 1978; Peirce et al. 2004). The representative chromosome substitution strain panel was B.P generated by J. Forejt, which has PWD/Ph chromosomes introgressed into the C57BL/6J background. The Northport HS derived from A/J, AKR/J, BALBc/J, CBA/J, C3H/HeJ, C57BL/6J, DBA/2J, and LP/J was used as the example of heterogeneous stock (Hitzemann et al. 1994). The Collaborative Cross is an RI line panel produced from the eight parental inbred strains A/J, C57BL6/J, 129S1/SvImJ, NOD/LtJ, NZO/HlLtJ, CAST/EiJ, PWK/PhJ, and WSB/EiJ (Threadgill et al. 2002; Churchill et al. 2004). Finally, since the emergence of the Mouse Phenome Project (Paigen and Eppig 2000), several panels of inbred strains have been considered for association studies (Bogue and Grubb 2004; Liao et al. 2004; McClurg et al. 2007; Payseur and Place 2007). The LSDP described by Payseur and Place was used as a representative because it is composed only of classical inbred strains, including A/J, AKR/J, BALB/cByJ, BTBR T +tf/tf, BUB/BnJ, CBA/J, CE/J, C3H/HeJ, C57BL/6J, C57BLKS/J, C57L/J, C57BR/cdJ, C58/J, DBA/2J, FVB/NJ, I/LnJ, KK/HIJ, LP/J, MA/MyJ, NOD/LtJ, NON/LtJ, NZB/B1NJ, NZW/LacJ, PL/J, RIIIS/J, SEA/GnJ, SJL/J, SM/J, SWR/J, and 129S1/SvImJ. Other inbred panels that also include wild-derived strains have not been useful for association mapping because of the large number of private polymorphisms contributed by strains derived from other subspecies.
Strain substitutions
Estimates of the polymorphism diversity captured by each resource represent best-case scenarios since they assume all diversity present in the parental strains is captured by the derived resources. Genetic diversity can be estimated directly in the BXD RI and the B.P CSS because the parental strains have been sequenced. In the remaining resources it was necessary to substitute sequenced strains for those that have not been sequenced. These substitutions were based on genetic similarity estimated using genotypes at SNPs distributed along the entire genome (Petkov et al. 2004). Five of the parental strains in the Northport HS have been sequenced and include A/J, AKR/J, C3H/HeJ, C57BL/6J, and DBA/2J. The remaining three strains were substituted by a sister substrain (BALBc/J was substituted by BALB/cBy), a related strain (LP/J was substituted by BTBR T + tf/J), or a Castle strain that will overestimate the diversity present in this panel (CBA/J was substituted by NZW/LacJ). Six of the parental strains in the Collaborative Cross have been sequenced: 129S1/SvImJ, A/J, C57BL/6J, NOD/LtJ, WSB/EiJ, and CAST/EiJ. The remaining two strains were substituted by strains from similar origins (NZO/HlLtJ was substituted by NZW/LacJ and PWK/PhJ was substituted by PWD/PhJ). Finally, for the LSDP we used all 12 classical inbred strains plus WSB/EiJ. Although the number of strains used for our analyses is significantly lower than in the original panel (Payseur and Place 2007), the WSB/EiJ strain is a larger contributor to the diversity than any single classical strain or group of classical inbred strains combined (Yang et al. 2007), suggesting that this will be an accurate representation of existing panels.
Results
The genetic diversity captured in the major mouse genetic resources depends on the number and identity of parental strains involved in their derivation, as well as the breeding design used to generate the resource (Fig. 1). With the resequencing of the mouse genome, there are new insights into the single nucleotide polymorphism (SNP) architecture captured by widely used mouse genetic resources. However, since the mouse genome resequencing project did not include every parental strain used in common genetic resources, we conservatively replaced the nonsequenced strains by an appropriate substitute, ensuring that our analysis of the SNP architecture does not underestimate the actual diversity present in existing resources. Resequencing to estimate the false-positive and false-negative rate in the Perlegen data has been reported (Yang et al. 2007). The missing variation is for the most part randomly distributed. However, resources such as the CC that include wild-derived strains will have underestimates of the true variation captured, while those lacking wild-derived strains will have overestimates because of the high false-negative SNP call rate in wild-derived strains. In all analyses, we considered that a polymorphic variant was captured if the two alleles are represented among the parental strains of a particular mouse genetic resource. However, it should be noted that the diversity present in the founder population for each resource represents the upper bound of diversity that can be captured by the derived resource. The actual diversity captured may be lower, particularly in small resources, due to genetic drift during generation of the resource. The color scheme used for the classical inbred strains in Fig. 1 reflects data that indicate that their genomes are largely derived from M. m. domesticus as recently determined (Yang et al. 2007).
