

Abstract
JG-03-14, a substituted pyrrole that inhibits microtubule polymerization, was screened against MCF-7 (p53 wild type), MDA-MB 231 (p53 mutant), MCF-7/caspase 3 and MCF-7/ADR (multidrug resistant) breast tumor cell lines. Cell viability and growth inhibition were assessed by the crystal violet dye assay. Apoptosis was evaluated by the TUNEL assay, cell cycle distribution by flow cytometry, autophagy by acridine orange staining of vesicle formation, and senescence based on β-galactosidase staining and cell morphology. Our studies indicate that exposure to JG-03-14, at a concentration of 500 nM, induces time dependent cell death in the MCF-7 and MDA-MB 231 cell lines. In MCF-7 cells, a residual surviving cell population was found to be senescent; in contrast, there was no surviving senescent population in treated MDA-MB 231 cells. No proliferative recovery was detected over a period of 15 days post-treatment in either cell line. Both the TUNEL assay and FLOW cytometry indicated a relatively limited degree of apoptosis (< 10%) in response to drug treatment in MCF-7 cells with more extensive apoptosis (but < 20%) in MDA-MB231 cells; acidic vacuole formation indicative of autophagic cell death was relatively extensive in both MCF-7 and MDA-MB231 cells. In addition, JG-03-14 induced the formation of a large hyperdiploid cell population in MDA-MB231 cells. JG-03-14 also demonstrated pronounced anti-proliferative activity in MCF-7/caspase 3 cells and in the MCF-7/ADR cell line. The observation that JG-03-14 promotes autophagic cell death and also retains activity in tumor cells expressing the multidrug resistance pump indicates that novel microtubule poisons of the substituted pyrroles class may hold promise in the treatment of breast cancer.
1. Introduction
A number of compounds that have either proven or potential activity against various diseases, including cancer, have been derived from marine organisms. The substituted pyrroles, which were synthesized based on similarity to marine derived compounds [1,2], have previously been demonstrated to have significant growth inhibitory activity against a variety of human tumor cell lines, with antiproliferative effects evident at nanomolar concentrations in some human breast tumor cell lines [3–6].
The development of synthetic or semisynthetic derivatives of the pyrroles is facilitated due to the relative ease with which these compounds can be synthesized and their functional groups manipulated [3]. One of these pyrroles, termed JG-03-14 (2,4-Dibromo-5-carbethoxy-3-(3,4 dimethoxyphenyl) pyrrole), was found to act as a microtubule poison and to bind at the colchicine binding site of tubulin [7] (and unpublished data).
The current investigation was designed to characterize the action of the substituted pyrrole, JG-03-14, in two human breast cancer cells lines expressing either wild type p53 (MCF-7 cells) or mutant p53 (MDA-MB231 cells). Our studies focused on the capacity of JG-03-14 to promote irreversible growth arrest and/or cell death, as well as to characterize the nature of the growth arrest/cell death response. We were also interested in evaluating the possibility that this compound would be effective in a tumor cell line expressing the multidrug resistant phenotype.
2. Materials and Methods
2.1 Materials
RPMI 1640 medium with L-glutamine, trypsin-EDTA (1X; 0.05% trypsin, 0.53 mM EDTA-4 Na), penicillin/streptomycin (10,000 units/ml penicillin and 10 mg/ml streptomycin), and fetal bovine serum were obtained from GIBCO Life Technologies (Gaithersburg, MD). Defined bovine calf serum was obtained from Hyclone Laboratories (Logan, UT). Reagents used for the TUNEL assay (terminal transferase, reaction buffer, and fluorescein-dUTP) were purchased from Boehringer Mannheim (Indianapolis, IN). X-gal was obtained from Gold Biotechnology (St. Louis, MO). The following materials were obtained from Sigma Chemical (St. Louis, MO): trypan blue solution, formaldehyde, acetic acid, albumin bovine (BSA), 6-diamidino-2-phenylindole (DAPI) and dimethyl sulfoxide (DMSO). Acridine orange was purchased from Invitrogen (Eugene, OR).
2.2 Cell Lines
The p53 wild-type MCF-7 human breast tumor cell line was obtained from NCI, Frederick, MD. MDA-MB-231 cells were obtained from ATCC. MCF-7/ADR cells were developed by Dr. Ken Cowan [8].
2.3 Cell Culture and Treatment
Cells were grown from frozen stocks in basal RPMI 1640 medium supplemented with 5% fetal calf serum, 5% bovine calf serum, 2 mM L-glutamine, and penicillin/streptomycin (0.5ml/100ml medium) at 37°C under a humidified, 5% CO2 atmosphere. Drug treatment involved continuous exposure to the compounds. For all cell culture experiments, cells were permitted to adhere for 24 hours following plating before the investigational compounds were administered.
2.4 Assessment of Viable Cell Number
Cell viability was determined by trypan blue exclusion at various time points beginning 24 hr after initiation of JG-03-14 treatment. Cells were harvested by trypsinization, stained with 0.4% trypan blue dye, and counted using phase contrast microscopy. Cells that excluded trypan blue dye were considered to be viable.
2.5 Crystal Violet Assay for viable cell number
The Crystal Violet assay as described by Wosikowski et al [9] was used to determine cell viability. Cells were seeded at approximately 10,000 cells per well in 48-well plates and allowed to adhere overnight. The following morning, cells were treated with the pyrrole compounds and maintained in drug containing media for 72 hours. Following the 72-hour exposure, cells were fixed in 100% methanol for 10 minutes, stained with 0.5% crystal violet solution in 25% methanol for 10 minutes, washed three times with 1X PBS to remove excess dye and allowed to dry overnight. The following morning, crystal violet containing cells were solubilized in 0.1 M sodium citrate in 50% ethanol, and an absorbance reading was taken at 540 nm.
2.6 TUNEL Assay for apoptosis
The method of Gavrieli et al. [10] was utilized as an independent assessment of apoptotic cell death in combined cytospins containing both adherent and non-adherent cells. Cells were fixed and the fragmented DNA in cells undergoing apoptosis was detected using the In Situ Cell Death Detection Kit (Boehringer-Mannheim), where strand breaks are end labeled with fluorescein dUTP by the enzyme terminal transferase. Cells were then washed, mounted in Vectashield and photographed using a Nikon fluorescent microscope. Three fields per condition were evaluated at a magnification of 10X.
2.7 Beta-Galactosidase histochemical staining for senescence
pH 6.0-dependent β-galactosidase expression was used as a marker for senescence along with senescent related morphology [11]. At the appropriate times after treatment, cells were washed twice with PBS and fixed with 2% formaldehyde, 0.2% glutaraldehyde for 5 min. The cells were then washed again with PBS and stained with a solution of 1mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactosidase in dimethylformamide (20mg/ml stock), 5mM potassium ferrocyanide, 5mM potassium ferricyanide, 150mM NaCl, 40mM citric acid/sodium phosphate, pH 6.0, and 2mM MgCl2. Following overnight incubation at 37°C, the cells were washed twice with PBS, and photographed with a light microscope. The extent of senescence was quantified based on the mean number of cells displaying both changes in morphology as well as blue-green staining for three fields (containing at least 50 cells per field) for each experimental condition.
