

Abstract
The dynein motor domain is composed of a tail, head, and stalk and is thought to generate a force to microtubules by swinging the tail against the head during its ATPase cycle. For this “power stroke,” dynein has to coordinate the tail swing with microtubule association/dissociation at the tip of the stalk. Although a detailed picture of the former process is emerging, the latter process remains to be elucidated. By using the single-headed recombinant motor domain of Dictyostelium cytoplasmic dynein, we address the questions of how the interaction of the motor domain with a microtubule is modulated by ATPase steps, how the two mechanical cycles (the microtubule association/dissociation and tail swing) are coordinated, and which ATPase site among the multiple sites in the motor domain regulates the coordination. Based on steady-state and pre-steady-state measurements, we demonstrate that the two mechanical cycles proceed synchronously at most of the intermediate states in the ATPase cycle: the motor domain in the poststroke state binds strongly to the microtubule with a Kd of ≈0.2 μM, whereas most of the motor domains in the prestroke state bind weakly to the microtubule with a Kd of >10 μM. However, our results suggest that the timings of the microtubule affinity change and tail swing are staggered at the recovery stroke step in which the tail swings from the poststroke to the prestroke position. The ATPase site in the AAA1 module of the motor domain was found to be responsible for the coordination of these two mechanical processes.
Keywords: AAA+ protein, ATPase cycle, microtubule affinity, motor protein, power stroke
Cytoplasmic dynein is an enormous motor complex (having a molecular mass of ≈1.2 MDa) that utilizes energy from ATP hydrolysis for minus-end-directed movements along a microtubule (MT) (1). This motor activity is critical for a wide variety of cellular processes within eukaryotic cells, including chromosome separation during mitosis, maintenance of the Golgi apparatus, and most intracellular retrograde transports along MTs (2–6). The cytoplasmic dynein complex is composed of two identical heavy chains and several smaller associated polypeptides (7). Among them, the heavy chain, which belongs to the AAA+ (ATPases associated with diverse cellular activities) superfamily (8), is responsible for the motor activities of dynein (9, 10). The heavy chain forms a ring-like head (11) composed of six tandemly linked AAA+ modules (AAA1–AAA6) (8), among which the first four (AAA1–AAA4) contain nucleotide-binding/hydrolysis sites (12–15). Multiple nucleotide-binding/hydrolysis sites within a ring structure are a common feature of the AAA+ superfamily proteins (see, for example, refs. 16 and 17 for reviews). From this head, the tail and stalk, which are unique features among the AAA+ proteins, emerge as elongated structures. The tail contributes to the dimerization of the heavy chains and association of the other smaller polypeptides (18, 19), whereas the stalk, which consists of an antiparallel coiled-coil of 10–15 nm in length, binds to an MT at a small globular region located at its distal tip (stalk head) (20–23).
Because of its large size and complex structure, the molecular mechanism of dynein force generation has remained unresolved. However, the “power stroke mechanism” is gradually emerging as a feasible model for force generation. Based on electron microscopic analyses of an axonemal dynein, it has been suggested that the dynein tail assumes two distinct positions, the prestroke and poststroke positions, against the head, depending on the nucleotide state, and thereby acts as a “lever arm” for the power stroke (24). Fluorescence resonance energy transfer (FRET) measurements of a recombinant dynein motor domain with GFP-based FRET sensors also have demonstrated that the tail assumes two distinct positions, presumably corresponding to those detected by the electron microscopic study (25). Furthermore, the coupling of intermediate states of the ATPase cycle and the two tail positions has been established by FRET measurements of the motor domains trapped in specific intermediate states (25). On the basis of these studies, the ATPase-dependent tail swing cycle for the power and recovery strokes has been proposed (Fig. 1) (25).
Fig. 1.

A scheme for the tail swing cycle of dynein coupled with its ATPase cycle. The indicated sequence of the ATPase cycle is based on previous kinetic studies on Tetrahymena outer-arm dynein (27–29). The nucleotides and mutations used to trap each intermediate state of the ATPase cycle are denoted in the second line. The two states of dynein tail position are indicated as D and D*. Here, D represents the motor domain in which the tail adopts the poststroke position, and D* represents the motor domain in which the tail adopts the prestroke position. The intermediate state D*-ADP given in parentheses is only tentatively assigned.
For successful power and recovery strokes, the cyclic tail swing must be tightly coordinated with another mechanically distinct process, i.e., a cyclic change in the affinity of dynein for MT. Therefore, to elucidate the power stroke cycle of the MT–dynein system, the cyclic change of the MT–dynein interaction should be integrated with the tail swing cycle. To date, there have been limited quantitative studies on the ATPase-dependent MT–dynein interaction because of the fact that only axonemal outer-arm dyneins carrying multiple motor domains have been available for biochemical analyses. The cooperative binding of these multiple motor domains to MTs has been an obstacle to the straightforward analyses of the MT–dynein binding. However, by exploiting a single-headed recombinant motor domain with full motor activities (10, 26), it now has become feasible to assess the interaction of a single dynein motor domain with an MT at both the steady-state and pre-steady-state levels. Here, we address the questions of how the dissociation constant of the single-headed motor domain from MTs changes depending on the ATPase steps; how the two distinct mechanical cycles, i.e., the tail swing and the affinity change of the MT–motor domain complex, are coordinated; and which ATPase site among the multiple sites regulates the coordination.