Fig. 1.
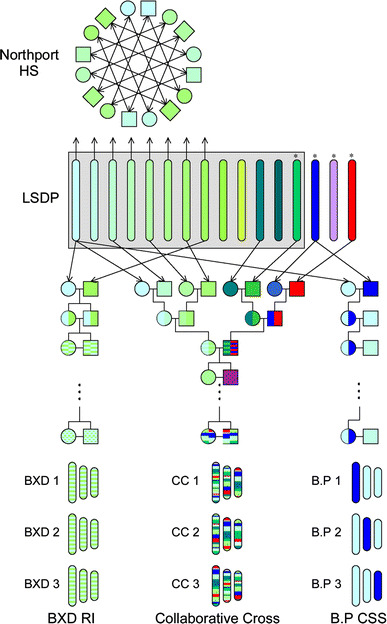
Parental strains and derivation of five major types of mouse genetic resources. Each of the sequenced strains is shown in a different color depending on the origin. The four wild-derived strains, denoted by asterisks, are CAST/EiJ (M. m. cataneus) in red, PWD/PhJ (M. m. muculus) in blue, MOLF/EiJ (M. m. molossinus) in purple, and WSB/EiJ (M. m. domesticus) in green. The remaining 12 classical laboratory strains are shown in green reflecting the predominant contribution of the M. m. domesticus subspecies to these strains (Yang et al. 2007). The shade of green denotes the different origin of the classical strains, with the darker shades denoting strains of Swiss origin (FVB/NJ and NOD/LtJ), the yellow-green denoting a strain of Asian origin (KK/HlJ), and intermediate shade denoting Castle or C57-related strains (129S1/SvImJ, A/J, AKR/J, BALB/cBy, C3H/HeJ, DBA/2J, BTBR T + tf/J, and NZW/LacJ) (Beck et al. 2000). The figure also shows schematically the derivation process for five types of resources, recombinant inbred lines (BXD); chromosome substitution strains (B.P), Collaborative Cross (CC), heterogeneous stocks (Northport HS), and laboratory strain diversity panel (LSDP)
Diversity captured is a function of the number of parental strains
Most resources used in genetic studies are derived from crosses involving two parental strains or multiples thereof in order to introduce equivalent variation from each parental strain. Therefore, we used the mouse genome resequencing data to determine the range (maximum, minimum, and average) of diversity captured in any theoretical resource involving any 2, 4, 8, and 16 parental strains (Fig. 2). As expected, on average the diversity captured increases with the number of parental strains involved. However, there is an extremely wide variation in the level of diversity captured within a given number of parental strains and a large overlap between the diversity that can be captured in resources with different number of parental strains. Our analysis reveals that the CC outperforms all combinations of two or four parental strains. However, an optimal set of fours strains would capture a similar, albeit lower, level of genetic diversity as what is present in the CC. We conclude that although the number of strains is an important factor in determining the level of diversity captured in a given resource, other factors such as the identity of the parental strains are of much greater consequence. This is illustrated by comparing the B.P CSS (two parental strains) with the Northport HS (eight parental strains). Because all Northport HS parental strains have a common ancestry, they contribute a relatively small amount of additional variation per strain. Conversely, because the two parental strains of the B.P CSS represent different subspecies, they capture over half of the known polymorphic sites within the mouse genome. Similarly, when the Northport HS is compared with the CC (also derived from eight parental strains), the level of diversity is almost threefold more in the CC (36% vs. 89%). This is expected since the CC has at least one representative from all three subspecies. The CC captures 89% of the variation in the mouse genome, which is close to the maximal amount of variation that can be captured by eight strains (97% by 129S1/SvImJ, CAST/EiJ, DBA/2J, FVB/NJ, KK/HIJ, MOLF/EiJ, PWD/PhJ, and WSB/EiJ).
Fig. 2.