2.8 Autophagy Detection with Acridine Orange Staining
As a marker of autophagy, the volume of the cellular acidic compartment was visualized by acridine orange staining [12]. Cells were seeded in T-25 flasks and treated as described above for the cell viability study. At the appropriate time points following treatment, cells were incubated with medium containing 1μg/ml acridine orange (Molecular Probes, Eugene, OR) for 15 min. The acridine orange was removed and fluorescent micrographs were taken using an inverted fluorescent microscope. Autophagy was quantified based on the mean number of cells displaying intense red staining for three fields (containing at least 50 cells per field) for each experimental condition.
2.9. Cell Cycle Analysis
Cells were seeded in 135 mm petri dishes at a density of 2 × 106 to 3 × 106 cells per dish and incubated with media or JG-03-14 at 500nM for the indicated times. Cell cycle analysis was performed by prodidium iodide staining (PIF) and analyzed by flow cytometry. Cells were harvested at appropriate times with trypsin and centrifuged at 1500 rpm for 2 minutes at 4°C. Cell pellets were resuspended in a PIF solution and maintained on ice. Approximately 1 × 106 cells per condition were collected for the cell cycle analysis. Prior to analysis, each sample was filtered through a 35 μm nylon mesh. Cells were analyzed with an EPICS 753 flow cytometer (Coulter Electronics, Hialeah, FL) using the 488 nm argon laser and standard optical emission filters. The resulting DNA distributions were analyzed for the proportion of cells in various stages of the cell cycle using Cytologic Software (Coulter Electronics). A minimum of 30,000 events were collected for each sample.
2. 10 Statistical Analysis
Data were analyzed using the student paired two-tailed “t” test.
3. Results
3.1 Effects of substituted pyrroles on viability of MCF-7 breast tumor cells
Figure 1A presents the structure of the compounds utilized for the studies described in this manuscript. Sensitivity of MCF-7 breast tumor cells to JG-03-14 and the related compounds JG-05-1, JG-05-2, JG-05-3, JG-05-4, JG-05-5, JG-05-6, JG-05-7, JG-05-8, and JG-05-10 was determined utilizing the Crystal Violet Assay [9] with continuous exposure of the cells to the indicated compounds for 72 hours prior to fixation and staining. Figures 1B and 1C show that compound JG-05-2 and JG-03-14 produced concentration dependent inhibition of cell growth in both MCF-7 and MDA-MB231 cells; Adriamycin, which is known to have antiproliferative and cytotoxic effects in this cell line [13, 14], was utilized as a positive control at a concentration of 100nM. in contrast, the other compounds tested were essentially ineffective at concentrations up through 500nM (data not shown). All subsequent studies were performed using JG-03-14 at a concentration of 500nM, a concentration that produced essentially equivalent antiproliferative effects (between 70–80% growth inhibition) in both the MCF-7 and MDA-MB231 cell lines.
Figure 1.
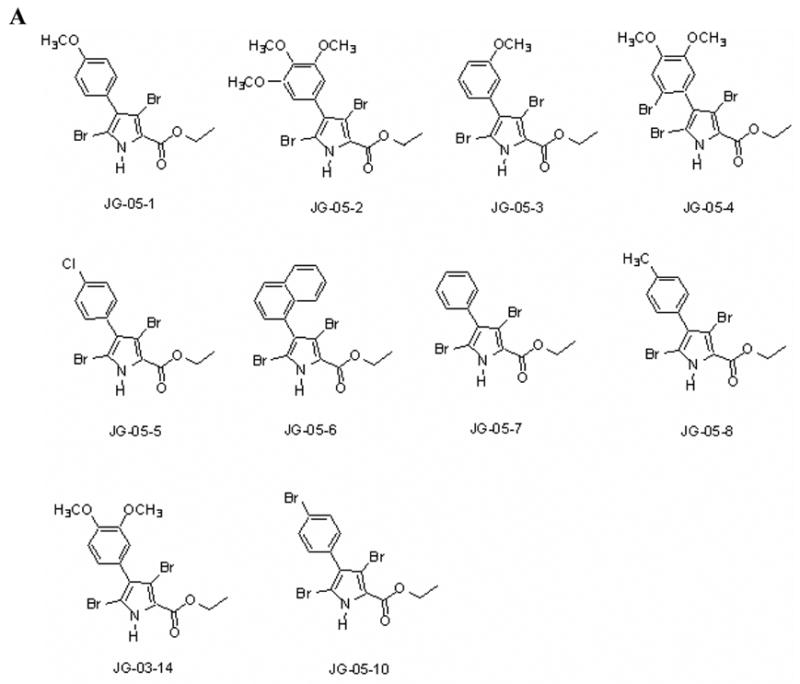
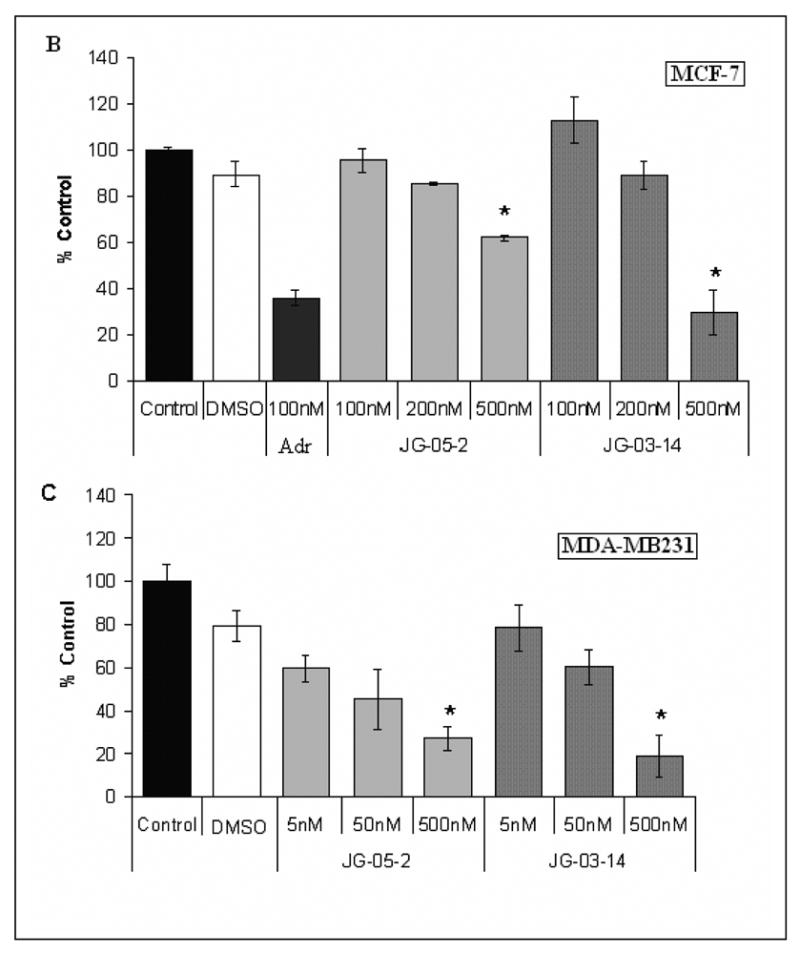
Antiproliferative activity of substituted pyrroles in MCF-7 and MDA-MB231 breast tumor cells. A. Structure of substituted pyrroles utilized for the current studies. B. Effects of substituted pyrroles on growth of MCF-7 cells. C. Effects of the substituted pyrroles on the growth of MDA-MB231 cells. Cells were treated with either JG-05-02 or JG-03-14. Cells were seeded in 48 well plates at a density of 10,000 cells per plate and exposed to the indicated compounds continuously for 72 hours. The solvent, DMSO, was utilized as a negative control and 100nM Adriamycin was used as a positive control for each assay. Values shown are for the only two active compounds screened and represent means ± standard errors for 3 replicate experiments. Growth inhibition at 500nM JG-03-14 was statistically significant compared to vehicle controls with p≤ 0.05 for both the MCF-7 and MDA-MB231 cell lines.