Results
Nucleotide States of the Dynein Motor Domain Modulate MT Affinity.
We previously have performed FRET measurements on a recombinant dynein motor domain in which the ATPase cycle was trapped at a particular intermediate state by the addition of nucleotides and/or the introduction of mutations, and we have demonstrated that the motor domain assumes one of the two distinct structures, represented here by D and D*, depending on the trapped state (Fig. 1) (25). D and D* represent dynein in which the tails are presumed to assume the poststroke and prestroke positions, respectively, observed in electron microscopic images (24).
In this study, we examined how a change in the affinity of the dynein motor domain for an MT is coupled with the ATPase and tail swing cycles. The affinity was quantitatively measured by the cosedimentation of MTs and motor domains trapped at a particular intermediate state of the ATPase cycle by means of nucleotide addition and/or mutation as described in our previous FRET study (25). We used the same single-headed motor domain (HFG380B2) that was used for the previous FRET measurements (25) to identify a correlation between the tail swing and the affinity change of the motor domain for an MT. This functional motor domain consists of a C-terminal 380-kDa portion of the Dictyostelium discoideum cytoplasmic dynein heavy chain that was fused with GFP at its N terminus and with blue fluorescent protein (BFP) at the interior of the AAA2 module.
We first examined the MT affinity of the motor domain in the apo state, i.e., in the absence of added adenine nucleotides, by measuring the concentration of free motor domain after precipitating the MT–motor domain complex by centrifugation. Here, the fluorescence of GFP fused to the motor domain was used for determining the protein concentration (for details, see Materials and Methods). By increasing the MT concentration with respect to a fixed amount of the motor domain, the binding curve shown in Fig. 2A was obtained. The results showed that almost all of the motor domain molecules in the apo state strongly bound to MT with a single dissociation constant (Kd) of ≈0.2 μM (Fig. 2A and Table 1). By increasing the concentration of the motor domain with respect to a fixed amount of MT, the number of tubulin dimers occupied by a single motor domain with a high affinity (Kd ≈0.2 μM) was determined to be ≈2 (Fig. 2C); this result is consistent with that of a previous study in which three-headed axonemal dynein bound to MT with a stoichiometry of one dynein per six tubulin dimers (30), i.e., one motor domain per two tubulin dimers.
Fig. 2.

Steady-state MT binding of the dynein motor domain. (A) Cosedimentation assays were performed with a constant concentration (0.05 μM) of HFG380B2 and various concentrations (up to ≈14 μM) of MT in the absence of added adenine nucleotides (filled red squares) or in the presence of 3.3 mM ATP (open blue squares) or 1 mM ATP plus Vi (filled green triangles). The smooth curves are the best fits of the data to hyperbolas with the dissociation constants (Kd) and maximum bindings (Bmax) given in Table 1. (B) The same assay as described in A also was performed with HFG380B2/E2022Q in the absence of added adenine nucleotides (filled red squares) or in the presence of 3.3 mM ATP (open blue squares). The smooth curves are the best fits of the data to hyperbolas with the Kd and Bmax given in Table 1. (C) To determine the binding stoichiometry of dynein to MTs (tubulin dimer), a cosedimentation assay was performed with various concentrations (up to ≈0.7 μM) of HFG380B2 and a constant concentration (1 μM) of MT in the absence of added adenine nucleotides. The smooth curve is the best fit of the data to a hyperbola with a Kd of 0.17 ± 0.01 μM and a stoichiometry of 0.50 ± 0.02.
Table 1.
Summary of the nucleotide-dependent MT binding of HFG380B2 and its mutants
| Dynein | Nucleotide | Kd, μM | Bmax |
|---|---|---|---|
| HFG380B2 | None | 0.24 ± 0.02 | 0.86 ± 0.01 |
| ATP | >10* | ND* | |
| ADP | 0.25 ± 0.02 | 0.87 ± 0.01 | |
| AMP-PNP | 0.22 ± 0.02 | 0.87 ± 0.01 | |
| ATP + Vi | >10* | ND* | |
| HFG380B2/E2022Q | None | 0.27 ± 0.02 | 0.90 ± 0.01 |
| ATP | >10 | ND | |
| HFG380B2/K1975T | None | 0.25 ± 0.02 | 0.85 ± 0.02 |
| ATP | 0.43 ± 0.03 | 0.86 ± 0.01 | |
| HFG380B2/K2675T | None | 0.49 ± 0.06 | 0.90 ± 0.02 |
| ATP | 1.2 ± 0.1 | 0.89 ± 0.03 |
ND, not determined.