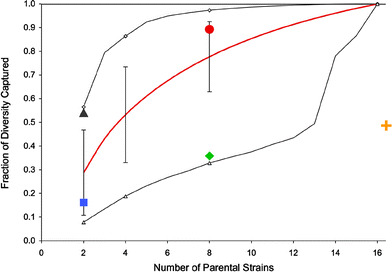
Genetic diversity captured as a function of the number of parental strains. Depicted are the ranges of genetic diversity that can be captured in resources with varying numbers of contributing parental strains based on the NIEHS resequencing data. The red line represents the average diversity captured and vertical bars represent the standard deviation. Open diamonds and open triangles represent the maximum and minimum diversity captured by 2, 4, 8, and 16 parental strains, respectively. In addition, the diversity captured in the BXD RI (blue square), the B.P CSS (gray triangle), the Northport HS (green diamond), the Collaborative Cross (red circle), and the LSDP (orange cross) is shown
Diversity captured is a function of the subspecific origin of the parental strains
A recent analysis of the mouse genome resequencing data demonstrates that over 92% of the genome of classical inbred strains is derived from the M. m. domesticus subspecies, and, unexpectedly, approximately 75% of the genome of MOLF/EiJ is of M. m. musculus origin (Yang et al. 2007). Based on these observations, it is possible to assign each of the 16 sequenced strains to a major subspecies (see Fig. 1 for assignments). After plotting the fraction of genetic diversity captured by strain sets of a given size (Fig. 3), it is clear that the distributions are multimodal. Furthermore, each lobe in these distributions perfectly clusters according to the number of subspecies represented among the parental strains (indicated by different shades of purple in Fig. 3). This indicates that the number of subspecies contributing to a particular resource is the major determinant of the level of genetic variation captured. This analysis also shows that the fraction of diversity captured by most existing resources is small, particularly those that have only one contributing subspecies like the BXD RI or Northport HS. We also analyzed other resources and found that the conclusions reached for BXD RI apply to other RI panels such as AXB/BXA (C57BL/6J and A/J), CXB (BALB/cByJ and C57BL/6J), AKXD (AKR/J and DBA/2J), and BXH (C57BL/6J and C3H/HeJ) (Table 1). Similarly, CSS derived from the introgression of A/J or 129S1/ImJ chromosomes into the C57BL/6J background (Nadeau et al. 2000) or the Boulder HS derived from C57BL/6, BALB/c, RIII, AKR, DBA/2, I, A/J, and C3H leads to similar results. While these genetic resources capture little variation because all of these strains are derived from the M. m. domesticus subspecies, the B.P CSS, B6.CAST CON, and LSDP fair better since they have representatives from two subspecies, M. m. domesticus and M. m. musculus or M. m. castaneous. This analysis also explains why the CC, with all three subspecies represented, dramatically outperforms other genetic resources in capturing genetic diversity.
Fig. 3.
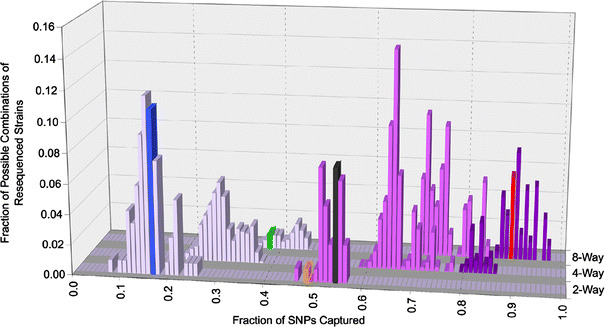
Genetic diversity captured as a function of the number and origin of parental strains. The individual diversity captured by every possible combination of two, four, and eight parental strains that can be generated among the sequenced strains is shown. The increasing number of subspecies (1–3) represented among the parental strains is denoted by an increasingly darker shade of purple. The diversity captured in the model resources is shown in their respective color as described in Fig. 2 (BXD RI, blue; B.P CSS, gray; Northport HS, green; LSDP, orange; CC, red). The LSDP is shown in the two-way cross for simplicity since there are more than eight strains involved
Table 1.