3.2 Growth arrest and cell death of MCF-7 cells after exposure to JG-03-14
In order to distinguish between the possible growth arrest and cell death effects of this compound, cell viability was monitored over a time frame of 5 days after initiating treatment with JG-03-14. Adriamycin was again used as a positive control; the effects of Adriamycin essentially overlapped with those of JG-03-14 in the MCF-7 cells (data not shown). Figure 2 indicates that during the first 72 hours of treatment, the number of viable MCF-7 cells showed a steady decline, indicative of cell death. The number of cells at 72 hours after JG-03-14 treatment was approximately 14% of control cells. Between days 3 and 5, cell number remained relatively constant, which is indicative of growth arrest and possibly senescence.
Figure 2.
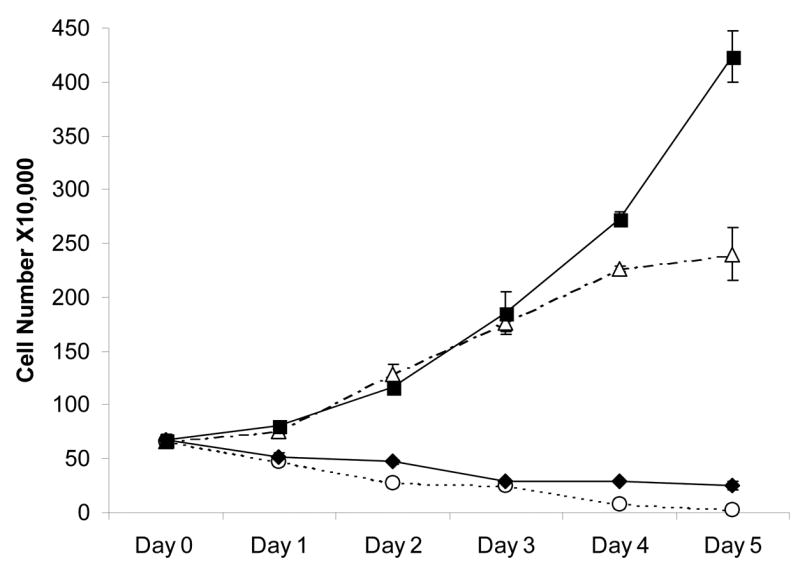
Temporal response of MCF-7 and MDA-MB231 cells to JG-03-14. Growth arrest and cell death were evaluated in response to a continuous 5-day treatment with 500nM JG-03-14. 100nM Adriamycin was used as a positive control (not shown). Cell viability was monitored by trypan blue exclusion. MDA-MB231 cells (dashed lines) were exposed to either control media (△) or JG-03-14 (○). MCF-7 cells (solid lines) were also either exposed to media (■) or JG-03-14 (◆). Data presented was pooled for three experiments ± standard errors.
3.3 Minimal Induction of Apoptosis by JG-03-14
The possibility that the cell death observed in MCF-7 cells after the first few days of exposure to JG-03-14 might be occurring through apoptosis was assessed using the TUNEL assay [10]. Cells treated with an elevated concentration of taxol (1μM) for three days, conditions that promoted extensive apoptosis, were used as a positive control. Despite the pronounced time dependent cell death noted during the first 72 hours of treatment, little fluorescent staining indicative of apoptosis was detected in the cells exposed to JG-03-14 (figure 3C). A further assessment of DNA fragmentation by analysis of the sub G1/G0 population using FACS analysis, shown in Figure 6, supports the conclusion of minimal apoptosis induced by JG-03-14 in MCF-7 cells.
Figure 3.
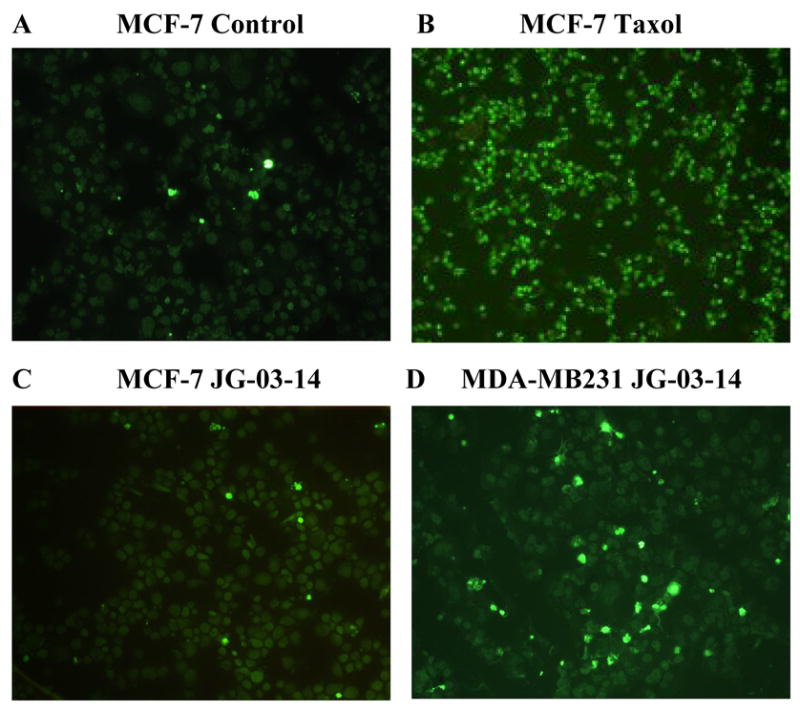
Determination of Apoptosis by the TUNEL assay. MCF-7 and MDA-MB231 cells were exposed to 500nM JG-03-14 for 5 days and monitored for apoptosis utilizing the TUNEL assay. Continuous exposure to Taxol (1μM) was used as a positive control for apoptosis. Apoptosis was monitored in combined cytospins of both adherent and non-adherent cells. Data presented are from one of two experiments with similar results. A. Untreated MCF-7 cells. B. Taxol, 1μM. C. MCF-7 cells after three days exposure to JG-03-14. D. MDA-MBA231 cells after three days exposure to JG-03-14. Magnification was 10X.