*The data indicated are those for the major population.
A similar strong binding of the motor domain to MTs was observed in the presence of ADP or adenosine 5′-[β,γ-imido]triphosphate (AMP-PNP) (Table 1), which traps the motor domain in the D-ADP or D-AMP-PNP state, respectively. The latter state would mimic the D-ATP state corresponding to the collision complex of the motor domain and ATP (25). Thus, the motor domain in the D (apo), D-ADP, and D-AMP-PNP (mimicking D-ATP) states tightly bound to MTs with a Kd of ≈0.2 μM.
In the presence of an excess amount of ATP, however, the binding curve exhibited biphasic behavior with a major weak-binding population (≈90%) and a minor strong-binding population (≈10%) (Fig. 2A), which may reflect the presence of two populations of motor domain molecules hydrolyzing ATP cyclically. By assuming two populations with different Kd values, the curve fitting demonstrated that the major population was in the weak-binding state with a Kd of >10 μM (Table 1), whereas the minor population was in the strong-binding state with a Kd of ≈0.1 μM. In the presence of ATP plus vanadate (Vi), which is assumed to trap dynein in the D*-ADP-Vi state that mimics the D*-ADP-Pi state (27), the motor domain molecules also bound to MTs as two populations: a major population (≈80%) with a Kd of >10 μM and a minor population (≈20%) with a Kd of ≈0.5 μM (Fig. 2A and Table 1). This result suggests that a majority of the motor domain in the D*-ADP-Pi state binds weakly to an MT.
We have demonstrated that dynein assumes two different ATP-bound states, D-ATP and D*-ATP, and that the latter state is trapped by introducing an E2022Q mutation in the Walker B motif of the AAA1 module (25). This mutation is expected to inhibit ATP hydrolysis, but not ATP-binding, at the AAA1 ATPase site. In the presence of ATP, the mutant is trapped in a single state, D*-ATP (Fig. 1) (25). The cosedimentation of HFG380B2/E2022Q with MTs in the absence of ATP showed that the mutant bound to MTs with a high affinity (Kd ≈ 0.2 μM) in a similar manner to HFG380B2 (Fig. 2B and Table 1). In the presence of ATP, however, the mutant in the D*-ATP state bound very weakly to MTs with a Kd of >10 μM (Fig. 2B and Table 1). There was no strong binding component in the binding curve. This result supports the notion that the minor strong binding component observed for HFG380B2 indicates heterogeneous populations of the motor domain in the presence of excess ATP or ATP plus Vi.
These steady-state measurements, combined with the previous FRET study (25), have demonstrated that during the ATPase cycle, the MT affinity and the tail position change synchronously: the motor domain in the intermediate state, wherein the tail is presumed to assume the prestroke position (D*-ATP or D*-ADP-Pi), weakly binds to MTs with a Kd of >10 μM, whereas the motor domain in the intermediate state, in which the tail is presumed to assume the poststroke position (D, D-ADP, or D-ATP), binds tightly to MTs with a Kd of ≈0.2 μM.
Staggered Timings of the ATP-Induced Dissociation of the MT–Motor Domain Complex and Tail Swing.
To perform the cyclic power stroke, dynein is required to undergo a recovery stroke at each cycle. For a successful recovery stroke, the motor domain has to be released from the MT before the tail swing from the poststroke to prestroke position. Therefore, there would be a transient state in which the motor domain is released from the MT, while its tail continues to adopt the poststroke position.
To detect such a transient state that has not been stably trapped by the addition of nucleotides or by mutation, we performed pre-steady-state measurements and determined the rate of the ATP-induced dissociation of the MT-HFG380B2 complex as well as the rate of the tail swing after the dissociation. We used the following kinetic scheme for the sequential dissociation and tail swing steps upon the addition of ATP to the MT–motor domain complex (MT-D):
 |
where MT-D-ATP is the collision complex in equilibrium with MT-D and ATP, and D+-ATP represents the transient state in which the motor domain binds only weakly to the MT and its tail assumes the poststroke state. kdis is the rate of the dissociation step and kiso is the rate of the isomerization (tail swing) step from the poststroke state (D+-ATP) to the prestroke state (D*-ATP). In this scheme, the dissociation and isomerization steps are assumed to be irreversible.