Genetic variation captured by widely accessible mouse genetic resources
| Name | Type | Parental strains | % of variation captured | Reference |
|---|---|---|---|---|
| BXD | RI | C57BL/6, DBA/2 | 16 | Taylor 1978; Peirce et al. 2004 |
| AKXD | RI | AKR, DBA/2 | 14 | Mucenski et al. 1986 |
| CXB | RI | BALB/cBy, C57BL/6 | 14 | Dux et al. 1978 |
| AXB/AXB | RI | C57BL/6, A | 15 | Nesbitt and Skamene 1984 |
| BXH | RI | C57BL/6, C3H/He | 16 | Watson et al. 1977 |
| CC | RI | C57BL/6, 129S1, NOD, A, NZO/HI, CAST, PWK, WSB | 89 | Threadgill et al. 2002; Churchill et al. 2004 |
| B.P | CSS | C57BL/6, PWD | 54 | J Forejt, personal communication |
| B.A | CSS | C57BL/6, A | 15 | Nadeau et al. 2000 |
| B.129 | CSS | C57BL/6, 129S1 | 16 | Nadeau, under development |
| B6.D2 | CON | C57BL/6, DBA/2 | 16 | Iakoubova et al. 2001 |
| B6.CAST | CON | C57BL/6, CAST | 51 | Iakoubova et al. 2001 |
| Northport | HS | C57BL/6, BALB/c, CBA, AKR, DBA/2, LP, A, C3H/He | 36 | Hitzemann et al. 1994 |
| Boulder | HS | C57BL/6, BALB/c, RIII, AKR, DBA/2, I, A, C3H | 36 | McClearn et al. 1970 |
| AcB | RCS | C57BL/6, A | 15 | Fortin et al. 2001 |
| BcA | RCS | C57BL/6, A | 15 | Fortin et al. 2001 |
| CcS | RCS | BALB/c, STS/A | 13 | Groot et al. 1992 |
| HcB | RCS | C3H, C57BL/10 | 16 | Demant and Hart 1986 |
| Diversity panel | LSDP | 30 strains | 49 | Payseur and Place 2007 |
Spatial distribution of the diversity varies significantly among resources
In addition to the total diversity captured, it is critical to consider how the variation captured in each resource is distributed across the genome. When such analyses are performed (Fig. 4), they reveal that the BXD RI, Northport HS, and LSDP genetic resources show a multimodal complex distribution with many intervals capturing very little variation and a variable number of intervals capturing a larger fraction of the available variation. In contrast, the B.P CSS and the CC have unimodal distributions centered on their respective genome-wide means (Fig. 2). It is also evident that the CC outperforms all other resources in uniformly capturing a large fraction of the available genetic variation.
Fig. 4.
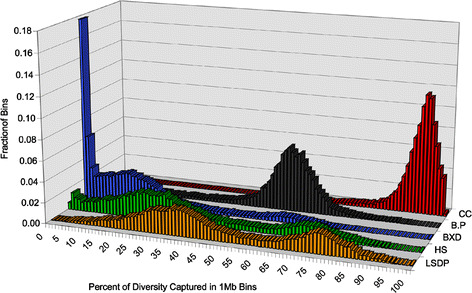
Frequency distribution of the genetic diversity captured in 1-Mb intervals across the entire genome. The percent of total SNPs captured in each interval was calculated for each resource before plotting the frequencies of total bins capturing similar levels of variation. The color scheme and the abbreviations are as described in Fig. 2 (BXD RI, blue; B.P CSS, gray; Northport HS, green; LSDP, orange; CC, red)
When the distribution of the variation captured is plotted in consecutive high-resolution intervals (Fig. 5), it becomes evident that only the CC maintains a uniformly high level of variation while all other resources vary dramatically from interval to interval. Such variation distributions destroy the uniformity required for systems biology analyses and leads to extended regions of blind spots with little or no variation in resources like the BXD RI panel. Most interestingly, blind spots are also present in the Northport HS, the B.P CSS, and the LSDP resources, although their locations vary among the resources. An important corollary is that blind spots are found in both gene-dense and gene-poor regions, creating potentially dramatic negative consequences when saturating the genome in the search for functional interactions among genes and phenotypes.
Fig. 5.