Figure 6.
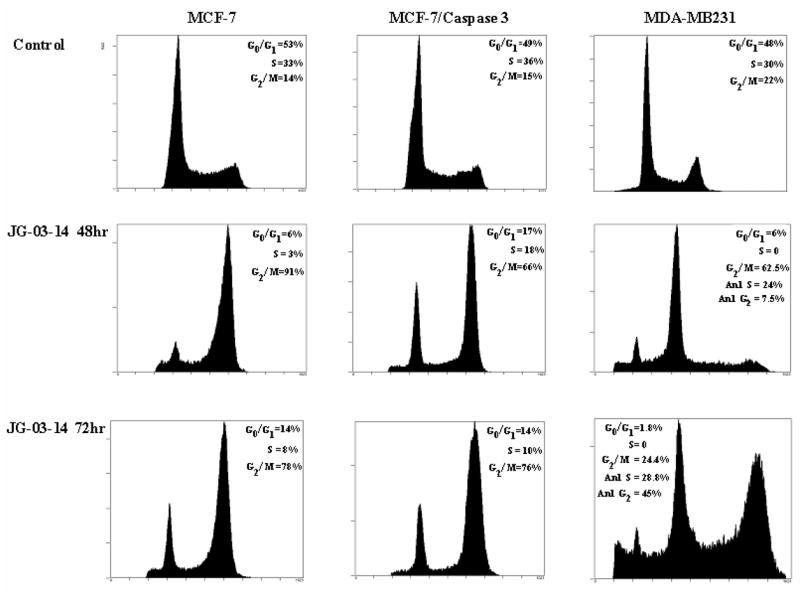
FACS analysis of breast tumor cells exposed to 500nM JG-03-14. MCF-7, MCF-7/Caspase 3, and MDA-MB231 cells were exposed to a 500nM concentration of JG-03-14 for 48 or 72 hours. At the appropriate time points, cells were harvested and stained with a propidium iodide staining (PIF) solution and quantified with a flow cytometer. Flow cytometry PIF voltages were altered for MDA-MB321 treatment groups compared to control groups in order to account for the increasing polyploidy seen at 48 and 72 hrs following drug exposure. 30,000-50,000 events per condition were collected for each histogram.
3.4 Induction of Autophagy by JG-03-14
In view of the minimal levels of apoptosis in MCF-7 cells with continuous exposure to 500nM JG-03-14, and since our recent findings indicated extensive promotion of autophagy by irradiation in MCF-7 cells [15], the possibility of autophagic vacuole formation associated with autophagic cell death was evaluated. Figure 4A indicates that there was significant autophagic vesicle formation in MCF-7 cells exposed to 500nM JG-03–14, based on acridine orange staining. Figure 4B indicates that up to 70% of the cell population appeared to be undergoing autophagy by the third day of treatment.
Figure 4.
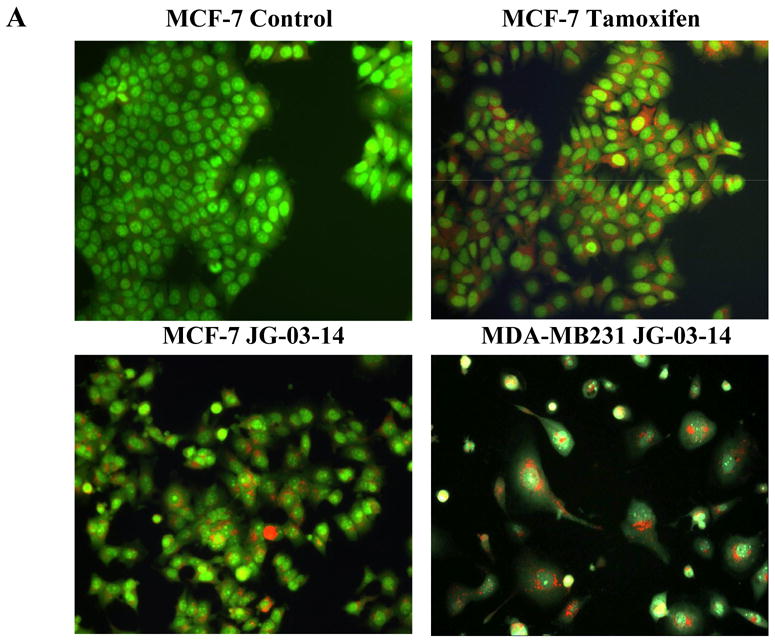
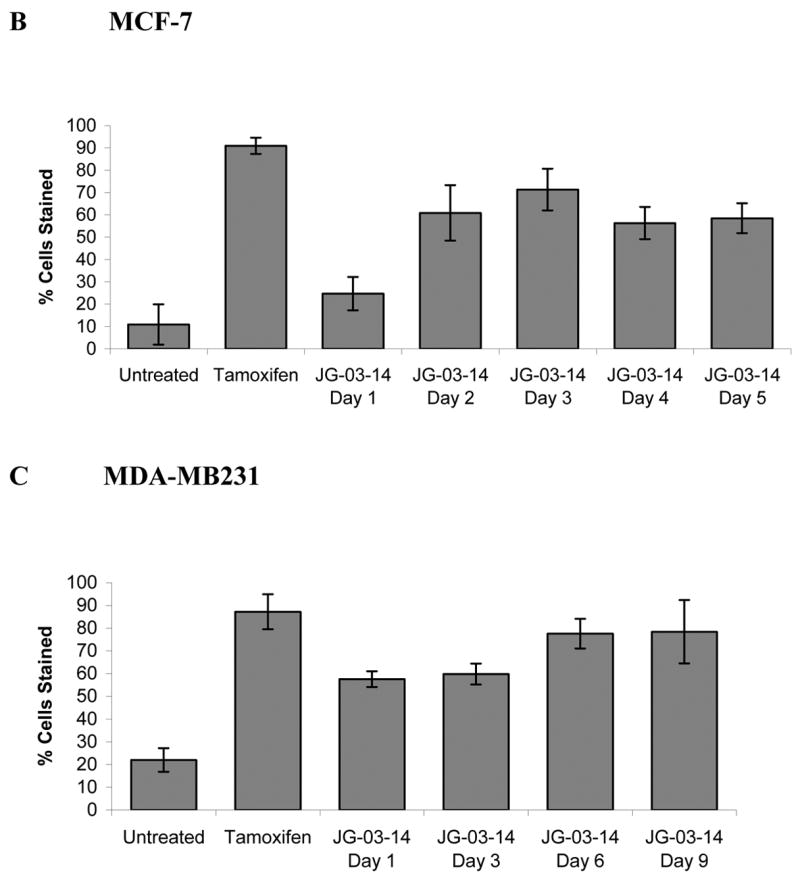
Determination of autophagy by acridine orange staining. A. MCF-7 or MDA-MB231 cells were exposed to 500nM JG-03-14 for 3 days, washed, and fluorescence monitored by fluorescence microscopy, where green staining reflects the background and bright orange staining occurs in acidic (autophagic) vacuoles. Tamoxifen (2.5μM) exposure for four days was used as a positive control. B. and C. The percent of the total cell population that stained for acridine orange was quantified. Data presented is representative of 3 replicate experiments ± standard errors (at a magnification of 20X). Autophagy was significantly greater than untreated controls (p≤ 0.05) for all conditions with the exception of day 1 for MCF-7 cells.