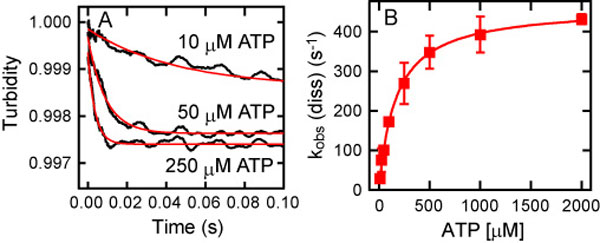
Upon the addition of ATP to the MT-HFG380B2 complex, its turbidity decreased because of dissociation of the complex (Fig. 3A). Time courses of the decrease in turbidity were well fitted to double exponentials. From the fast phase occupying a major portion of the amplitude change (>70%), a pseudo-first-order rate kobs (dis) was determined. The kobs (dis) values at various ATP concentrations are described by
where K is an equilibrium constant for collision complex formation. Plotting kobs (dis) against ATP concentration gave a hyperbolic curve with a maximum rate of 310 s−1 (Fig. 3B), which is equal to kdis. This rapid dissociation of the MT–motor domain complex on the addition of ATP is consistent with the previous measurement of the ATP-induced dissociation of an MT–axonemal dynein complex (31). As shown in Fig. 3B, the slow phase of the turbidity change occupied a minor amplitude (<30%), and its rate was too slow (<40 s−1) to be involved in the MT-activated ATPase cycle (kcat of ≈100 s−1; ref. 25). It is worth noting that the dissociation of the “wild-type” motor domain HFB380, which carries only the N-terminal tandem tags (32), from an MT was more rapid (kdis of 460 s−1) and exhibited no slow phase [supporting information (SI) Fig. 7], suggesting that the insertion of GFP/BFP FRET sensors slowed down this transient step and that the slow component would be an artifact related to these sensors, but it is unrelated to the dissociation process.
Fig. 3.

ATP-induced dissociation of the MT-HFG380B2 complex. (A) Representative time courses of the ATP-induced dissociation of the MT-HFG380B2 complex (1 μM MT and 0.2 μM HFG380B2) after mixing with 10, 50, and 250 μM ATP (jagged black curves). The smooth red curves are fits to double exponentials with rate constants of 23 s−1 and 3.4 s−1 (10 μM ATP), 78 s−1 and 12 s−1 (50 μM ATP), and 230 s−1 and 27 s−1 (250 μM ATP). (B) The observed rates (left scale) of the fast (filled red squares) and slow (open blue squares) phases of the ATP-induced dissociation as a function of ATP concentration. The rates of the fast phase were fitted to a hyperbola (red curve), giving kdis and K values of 310 s−1 and 7.5 × 103 M−1, respectively. The rates of the slow phase also were fitted to a hyperbola (blue curve) with the extrapolated maximum rate of 31 s−1. The gray line plots the percentage of the fast-phase amplitude against the total amplitude (right scale) in the double-exponential fitting. Error bars represent standard deviations.
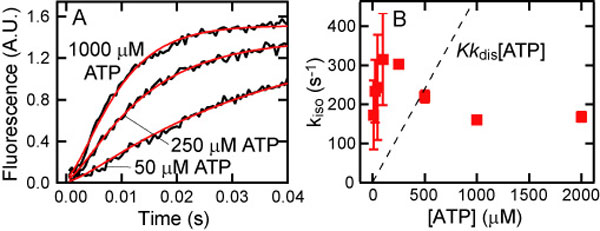
Next, we followed the ATP-induced tail swing detected by FRET, starting from the MT-HFG380B2 complex. On addition of ATP to the MT-HFG380B2 complex, the FRET signal changed rapidly because of the change in tail position from poststroke to prestroke, as reported previously for free HFG380B2 (25, 33) (Fig. 4A). By assuming the sequential steps described above, the time course of FRET change on the addition of ATP was analyzed by using the following equation,
 |
where A is the signal amplitude. The rate of isomerization kiso was calculated by fitting the time course to this equation with the known kobs (dis) value at each ATP concentration. The kiso values thus obtained were plotted against ATP concentration (Fig. 4B), showing that the rate (≈200 s−1) was virtually independent of ATP concentration as expected from the proposed scheme.
Fig. 4.

ATP-induced isomerization of HFG380B2 bound to MT. (A) Representative time courses of the ATP-induced isomerization of 0.05 μM HFG380B2 in the presence of 1 μM MT after mixing with 10, 50, and 250 μM ATP (jagged black curves). The smooth red curves are fits to the equations for the two-step reaction (see text) with kiso values of 270 s−1 (10 μM ATP), 140 s−1 (50 μM ATP), and 200 s−1 (250 μM ATP). (B) The kiso values are plotted against ATP concentration. Error bars represent standard deviations.
Similar measurements were performed on the complex of an MT and the HFG380B2/E2022Q mutant because the mutant is trapped at a single state, D*-ATP, on the addition of ATP, and therefore the kinetics for the ATP-induced changes may be more straightforward. The ATP-induced decrease in turbidity and FRET change of the MT-HFG380B2/E2022Q complex were, however, very similar to those of the MT-HFG380B2 complex (SI Figs. 8 and 9), giving kdis and kiso values of 320 s−1 and ≈180 s−1, respectively.