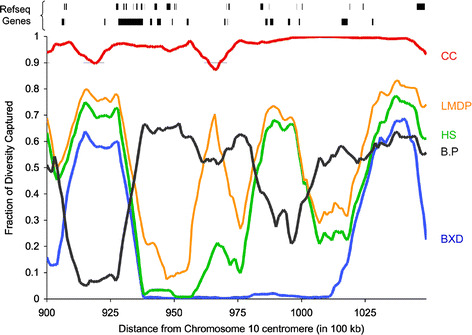
Genetic diversity captured in consecutive intervals in a 15-Mb region on mouse chromosome 10. The distribution of diversity captured by each resource is shown. Plots are generated from 1-Mb windows with 0.9-Mb overlap on mouse chromosome 10 from position 90 Mb to position 105 Mb. The location of Refseq genes is also shown (top). The color scheme and the abbreviations are as described in Fig. 2 (BXD RI, blue; B.P CSS, gray; Northport HS, green; LSDP, orange; CC, red)
Allele frequency of the variation captured
In addition to the level and distribution of the variation captured, the frequency of the minor alleles can impact the utility of a particular genetic resource. Therefore, to compare this characteristic among the different genetic resources, we determined the allele frequency present in the 8.3 million SNPs reported for the mouse genome resequencing project in the different resources considered in this study (Fig. 6). For reference we also added the distribution of allele frequencies reported for human populations (pink bars in Fig. 6) (Kruglyak and Nickerson 2001) and the fraction of SNPs that are not captured in each mouse genetic resource or that have very low allele frequency (≤1%) in humans. Resources fall into two distinct groups, with the BXD RI and B.P CSS having uniformly 50% allele frequency at the captured variants, as would occur with any resource that is equally derived from two parental strains. Conversely, the Northport HS, the LSDP, and the CC have a true distribution in which the fraction of SNPs captured decreases as the minor allele frequency increases. Among the latter group, the CC retains the most desirable distribution because the total number of variants with high minor allele frequency is significantly higher than that found in either the Northport HS or the LSDP. Interestingly, even though the CC is derived from only eight parental strains, the allele frequency distribution is remarkably similar to that observed in humans.
Fig. 6.

Minor allele frequency distribution. The frequency distribution of the minor SNPs in four equal quintiles is shown. The approximate frequency of human SNPs is shown in pink along with an additional class for SNPs with a minor allele frequency of zero (i.e., SNPs that are not informative in a given resource or those present at less than 0.01 frequency in humans) (Kruglyak and Nickerson 2001). The color scheme and the abbreviations are as described in Fig. 2 (BXD RI, blue; B.P CSS, gray; Northport HS, green; LSDP, orange; CC, red)
Discussion
The recent explosion in genetic variation data for mice made possible by the resequencing of 15 mouse inbred strains (http://www.niehs.nih.gov/crg/cprc.htm) allows us to accurately determine and compare the polymorphic architecture of different mouse genetic resources. The most widely used resources suffer from very low rates of polymorphism capture (all extant RI lines, RCS, and the B.A and B.129 CSS) or medium levels of polymorphism capture that is nonuniformly distributed (B.P CSS, B6.CAST CON, Northport, and Boulder HS, and the LSDP). Although the proportion of the genome being interrogated with these resources does not limit their use for discovering subsets of functional gene variants controlling specific phenotypes, it greatly impairs their utility for genome-wide systems biological analyses. In addition, differences in allele frequency among the resources impact the relative allele strength that can be detected, with a consequential effect on the number of functional gene variants that can be detected by a particular resource. The common ancestry, dominated by M. m. domesticus, of many the strains that have contributed to most mouse genetic resources has resulted in a dramatic reduction in the pool of available gene variants for genome-wide discovery and, more importantly, may complicate their use for systems-level analyses of mammalian biology that is dependent on high levels of uniformly distributed genetic variation.
The CC represents a resource that has optimal polymorphism architecture for system biological applications. In particular, the uniform distribution of the high level of variation captured is ideal to support global analysis of complex biological systems that is most efficiently achieved using experimental designs that employ multifactorial perturbations (Fisher 1935). Although the allele frequency distribution in the CC is not necessarily the best to detect the effects of any particular polymorphism, it is representative of natural populations and should outperform all resources for trait correlation analysis, which is the foundation of systems genetics, and all but the resources with only two parental strains in detection of specific gene functional variants. However, the resources with only two parental strains capture much lower levels of available polymorphisms, and the captured polymorphisms are not uniformly distributed, greatly reducing their genome-wide utility for systems biology applications. With the shift in complex trait gene discovery to humans that has been made possible by affordable high-density genotyping of large numbers of phenotyped individuals, the mouse will be taking a new role in biological research, that of a model to support mammalian systems biology investigations. Our analyses demonstrate that the CC represents a dramatic improvement over other genetic resources since it is the only resource that can serve this role based on the level, distribution, and allele frequency of captured polymorphisms. The overall performance of the CC is particularly remarkable given that the original choice of parental strains represented a compromise between the practical desire to take advantage of existing resources such as genome sequence, mapping panels, and ES cell lines and the ultimate goal of maximizing diversity (Churchill et al. 2004).