3.5 Detection of senescence in the residual surviving cell population
In view of previous studies where the hallmarks of senescence were detected in residual cells surviving after treatment with the vitamin D3 analogue, EB 1089 and ionizing radiation [16], we evaluated whether the residual population after treatment with 500nM of JG-03-14 was senescent, based on staining for beta-galactosidase [11], cell expansion, and flattening. Figure 5A shows clear evidence of a senescent population upon treatment of MCF-7 cells with JG-03-1,. Figure 5B provides quantification of senescence with maximal staining evident by the sixth day of continuous drug exposure and indicates that up to 80% of the residual cell population was in a senescent state. At lower doses (50, 100, and 200nM), JG-03-14 failed to produce any significant staining or morphological changes associated with senescence (data not shown).
Figure 5.
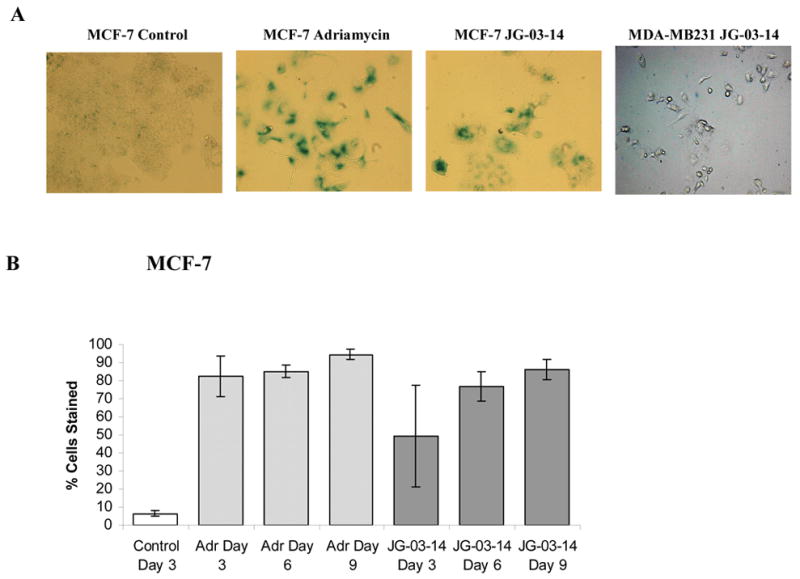
Senescence in residual surviving cells after JG-03-14 treatment for six days. A. MCF-7 or MDA-MB231 cells seeded in 6 well plates at a density of 2 × 105cells/flask were continuously exposed to 500nM JG-03-14. Cells were fixed and suspended in 1X PBS and stained with Beta-Galactosidase. Data shown is for Day 6. Exposure to 0.75 μM doxorubicin for 2 hours was utilized as a positive control for senescence. All treatment groups were photographed at a 20X magnification; untreated groups were photographed at a 10X magnification. Data presented is for one of three experiments with similar results. B. Quantification of beta-gal staining ± standard errors. Control cells were allowed to grow for three days and adriamycin-treated cells were utilized as a positive control. Data is presented as the mean ± standard error for three experiments at a magnification of 20X. (Quantification was assessed with fields at 10X in order to obtain 50 cells per field). The extent of senescence staining in MCF-7 cells was significant (p≤ 0.05 compared to the untreated group) for all days of treatment with JG-03-14 with the exception of day 3.
3.6 Lack of proliferative recovery in MCF-7 cells exposed to JG-03-14
One desirable element of drug action in addition to primary cell killing or growth inhibitory effects is the ability to suppress the capacity of the tumor cell to recover proliferative capacity. In previous work, we have demonstrated proliferative recovery of MCF-7 cells after exposure to either ionizing radiation or adriamycin [16, 17, 18]. However, even after 15 days there was no evidence of a proliferating cell population in MCF-7 cells exposed to JG-03-14 (data not shown).
3.7 Drug effects in MDA-MB231 breast tumor cells
Although MCF-7 and MDA-MB231 cells are non-isogenic cell lines, the fact that MDA-MB231 cells are mutant in p53 permitted us to determine whether the presence of p53 is a prerequisite for sensitivity to JG-03-14 as well whether the absence of functional p53 might alter the nature of the cell death response. Figure 1C indicates that, as with the MCF-7 cells, JG-05-2 and JG-03-14 inhibited growth of the MDA-MB231 cells while the other analogs were essentially ineffective (data not shown). Data from clonogenic survival studies, shown in Table 1, indicated that both MDA-MB231 cells and MCF-7 cells are highly sensitive to JG-03-14, with almost complete abrogation of reproductive capacity at a drug concentration of 500nM. Time course studies presented in Figure 2 indicated that MDA-MB231 cells responded to JG-03-14 in a fashion that was generally similar to the MCF-7 cells, with a gradual decline in the number of viable cells over time. However, unlike the studies with MCF-7 cells, there were essentially no surviving cells detected after five days treatment of MDA-MB231 cells with JG-03-14. As expected, due to the extensive cell death, there was no evidence of proliferative recovery evident after 15 days (data not shown).
Table 1. Clonogenic Survival in cells exposed to 500nM JG-03-14.
Clonogenic survival of MCF-7 and MDA-MB231 cells following treatment with a 500nm concentration of JG-03-14. Cells were seeded at 200 cells per dish for control groups and 5000 cells per dish for treatment groups. JG-03-14 groups were exposed continuously for 14 days then stained with a dilute crystal violet solution to count colonies. Shown is the percent clonogenic survival mean for three different experiments in both MCF-7 and MDA-MB231 cell lines ± standard errors.
| % Clonogenic Survival ± SD | ||
|---|---|---|
| Control | JG-03-14 500nM | |
| MDA-MB231 | 96.8 ± 15 | 2.5 ± 2.04 |
| MCF-7 | 50.75 ± 6.11 | 0.3 ± 0.2 |
3.8 Mode of cell death in MDA-MB231 cells
As with the studies in MCF-7 cells, we evaluated both apoptosis and autophagy after treatment with JG-03-14. Figures 3D indicates that a significant amount of apoptosis appeared three days after treatment. MDA-MB231 cells also showed quite extensive acridine orange staining indicative of autophagic cell death (Figure 4A and 4C). In contrast to the findings in MCF-7 cells, MDA-MB231 cells failed to stain for β-galactosidase or demonstrate morphological alterations ( enlargement and flattening) associated with senescence (Figure 5A, far right).