Although the sequential steps are assumed for the dissociation of the MT–motor domain complex and the tail swing in the above analyses, it also is possible that these two processes proceed in parallel. These sequential and parallel schemes are not distinguishable based only on the observed time courses of FRET change. When the parallel scheme is assumed, the time course gives a pseudo-first-order rate kobs (iso) by fitting it to a single exponential (33). By plotting kobs (iso) against ATP concentration (SI Fig. 10), a rate for the single-step transition from the poststroke (MT-D-ATP) to the prestroke (D*-ATP) state was calculated as 160 s−1. The results reveal that, even for the parallel scheme, the dissociation is more rapid (310 s−1) than the tail swing (160 s−1). Therefore, in both the sequential and parallel schemes, the dissociation step—either obligatorily (the sequential scheme) or frequently (the parallel scheme)—precedes the tail swing step for a successful recovery stroke.
The AAA1–ATPase Cycle Regulates the Coordination of MT-Affinity Change and Tail Swing.
We previously have demonstrated that ATP hydrolysis in the AAA1 and AAA3 modules is critical for MT sliding (34) and that the AAA1 ATPase is probably responsible for the tail swing (25). To determine which site is responsible for the ATPase-dependent MT-affinity change, we introduced Walker A mutations into the AAA1 and AAA3 modules of HFG380B2 (K1975T and K2675T, respectively). By introducing Walker A mutations, ATP binding to the AAA modules is expected to be blocked (35, 36). We first examined the steady-state binding of these mutants to MTs in the presence and absence of ATP using the cosedimentation assay. As shown in Fig. 5 A and B, both mutants tightly bound to MTs in the presence and absence of ATP. Although the HFB380B2/K2675T mutant exhibited a slight ATP-dependent change (Kd = 0.5 μM in the absence of ATP and 1.2 μM in its presence), it was considerably less than that of HFG380B2 (0.2 μM and >10 μM, respectively). The results thus appear to indicate that both the AAA1 and AAA3 ATPase sites are important for the ATPase-dependent changes in MT affinity as proposed in previous studies (26, 34, 37). However, the effect of each of the Walker A mutations on the steady-state binding was not direct because of the tight coupling of these two ATPase sites (25, 34). Therefore, to further examine the direct effects of mutations without the strong coupling of AAA1 and AAA3 ATPase cycles, we performed pre-steady-state measurements of the ATP-induced dissociation of MT-HFG380B2/K1975T and MT-HFG380B2/K2675T complexes.
Fig. 5.

Effect of Walker A mutations on the ATP-induced dissociation. (A and B) Cosedimentation assays of HFG380B2/1975T (A) and HFG380B2/K2675T (B) with MT in the absence of added adenine nucleotide (filled red squares) or in the presence of 3.3 mM ATP (open blue squares). The smooth curves are the best fits of the data to hyperbolas with the dissociation constants (Kd) and maximum bindings (Bmax) given in Table 1. (C) Representative time courses of the ATP-induced dissociation of the complex of MT and HFG380B2, HFG380B2/K1975T, or HFG380B2/K2675T after mixing with 1 mM ATP. For the K2675T mutant, the experiments also were performed with higher concentrations of MT (2 and 3 μM) in addition to 1 μM to make the reassociation process clearly observable.
When the MT-HFG380B2/K1975T complex was mixed rapidly with ATP, dissociation of the complex was never detected (Fig. 5C). In contrast, the MT-HFG380B2/K2675T complex rapidly dissociated on the addition of ATP and then reassociated to enter into a new steady state (Fig. 5C), as expected from the steady-state binding shown in Fig. 5B. At a higher MT concentration (3 μM), the complex dissociated and then quickly reassociated to form a stable complex, whereas at a lower MT concentration (1 μM), the complex dissociated and then only slowly reassociated. These results demonstrate that the inhibition of ATP binding to the AAA1 ATPase site by the K1975T mutation completely blocked the ATP-induced dissociation of the complex, whereas inhibition of ATP binding to the AAA3 ATPase site by the K2675T mutation did not; these observations indicate that the AAA1 site, but not the AAA3 site, is responsible for the ATPase-dependent MT-affinity change.
Discussion
The nucleotide-induced structural changes underlying the two mechanical processes, ATPase-dependent MT-affinity change and tail swing, would be propagated through two distinct pathways: one is directed from the ATPase site(s) in the head to the stalk for the MT-affinity change (38), whereas the other is directed to the interface between the head and the tail for the tail swing. The coordination of these processes is tightly regulated by ATPase steps for the successful power and recovery strokes required for the multiple stepping of dynein along the MT.
We previously have characterized the ATPase-dependent tail swing cycle by FRET measurements (Fig. 1) (25, 33). However, the other mechanical cycle essential for the force generation, the ATPase-dependent change in motor domain affinity for MTs, has remained unresolved. Here, using steady-state and pre-steady-state measurements, we have demonstrated the following:
There are at least two distinct states of the MT–motor domain complex with very different dissociation constant (Kd), the strong-binding state with a Kd of ≈0.2 μM and the weak-binding state with a Kd of >10 μM, depending on the nucleotide state of the motor domain.
At most of intermediate states in the ATPase cycle, the MT affinity synchronizes with the tail position: the motor domain whose tail is presumed to assume the prestroke position is virtually dissociated from the MT, whereas the motor domain whose tail is presumed to assume the poststroke position forms a tight complex with the MT.