Acknowledgments
This work resulted from a collaborative effort by members of the UNC Computational Genetics Workgroup and was supported in part by the National Institute of General Medical Sciences as part of the Center of Excellence in Systems Biology (1P50 GM076468 to FPMV), the National Science Foundation (IIS 0448392 to WW), the Environmental Protection Agency (STAR RD832720 to WW), the Barry M. Goldwater Scholarship to AR, and the National Cancer Institute (1U01 CA105417 to DWT). Center support from the National Cancer Institute (5P30 CA016086), the National Institute of Environmental Health Sciences (2P30 ES010126), and the National Institute of Diabetes and Digestive and Kidney Diseases (5P30 DK034987) supported the collaborative environment.
References
- Bailey DW. Recombinant-inbred strains. An aid to finding identity, linkage, and function of histocompatibility and other genes. Transplantation. 1971;11(3):325–327. doi: 10.1097/00007890-197103000-00013. [DOI] [PubMed] [Google Scholar]
- Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, et al. Genealogies of mouse inbred strains. Nat Genet. 2000;24(1):23–25. doi: 10.1038/71641. [DOI] [PubMed] [Google Scholar]
- Bogue MA, Grubb SC. The Mouse Phenome Project. Genetica. 2004;122(1):71–74. doi: 10.1007/s10709-004-1438-4. [DOI] [PubMed] [Google Scholar]
- Broman KW. The genomes of recombinant inbred lines. Genetics. 2005;169(2):1133–1146. doi: 10.1534/genetics.104.035212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36(11):1133–1137. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Demant P, Hart AA. Recombinant congenic strains–a new tool for analyzing genetic traits determined by more than one gene. Immunogenetics. 1986;24:416–422. doi: 10.1007/BF00377961. [DOI] [PubMed] [Google Scholar]
- Dux A, Mühlbock O, Bailey DW. Genetic analyses of differences in incidence of mammary tumors and reticulum cell neoplasms with the use of recombinant inbred lines of mice. J Natl Cancer Inst. 1978;61:1125–1129. [PubMed] [Google Scholar]
- Fisher RA. The design of experiments. Edinburgh: Oliver and Boyd; 1935. [Google Scholar]
- Groot PC, Moen CJ, Dietrich W, Stoye JP, Lander ES, et al. The recombinant congenic strains for analysis of multigenic traits: genetic composition. FASEB J. 1992;6:2826–1835. doi: 10.1096/fasebj.6.10.1634045. [DOI] [PubMed] [Google Scholar]
- Fortin A, Diez E, Rochefort D, Laroche L, Malo D, et al. Recombinant congenic strains derived from A/J and C57BL/6J: a tool for genetic dissection of complex traits. Genomics. 2001;74:21–35. doi: 10.1006/geno.2001.6528. [DOI] [PubMed] [Google Scholar]
- Grupe A, Germer S, Usuka J, Aud D, Belknap JK, et al. In silico mapping of complex disease-related traits in mice. Science. 2001;292(5523):1915–1918. doi: 10.1126/science.1058889. [DOI] [PubMed] [Google Scholar]
- Hitzemann B, Dains K, Kanes S, Hitzemann R. Further studies on the relationship between dopamine cell density and haloperidol-induced catalepsy. J Pharmacol Exp Ther. 1994;271:969–976. [PubMed] [Google Scholar]
- Hudgins CC, Steinberg RT, Klinman DM, Reeves MJ, Steinberg AD. Studies of consomic mice bearing the Y chromosome of the BXSB mouse. J Immunol. 1985;134(6):3849–3854. [PubMed] [Google Scholar]
- Iakoubova OA, Olsson CL, Dains KM, Ross DA, Andalibi A, et al. Genome-tagged mice (GTM): two sets of genome-wide congenic strains. Genomics. 2001;74:89–104. doi: 10.1006/geno.2000.6497. [DOI] [PubMed] [Google Scholar]
- Ideraabdullah FY, de la Casa-Esperon E, Bell TA, Detwiler DA, Magnuson T, et al. Genetic and haplotype diversity among wild-derived mouse inbred strains. Genome Res. 2004;14(10A):1880–1887. doi: 10.1101/gr.2519704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27(3):234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- Liao G, Wang J, Guo J, Allard J, Cheng J, et al. In silico genetics: identification of a functional element regulating H2-Ealpha gene expression. Science. 2004;306(5696):690–695. doi: 10.1126/science.1100636. [DOI] [PubMed] [Google Scholar]
- Lyon MF, Rastan S, Brown SDM, editors. Genetic variants and strains of the laboratory mouse. Oxford: Oxford University Press; 1996. [Google Scholar]