3.9 Sensitivity to JG-03-14 in MCF-7/caspase 3 cells
MCF-7 cells lack caspase 3 [19], which has been associated with resistance to apoptosis and possibly reduced sensitivity to chemotherapy [20, 21]. Consequently, sensitivity to 500nM JG-03-14 was assessed in MCF-7/caspase 3 cells [15]. Growth as a percent of untreated controls was essentially similar in the MCF-7/caspase 3 cells (47.3 ± 15.9%) and in MCF-7 cells (29.5 ± 9.4%) exposed to JG-03-14.
3.10. FACS analysis of breast tumor cells treated with JG-03-14
Given the evidence that JG-03-14 is likely to be acting as a microtubule poison [7], cell cycle distribution after drug treatment was determined by FACS analysis in MCF-7, MCF-7/Caspase 3, and MDA-MB231 cells. Incubation with JG-03-14 resulted in a substantial G2/M block, consistent with its proposed microtubule destabilizing properties (Figure 6). In the MCF-7 and MCF-7/Caspase 3 cells, this G2/M block persisted for 72 hours. Interestingly, the p53 mutant MDA-MB231 cell line continued to replicate DNA following G2/M arrest, resulting in development of a hyperdiploid population (polyploidy). This effect was first evident at 48 hours and became quite pronounced after 72 hours of treatment, where a full 75% of the remaining cells were polyploid. FACS analysis confirmed a minimal degree of apoptosis (a sub G1 cell population) in both the MCF-7 and MCF-7/caspase 3 cell lines following both the 48 and 72 hour incubation periods with 500nM JG-03-14. A moderate apoptotic population was detected in MDA-MB231 cells, consistent with the results of the TUNEL assay; however, due to the extensive noise generated as a consequence of the dead and dying cells, apoptotic events could not be accurately quantified.
3. 11 Sensitivity to JG-03-14 in multidrug resistant tumor cells
One common problem with chemotherapy of breast cancer, as well as other malignancies, is drug resistance that is frequently expressed through several different mechanisms. The first is through multidrug resistance (mdr) pumps. These mdr cell lines show cross-resistance to multiple structurally unrelated drugs including anthracyclines, the vinca alkaloids, the epipodophyllotoxins, actinomycin D, and colchicines [22]. The lack of drug accumulation from efflux pumps in mdr cells is the result of overexpression of a cellular membrane protein called P-glycoprotein (Pgp) [23]. A second type of multidrug resistance is associated with overexpression of the glutathione transferase and glutathione peroxidase. These multidrug resistant cells are associated with the appearance of an anionic isozyme similar to the glutathione transferase seen in human placenta [8]. MCF-7/ADR cells are known to posses both efflux pumps as well as increased levels of glutathione transferase and peroxidase [8]. In this context, we evaluated sensitivity to JG-03-14 in multidrug resistant MCF-7/ADR cells. Table II indicates that the MCF-7/ADR cells maintained sensitivity to JG-03-14 while demonstrating resistance to adriamycin. This observation demonstrates that neither common mechanism of resistance to conventional chemotherapeutics was a barrier to the cytotoxicity of JG-03-14.
Table 2. Sensitivity to adriamycin and JG-03-14 inMCF-7/ADR cells.
Antiproliferative activity of substituted pyrroles in MCF-7/ADR breast tumor cells. Cells were seeded in 48 well plates at a density of 10,000 cells per plate and exposed to the indicated compounds continuously for 72 hours. The solvent, DMSO, was utilized as a negative control for each assay (not shown). After 72 hours, plates were fixed with methanol and stained with crystal violet. Plates were allowed to dry overnight then solubilized in 0.1M sodium citrate and an absorbance reading was taking at 570 nanometers. 100nM Adriamycin was used as a negative control for each assay. MCF-7 crystal violet data from figure 1B is shown for comparison. Values shown are averages of percent control for three experiments ± standard errors.
| Adr
|
JG-03-14
|
||||
|---|---|---|---|---|---|
| Control | 100nM | 125 nM | 250 nM | 500 nM | |
| MCF-7/ADR | 100 ± 11 | 124 ± 21 | 96 ± 9 | 52 ± 4 | 19 ± 3 |
|
| |||||
| Adr
|
JG-03-14
|
||||
| Control | 100nM | 100nM | 200nM | 500nM | |
|
| |||||
| MCF-7 | 100 ± 1 | 37 ± 1.7 | 112 ± 10 | 89 ± 6 | 30 ± 9 |
4. Discussion
Of the 10 compounds shown in Figure 1A, both MCF-7 cells and MDA-MB231 cells were most sensitive to JG-03-14, with growth inhibition of 70% and 82%, respectively, at a concentration of 500nM. At this concentration, clonogenic survival of both MCF-7 and MDA-MB231 cells was suppressed by greater than 90%, indicating that JG-03-14 effectively eliminates the self-renewal capacity of the breast tumor cells. These observations are consistent with the lack of proliferative recovery in both cell lines.
The observation that MDA-MB231 cells demonstrated sensitivity to JG-03-14 strongly suggests that lack of functional p53 is not a barrier to drug action. Although the absence of functional 53 has been considered to confer resistance to chemotherapy [24,25], there is extensive evidence for sensitization to chemotherapeutic agents, including the microtubule poison, paclitaxel, in p53 deficient cells [26–30]. In the case of paclitaxel, sensitization in p53 mutant cells is thought to be due to the capacity of functional p53 to regulate cell cycle progression following spindle checkpoint arrest. Microtubule poisoning initially results in a G2/M arrest, where activation of the spindle assembly checkpoint prevents progression through mitosis [31]. However, cancer cells are frequently able to adapt and undergo mitotic slippage out of G2/M arrest [32]. While cells which possess wild type p53 can subsequently arrest at G0/G1 post mitotic slippage, a response that is mediated by p21/WAF1[33], cells lacking functional p53 are thought to continue synthesizing DNA after mitotic slippage without cytokinesis, resulting in polyploidy [34,35,36].
In our studies, a substantial fraction of the p53 mutant cells became hyperdiploid following 72 hours of drug treatment, strongly suggesting that MDA-MB231 cells undergo mitotic slippage and re-replicate their DNA following a failed mitosis. The development of a hyperdiploid (polyploid) population was not observed in the analysis of MCF-7 or MCF-7/Caspase 3 cells, and appears to indicate that the (residual surviving) p53 wild type cells preferentially undergo senescence following JG-03-14 treatment, similar to our previous observations with adriamycin [37]. Previous studies have indicated that p53 status can play an important role in determining whether cells rereplicate DNA following spindle checkpoint-mediated arrest [33–36]. However, a fuller understanding of the role of p53 in the response to JG-03-14 will require the utilization of isogenic cell lines with a knockdown of p53 function.