At the recovery stroke corresponding to the poststroke to prestroke transition, the timings of the two processes are staggered: the dissociation of the MT–motor domain complex precedes the tail swing to ensure a successful recovery stroke for priming the power stroke.
These affinity changes were determined by ATPase steps at the AAA1 ATPase site in a manner similar to the tail swing.
Based on these results, we propose a scheme to describe how the tail swing, the MT-affinity change, and ATPase steps at the AAA1 ATPase site are coordinated (Fig. 6), although the latter stage of the scheme still is tentative because the ATPase steps after ATP hydrolysis have yet to be fully characterized.
Fig. 6.

A scheme for the major pathway of the MT–dynein complex ATPase cycle. Note that the intermediate states shown here are those in the AAA1 ATPase site. The two states of dynein tail position are indicated as D and D* as in Fig. 1. D+ represents the motor domain in the weak-binding state whose tail assumes the poststroke position. The two states of the binding affinity of the motor domain to MTs, the strong-binding and weak-binding states, also are indicated as the tightly associated complex (e.g., MT-D) and the weakly interacting MT and the motor domain (e.g., MT+D*-ATP), respectively. The intermediate state MT+D*-ADP given in parentheses is only tentatively assigned.
An MT-D-ATP collision complex initially is formed after the rapid binding of ATP to the MT-D complex. In the MT-D-ATP complex, the motor domain with bound ATP tightly associates with the MT, whereas the tail continues to assume the poststroke position. This complex is likely to be mimicked by MT-D-AMP-PNP, considering that the motor domain binds tightly to the MT in the presence of AMP-PNP (Kd = ≈0.2 μM) with its tail in the poststroke position (25), similar to the case of the collision complex MT-D-ATP.
The motor domain then rapidly dissociates from the MT (≈460 s−1 for the wild-type motor domain, HFB380) to assume the transient D+-ATP state, wherein the motor domain is in the weak-binding state and its tail continues to adopt the poststroke position.
This dissociation step is followed by an isomerization step that generates the D*-ATP state with a rate of ≈200 s−1, such that the tail now assumes the prestroke position. Here, we have to consider the possibility that the dissociation and the tail swing proceed in parallel. In this parallel scheme, the rate of isomerization from MT-D-ATP to D*-ATP is ≈160 s−1, whereas the rate of dissociation of the MT-D-ATP complex is ≈310 s−1. This parallel scheme is less likely, however, considering that the rates of dissociation and isomerization in this scheme will result in an abortive stroke with a frequency of more than one-third. In either case, the dissociation of the MT–motor domain complex (obligatorily or frequently) precedes the tail swing.
After ATP hydrolysis, the D*-ATP state is converted to the D*-ADP-Pi state, which is mimicked by the D*-ADP-Vi state generated in the presence of ATP plus Vi (27). From the transient D+-ATP state to the D*-ADP-Pi state, the motor domain remains virtually dissociated from the MT.
The D*-ADP-Pi state finally is converted to the MT-D-ADP state through other intermediate state(s), if any, such as the D*-ADP (28, 29). This prestroke to poststroke transition would correspond to the power stroke.
After ADP release from MT-D-ADP, the cycle is reset for a further round.
For the successful power stroke at step V in the above scheme, it is expected that the motor domain initially binds to the MT and then the tail swings from the prestroke to poststroke position for active translocation of the bound MT. Although, because of technical difficulties, the reassociation of the motor domain to the MT was not directly monitored in this study, our previous finding that the prestroke to poststroke transition is accelerated by the presence of the MT (33) implies that the reassociation of dynein with the MT occurs before the tail swing from the prestroke to the poststroke position. Thus, step V in the present scheme may be composed of multiple steps. To further dissect this power stroke process, a detailed characterization of the ATPase steps after ATP hydrolysis is required. Because there are multiple ATPase sites running simultaneously in the motor domain, it will be technically challenging to detect Pi and ADP releases specifically from the AAA1 ATPase site for the identification and characterization of the substeps between the D*-ADP-Pi and MT-D-ADP states.
Materials and Methods
Construction, Expression, and Purification.
The expression construct for the C-terminal 380-kDa motor domain of D. discoideum cytoplasmic dynein heavy chain fused with His6-FLAG-GFP at its N terminus and BFP at the interior of the AAA2 module (HFG380B2) and that fused with a His6-FLAG-biotinylation sequence at its N terminus (HFB380) have been described previously (25, 32). Point mutations within HFG380B2 were introduced with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's instructions. A plasmid carrying each of the expression constructs was introduced into D. discoideum cells, and the recombinant proteins were expressed and purified as described (34). Tubulin was purified from porcine brain (39). The protein concentrations were determined by the Bradford method (40) using BSA as a standard.
Steady-State MT Binding Assays.