- Markel PD, Bennett B, Beeson MA, Gordon L, Simpson VJ, et al. Strain distribution patterns for genetic markers in the LSXSS recombinant-inbred series. Mamm Genome. 1996;7(6):408–412. doi: 10.1007/s003359900122. [DOI] [PubMed] [Google Scholar]
- McClearn GE, Wilson JR, Meredith W. The use of isogenic and heterogenic mouse stocks in behavioral research. In: Lindzey G, Thiessen DD, editors. Contribution to behavior genetic analysis. The mouse as a prototype. New York: Appleton-Century-Crofts; 1970. pp. 3–32. [Google Scholar]
- McClurg P, Janes J, Wu C, Delano DL, Walker JR, et al. Genome-wide association analysis in diverse inbred mice: power and population structure. Genetics. 2007;176(1):675–683. doi: 10.1534/genetics.106.066241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucenski ML, Taylor BA, Jenkins NA, Copeland NG. AKXD recombinant inbred strains: models for studying the molecular genetics basis of murine lymphomas. Mol Cell Biol. 1986;6:4236–4243. doi: 10.1128/mcb.6.12.4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH, Singer JB, Matin A, Lander ES. Analysing complex genetic traits with chromosome substitution strains. Nat Genet. 2000;24(3):221–225. doi: 10.1038/73427. [DOI] [PubMed] [Google Scholar]
- Nesbitt MN, Skamene E. Recombinant inbred mouse strains derived from A/J and C57BL/6J: a tool for the study of genetic mechanisms in host resistance to infection and malignancy. J Leukoc Biol. 1984;36:357–364. doi: 10.1002/jlb.36.3.357. [DOI] [PubMed] [Google Scholar]
- Paigen K. One hundred years of mouse genetics: an intellectual history. II. The molecular revolution (1981–2002) Genetics. 2003;163(4):1227–1235. doi: 10.1093/genetics/163.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen K, Eppig JT. A mouse phenome project. Mamm Genome. 2000;11(9):715–717. doi: 10.1007/s003350010152. [DOI] [PubMed] [Google Scholar]
- Payseur BA, Place M. Prospects for association mapping in classical inbred mouse strains. Genetics. 2007;175(4):1999–2008. doi: 10.1534/genetics.106.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce JL, Lu L, Gu J, Silver LM, Williams RW. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, et al. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004;14(9):1806–1811. doi: 10.1101/gr.2825804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher MT, McClurg P, Batalov S, Su AI, Barnes SW, et al. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2004;2(12):e393. doi: 10.1371/journal.pbio.0020393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A, McMillan L, Wang W, Parker J, Rusyn I, et al. Inferring missing genotypes in large SNP panels using fast nearest-neighbor searches over sliding windows. Bioinformatics. 2007;23:i401–i407. doi: 10.1093/bioinformatics/btm220. [DOI] [PubMed] [Google Scholar]
- Silver L. Mouse Genetics: Concepts and Applications. Oxford: Oxford University Press; 1995. [Google Scholar]
- Taylor BA. Recombinant inbred strains: use in gene mapping. Origins of inbred mice. New York: Academic Press; 1978. [Google Scholar]
- Threadgill DW, Hunter KW, Williams RW. Genetic dissection of complex and quantitative traits: from fantasy to reality via a community effort. Mamm Genome. 2002;13(4):175–178. doi: 10.1007/s00335-001-4001-y. [DOI] [PubMed] [Google Scholar]
- Valdar W, Flint J, Mott R. Simulating the collaborative cross: power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics. 2006;172(3):1783–1797. doi: 10.1534/genetics.104.039313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J, Riblet R, Taylor BA. The response of recombinant inbred strains of mice to bacterial lipopolysaccharides. J Immunol. 1977;118:2088–2093. [PubMed] [Google Scholar]
- Williams RW, Gu J, Qi S, Lu L. The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2(11):RESEARCH0046. doi: 10.1186/gb-2001-2-11-research0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Flint J, Mott R. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics. 2005;171(2):673–681. doi: 10.1534/genetics.104.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F (2007) On the subspecific origin of the laboratory mouse. Nat Genet 39(9) (in press) [DOI] [PubMed]