It remains to be determined whether the senescence detected in the residual MCF-7 cell population following JG-03-14 treatment is dependent on p53. To our knowledge, only one other study has reported a senescence response to microtubule poisons, also in cells with functional p53 [38]. The absence of a residual senescent MDA-MB231 population could simply reflect the more extensive cell killing evident after exposure of MDA-MB231 cells to JG-03-14. In the MCF-7 cell line, the residual senescent population persisted for 15 days post treatment, but the lack of proliferative recovery strongly suggests that the senescent state is permanent and irreversible.
As indicated above, the absence of functional p53 appears to allow for a hyperdiploid state, which may ultimately lead to increased cell death as the cells with hyperdiploid DNA fail to divide. The apparent difference in the extent of apoptosis observed in the MDA-MB231 and MCF-7 cells does not appear to be solely a function of the expression of caspase 3 in MDA-MB231 cells and not in MCF-7 cells [19], since the MCF-7/caspase 3 cells do not appear to undergo apoptosis after JG-03-14. It is further worth noting that extensive autophagy occurs in the MDA-MB231 cells, despite the fact that autophagy is frequently thought to be dependent on functional p53 [39].
JG-05-02 and JG-03-14, the compounds demonstrating activity against the MCF-7 and MDA-MB231 cells, share a partial homology to the drug colchicine. According to molecular modeling studies [8 and unpublished data], JG-03-14 is likely to act as a microtubule poison by binding at the colchicine-binding site on tubulin. An increase in the number of methoxy groups on the phenol ring structure of these functional pyrroles appears to increase the binding affinity of the compound for the colchicine-binding site, which may be responsible for their increased cytotoxicity to the breast tumor cell as compared to the other pyrroles studied.
One further implication for the potential utility of JG-03-14 relates to its ability to bind tubulin. Similar tubulin binding agents, such as the combretastatin family of drugs including CA-4-P, are known to be vascular disrupting agents (VDA) [40]. The tumor vasculature is an exciting target for therapy because most tumor cells rely on intact vascular support for growth and survival [41]. These compounds cause an extensive shutdown of the vascular supply to the tumor, interfering with both tumor growth and metastatic spread. Several vascular disrupting agents are currently in clinical trials for the treatment of cancer, and the possibility that JG-03-14 might also disrupt tumor vasculature appears worthy of future consideration.
The fact that JG-03-14 appears to be active in multidrug resistant tumor cells further argues for its potential utility and the possibility that this agent could circumvent multidrug resistance. It should, however, be noted, that recent studies indicate that the MCF-7/ADR cells are not derived from breast cancer but are likely to be an ovarian tumor line [42]. Nevertheless, this fact does not undermine the validity of our observations that JG-03-14 does not appear to be a substrate for the multidrug resistant pump in this cell line. Similar findings have recently reported by others [7].
Finally, studies in a (prostate) tumor xenograft model system indicate that JG-03-14 can be an effective antitumor agent with minimal toxicity to the test animal [7]. These are encouraging findings that indicate that JG-03-14 or its analogs might ultimately have utility as antitumor drugs in the clinic.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Furstner A, Weintritt H, Hupperts A. A new, titanium-mediated approach to pyrroles: first synthesis of lukianol A and lamellarin 0 dimethyl ether. J Org Chem. 1995;60:6637–41. [Google Scholar]
- 2.Edstrom E, Wei Y. Synthesis of a novel pyrrolo[2,3-d]pyrimidine alkaloid, Rigidin. Org Chem. 1995;58:403–7. [Google Scholar]
- 3.Gupton J, Burham B, Krumpe K, Du K, Sikorski J, Warren A, et al. Synthesis and cytotoxicity of 2,4-disubstituted and 2,3,4-trisubsitituted brominated pyrroles in murine and human cultured tumor cells. Arch Pharm Pharm Med Che. 2000;333:3–9. doi: 10.1002/(sici)1521-4184(200001)333:1<3::aid-ardp3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 4.Burnham B, Gupton J, Krumpe K, Webb T, Shuford J, Bowers B, et al. Cytotoxicity of substituted alkyl-3,4-bis(4-methoxyphenyl)pyrrole-2-carboxylates in L1210 lymphoid leukemia cells. Arch Pharm Pharm Med Chem. 1998;331:337–41. doi: 10.1002/(sici)1521-4184(199811)331:11<337::aid-ardp337>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Gupton J, Burham B, Byrd Krumpe K, Stokes C, Shuford J, et al. The cytotoxicity and mode of action of 2,3,4-trisubstituted pyrroles and related derivatives in human Tmolt4 leukemia cells. Pharmazie. 1999;9:691–7. [PubMed] [Google Scholar]
- 6.Evans M, Smith D, Holub J, Argenti A, Hoff M, Dalglish G, et al. Synthesis and cytotoxicity of substituted ethyl 2-phenyacyl-3-phenylpyrrole-4-carboxylates. Arch Pharm Pharm Med Chem. 2003;3:1–10. doi: 10.1002/ardp.200390018. [DOI] [PubMed] [Google Scholar]
- 7.Mooberry SL, Weiderhold KN, Dakshanamurthy S, Hamel E, Banner EJ, Kharlamova A, et al. Identification and characterization of a new tubulin-binding tetrasubstituted brominated pyrroles. Mol Pharmacol. 2007 doi: 10.1124/mol.107.034876. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 8.Batist G, Tulpule A, Sinha B, Katki A, Myers C, Cowan K. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J Biol Chem. 1986;261:15544–49. [PubMed] [Google Scholar]
- 9.Wosikowski K, Kung W, Hasmann M, Loser R, Eppenberger U. Inhibition of growth-factor-activated proliferation by anti-oestrogen and effects of on early gene expression of MCF-7 cells. Int J Cancer. 1993;53:290–7. doi: 10.1002/ijc.2910530220. [DOI] [PubMed] [Google Scholar]
- 10.Gavrieli Y, Sherman Y, Ben-Sasson S. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Nat Acad Sci. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphika E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–44. [PubMed] [Google Scholar]
- 13.Fornari FA, Jr, Jarvis WD, Grant S, Orr MS, Randolph JK, Gewirtz DA. Growth arrest and non-apoptotic cell death associated with the suppression of c-myc expression in MCF-7 breast tumor cells following acute exposure to doxorubicin. Biochem Pharmacol. 1996;51:931–40. doi: 10.1016/0006-2952(96)00050-0. [DOI] [PubMed] [Google Scholar]