Steady-state binding assays were performed by cosedimentation in a dynein assay buffer (10 mM K-Pipes, 50 mM potassium acetate, 4 mM MgSO4, 1 mM EGTA, and 1 mM DTT, pH 7.0) containing 5 μM free paclitaxel and 250 μM GTP at 25°C. In the experiments performed at a constant dynein concentration, 0.05 μM HFG380B2 or its mutant was mixed with various concentrations (up to ≈14 μM) of paclitaxel-stabilized MT in the absence of added adenine nucleotides or in the presence of 3.3 mM ATP, 1 mM ADP, 1 mM AMP-PNP, or 1 mM ATP plus Vi. After a 5-min incubation, the mixtures were centrifuged at 100,000 × g for 10 min. The supernatants were carefully removed, and the concentrations of the recombinant protein in the supernatants were determined by measuring the fluorescence intensities of GFP (excitation at 488 nm, emission at 510 nm) with reference to a standard recombinant protein whose concentration had been determined previously by the Bradford method (40). Because of light scattering by MTs, we were unable to accurately determine the protein concentrations of the pellets by fluorescence, thus these concentrations were determined indirectly by subtracting the protein concentrations in the supernatants from those present in the inputs. The results of the cosedimentation assays also were verified by SDS/PAGE analyses.
To determine the binding stoichiometry, a cosedimentation assay was performed using various concentrations (up to ≈0.7 μM) of HFG380B2 and a constant concentration (1 μM) of MT in the absence of added adenine nucleotides.
Kinetic Measurements.
Pre-steady-state kinetic experiments were performed with an SX-18MV stopped-flow apparatus (Applied Photophysics, Leatherhead, U.K.) in the dynein assay buffer containing 5 μM free paclitaxel, 50 μM ADP, and 250 μM GTP at 25°C. ATP-induced dissociation of the MT–dynein complex (1 μM paclitaxel-stabilized MT and 0.2 μM HFG380B2, its mutant, or HFB380) was observed by monitoring the changes in turbidity at 340 nm after mixing with 10–2,000 μM ATP. Each trace was fitted to a double exponential (for HFG380B2 or its mutant) or a single exponential (for HFB380) after subtracting a blank trace, which was obtained by monitoring the turbidity of 1 μM MT after mixing the assay buffer. For HFG380B2/K2675T, the experiment also was performed with 2 or 3 μM MT, in which the blank trace was obtained by monitoring the turbidity of 2 or 3 μM MT, respectively, after mixing the assay buffer. The ATP-induced tail swing of dynein was followed by FRET measurement in the stopped-flow apparatus. The FRET change of 0.05 μM HFG380B2 in the presence of 1 μM MT was observed by exciting the donor (BFP) at 380 nm and monitoring the emission intensity of the accepter (GFP) with a 515-nm long-pass filter after mixing with 10–2,000 μM ATP. Each trace was fitted to a single exponential. All concentrations given are final after mixing.
Supplementary Material
Acknowledgments
This work was supported by a grant-in-aid for scientific research (S) from the Japan Society for the Promotion of Science (JSPS); a grant-in-aid for scientific research on priority areas from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); a grant-in-aid for young scientists (B) from MEXT; a Core Research for Evolutional Science and Technology (CREST) program grant from the Japan Science and Technology Agency (JST); and a grant-in-aid for JSPS fellows from JSPS.
Abbreviations
- MT
microtubule
- AAA
ATPases associated with diverse cellular activities
- BFP
blue fluorescent protein
- AMP-PNP
5′-[β,γ-imido]triphosphate
- Vi
vanadate.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702370104/DC1.
References
- 1.Paschal BM, Vallee RB. Nature. 1987;330:181–183. doi: 10.1038/330181a0. [DOI] [PubMed] [Google Scholar]
- 2.Holzbaur EL, Vallee RB. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- 3.Vallee RB, Sheetz MP. Science. 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- 4.Hirokawa N. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 5.Karki S, Holzbaur EL. Curr Opin Cell Biol. 1999;11:45–53. doi: 10.1016/s0955-0674(99)80006-4. [DOI] [PubMed] [Google Scholar]
- 6.Vale RD. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 7.King SJ, Bonilla M, Rodgers ME, Schroer TA. Protein Sci. 2002;11:1239–1250. doi: 10.1110/ps.2520102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neuwald AF, Aravind L, Spouge JL, Koonin EV. Genome Res. 1999;9:27–43. [PubMed] [Google Scholar]
- 9.Koonce MP, Samso M. Mol Biol Cell. 1996;7:935–948. doi: 10.1091/mbc.7.6.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishiura M, Kon T, Shiroguchi K, Ohkura R, Shima T, Toyoshima YY, Sutoh K. J Biol Chem. 2004;279:22799–22802. doi: 10.1074/jbc.M313362200. [DOI] [PubMed] [Google Scholar]
- 11.Samso M, Radermacher M, Frank J, Koonce MP. J Mol Biol. 1998;276:927–937. doi: 10.1006/jmbi.1997.1584. [DOI] [PubMed] [Google Scholar]
- 12.Gibbons IR, Gibbons BH, Mocz G, Asai DJ. Nature. 1991;352:640–643. doi: 10.1038/352640a0. [DOI] [PubMed] [Google Scholar]
- 13.Ogawa K. Nature. 1991;352:643–645. doi: 10.1038/352643a0. [DOI] [PubMed] [Google Scholar]
- 14.Koonce MP, Grissom PM, McIntosh JR. J Cell Biol. 1992;119:1597–1604. doi: 10.1083/jcb.119.6.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikami A, Paschal BM, Mazumdar M, Vallee RB. Neuron. 1993;10:787–796. doi: 10.1016/0896-6273(93)90195-w. [DOI] [PubMed] [Google Scholar]
- 16.Vale RD. J Cell Biol. 2000;150:F13–F19. doi: 10.1083/jcb.150.1.f13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogura T, Wilkinson AJ. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 18.Habura A, Tikhonenko I, Chisholm RL, Koonce MP. J Biol Chem. 1999;274:15447–15453. doi: 10.1074/jbc.274.22.15447. [DOI] [PubMed] [Google Scholar]
- 19.Tynan SH, Gee MA, Vallee RB. J Biol Chem. 2000;275:32769–32774. doi: 10.1074/jbc.M001537200. [DOI] [PubMed] [Google Scholar]
- 20.Goodenough U, Heuser J. J Mol Biol. 1984;180:1083–1118. doi: 10.1016/0022-2836(84)90272-9. [DOI] [PubMed] [Google Scholar]
- 21.Gee MA, Heuser JE, Vallee RB. Nature. 1997;390:636–639. doi: 10.1038/37663. [DOI] [PubMed] [Google Scholar]
- 22.Koonce MP. J Biol Chem. 1997;272:19714–19718. doi: 10.1074/jbc.272.32.19714. [DOI] [PubMed] [Google Scholar]
- 23.Koonce MP, Tikhonenko I. Mol Biol Cell. 2000;11:523–529. doi: 10.1091/mbc.11.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burgess SA, Walker ML, Sakakibara H, Knight PJ, Oiwa K. Nature. 2003;421:715–718. doi: 10.1038/nature01377. [DOI] [PubMed] [Google Scholar]
- 25.Kon T, Mogami T, Ohkura R, Nishiura M, Sutoh K. Nat Struct Mol Biol. 2005;12:513–519. doi: 10.1038/nsmb930. [DOI] [PubMed] [Google Scholar]
- 26.Reck-Peterson SL, Vale RD. Proc Natl Acad Sci USA. 2004;101:1491–1495. doi: 10.1073/pnas.2637011100. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Shimizu T, Johnson KA. J Biol Chem. 1983;258:13833–13840. [PubMed] [Google Scholar]
- 28.Holzbaur EL, Johnson KA. Biochemistry. 1989;28:5577–5585. doi: 10.1021/bi00439a036. [DOI] [PubMed] [Google Scholar]
- 29.Holzbaur EL, Johnson KA. Biochemistry. 1989;28:7010–7016. doi: 10.1021/bi00443a034. [DOI] [PubMed] [Google Scholar]
- 30.Porter ME, Johnson KA. J Biol Chem. 1983;258:6575–6581. [PubMed] [Google Scholar]
- 31.Porter ME, Johnson KA. J Biol Chem. 1983;258:6582–6587. [PubMed] [Google Scholar]
- 32.Shima T, Imamula K, Kon T, Ohkura R, Sutoh K. J Struct Biol. 2006;156:182–189. doi: 10.1016/j.jsb.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Mogami T, Kon T, Ito K, Sutoh K. J Biol Chem. 2007;282:21639–21644. doi: 10.1074/jbc.M701914200. [DOI] [PubMed] [Google Scholar]
- 34.Kon T, Nishiura M, Ohkura R, Toyoshima YY, Sutoh K. Biochemistry. 2004;43:11266–11274. doi: 10.1021/bi048985a. [DOI] [PubMed] [Google Scholar]
- 35.Whiteheart SW, Rossnagel K, Buhrow SA, Brunner M, Jaenicke R, Rothman JE. J Cell Biol. 1994;126:945–954. doi: 10.1083/jcb.126.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Babst M, Wendland B, Estepa EJ, Emr SD. EMBO J. 1998;17:2982–2993. doi: 10.1093/emboj/17.11.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silvanovich A, Li MG, Serr M, Mische S, Hays TS. Mol Biol Cell. 2003;14:1355–1365. doi: 10.1091/mbc.E02-10-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibbons IR, Garbarino JE, Tan CE, Reck-Peterson SL, Vale RD, Carter AP. J Biol Chem. 2005;280:23960–23965. doi: 10.1074/jbc.M501636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sloboda RD, Rosenbaum JL. Methods Enzymol. 1982;85:409–416. doi: 10.1016/0076-6879(82)85041-6. [DOI] [PubMed] [Google Scholar]
- 40.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}