- 14.Fornari FA, Jr, Jarvis WD, Grant S, Orr MS, Randolph JK, Gewirtz DA. Induction of differentiation and growth arrest associated with nascent (nonoligosomal) DNA fragmentation and reduced c-myc expression in MCF-7 human breast tumor cells after continuous exposure to a sublethal concentration of doxorubicin. Cell Growth Differ. 1994;5:723–33. [PubMed] [Google Scholar]
- 15.DeMasters G, Di Xu, Newsham I, Shiu R, Gewirtz D. Potentiation of radiation sensitivity in breast tumor cells by the vitamin D analog, EB 1089, through promotion of autophagy and interference with proliferative recovery. Mol Cancer Ther. 2006;5:2786–97. doi: 10.1158/1535-7163.MCT-06-0316. [DOI] [PubMed] [Google Scholar]
- 16.DeMasters GA, Gupta MS, Jones JR, Cabot M, Wang H, Gennings C, et al. Potentiation of cell killing by fractionated radiation and suppression of proliferative recovery in MCF-7 breast tumor cells by the Vitamin D3 analog EB 1089. J Steroid Biochem Mol Biol. 2004;92:365–74. doi: 10.1016/j.jsbmb.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Jones K, Elmore L, Jackson-Cook C, DeMasters G, Povirk LF, Holt SE, et al. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumor cells. Int J Radiat Biol. 2005;81:445–58. doi: 10.1080/09553000500168549. [DOI] [PubMed] [Google Scholar]
- 18.Elmore LW, Di X, Dumur C, Holt SE, Gewirtz DA. Evasion of a single-step, chemotherapy-induced senescence in breast cancer cells: implications for treatment response. Clin Cancer Res. 2005;11:2637–43. doi: 10.1158/1078-0432.CCR-04-1462. [DOI] [PubMed] [Google Scholar]
- 19.Kurokawa H, Nishio K, Fukumoto H, Tomonari A, Suzuki T, Saijo N. Alteration of caspase-3 (CPP32/Yama/apopain) in wild-type MCF-7, breast cancer cells. Oncol Rep. 1999;6:33–7. doi: 10.3892/or.6.1.33. [DOI] [PubMed] [Google Scholar]
- 20.Devarajan E, Sahin A, Chen J, Krishnamurthy R, Aggarwal N, Brun AM, et al. Down-regulation of caspase 3 in breast cancer: a possible mechanism of chemoresistance. Oncogene. 2002;21:8843–51. doi: 10.1038/sj.onc.1206044. [DOI] [PubMed] [Google Scholar]
- 21.Yang XH, Sladek T, Liu X, Butler B, Froelich C, Thor A. Reconstitution of Caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Res. 2001;61:348–54. [PubMed] [Google Scholar]
- 22.Chen Y, Mickley L, Schwartz A, Acton E, Hwang J, Fojo A. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J Biol Chem. 1990;265:10073–80. [PubMed] [Google Scholar]
- 23.Juliano J, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochem Biophys Acta. 1976;455:152–62. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 24.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–67. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 25.Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, et al. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–10. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 26.Hawkins D, Demers W, Galloway D. Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res. 1996;56:892–8. [PubMed] [Google Scholar]
- 27.Vikhanskaya FS, Vignati P, Beccaglia C, Ottoboni P, Russo M, Broggini M. Inactivation of p53 in a human ovarian cancer cell line increases the sensitivity to paclitaxel by inducing G2/M arrest and apoptosis. Exp Cell Res. 1998;241:96–101. doi: 10.1006/excr.1998.4018. [DOI] [PubMed] [Google Scholar]
- 28.Wagenknecht R, Grimmel B, Dichgans C, Weller M. Taxol mediated augmentation of CD95 ligand-induced apoptosis of human malignant glioma cells. Association with bcl-2 phosphorylation but neither activation of p53 nor G2/M cell cycle arrest. Br J Cancer. 1998;77:404–11. doi: 10.1038/bjc.1998.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safran H, King T, Choy H, Gollerkeri A, Kwakwa H, Lopez F, et al. A p53 mutations do not predict response to paclitaxel/radiation for non-small cell lung carcinoma. Cancer. 1996;78:1203–10. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1203::AID-CNCR6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Vasey P, Jones N, Jenkins S, Dive C, Brown R. Cisplatin, camptothecin and taxol sensitivities of cells with p53-associated multidrug resistance. Mol Pharm. 1996;50:1536–40. [PubMed] [Google Scholar]
- 31.Logarinho E, Bousbaa H, Dias JM, Lopes C, Amorim I, Antunes-Martins A, et al. Different spindle checkpoint proteins monitor microtubule attachment and tension at kinetochores in Drosophila cells. Journal Cell Science. 2004;117:1757–71. doi: 10.1242/jcs.01033. [DOI] [PubMed] [Google Scholar]
- 32.Azeddine E, Cunha M, Kirsch-Volders M. Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis-block assay. Mutagenesis. 1998;13:193–8. doi: 10.1093/mutage/13.2.193. [DOI] [PubMed] [Google Scholar]
- 33.Stewart Z, Mays D, Pietenpol J. Defective G1-S cell cycle checkpoint function sensitizes cells to microtubule inhibitor-induced apoptosis. Cancer Res. 1999;59:3831–37. [PubMed] [Google Scholar]
- 34.Notterman D, Young S, Wainger B, Levine AJ. Prevention of mammalian DNA reduplication, following the release from the mitotic spindle checkpoint, requires p53 protein, but not p53-mediated transcriptional activity. Oncogene. 1998;17:2743–51. doi: 10.1038/sj.onc.1202210. [DOI] [PubMed] [Google Scholar]
- 35.Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressor prevents tetraploidization. Genes Dev. 2006;20:2687–700. doi: 10.1101/gad.1447006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cross S, Sanchez C, Morgan C, Schinake M, Ramel S, Idzerda R, et al. A p53-dependent mouse spindle checkpoint. Science. 1995;267:1353–6. doi: 10.1126/science.7871434. [DOI] [PubMed] [Google Scholar]
- 37.Elmore L, Rehder C, Di X, McChesney P, Jackson-Cook C, Gewirtz D, et al. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–15. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 38.Klein LE, Freeze BS, Smith AB, 3rd, Horwitz SB. The microtubule stabilizing agent discodermolide is a potent inducer of accelerated cell senescence. Cell Cycle. 2005;4:501–7. doi: 10.4161/cc.4.3.1550. [DOI] [PubMed] [Google Scholar]
- 39.Jin S. p53, autophagy and tumor suppression. Autophagy. 2005;1:171–3. doi: 10.4161/auto.1.3.2051. [DOI] [PubMed] [Google Scholar]
- 40.Tozer G, Prize V, Wilson J, Locke R, Vojnovic B, Stratford M, et al. Combretastatin A-4 phosphate as a tumor vascular targeting agent: early effects in tumors and normal tissues. Cancer Res. 1999;59:1626–34. [PubMed] [Google Scholar]
- 41.Folkman J. Angiogenesis and angiogenesis inhibition: an overview. In: Goldberg I, Rosen E, editors. Regulation of Angiogenesis. Vol. 9. Birkhäuser Verlag; 1997. pp. 1–8. [Google Scholar]
- 42.Liscovitch M, Ravid D. A case study in misidentification of cancer cell lines: MCF-7/AdR cells (redesignated NCI/ADR-RES) are derived from OVCAR-8 human ovarian carcinoma cells. Cancer Lett. 2007;245:350–2. doi: 10.1016/j.canlet.2006.01.013. [DOI] [PubMed] [Google Scholar]