

Abstract
IL-4 and IL-13 are up-regulated during in vivo responses to many nematode parasites, but increasing evidence suggests that increases in IL-13 can also occur independently of the IL-4-dominant Th2 response. Blocking B7 after Trichuris muris inoculation inhibits resistance and IL-4 elevations, instead resulting in an IFN-γ-dominant response associated with susceptibility. However, blocking IFN-γ under these conditions restores IL-13-dependent resistance. In this study, we examined the mechanism of IL-13 up-regulation and associated protection during this in vivo immune response. CD4+ T cells and DX5+TCR− cells were identified as the major producers of IL-13, and the DX5+TCR− cells were phenotyped as NK cells, since they expressed CD11b, IL-2Rβ and Ly49C but not c-kit or FcεRI. NK cell-derived IL-13 elevations were Tcell-dependent, as CD4+ Tcell depletion blocked IL-13 production by mesenteric lymph node cells and induced susceptibility. IL-13 expression was increased independently of IL-12; however, blocking IL-18 function inhibited IL-13 production and increased susceptibility. These results indicate that CD4+ T cells and NK cells are the major sources of IL-13 during the in vivo Th1 response induced by B7 blockade and that under these conditions, IL-18 is specifically required for the in vivo up-regulation of IL-13 production and associated host protection.
Keywords: Cytokines, Natural killer cells, Parasitichelminth, Rodent, T helper cells
Introduction
The host protective response that develops following infection varies greatly with the particular pathogen. The type 2 immune response, associated with high levels of IL-4, IL-13 and other Th2 cytokines, provides protection against intestinal nematode parasites [1–3], while type 1 immunity, associated with an IFN-γ-dominant response, mediates resistance against many viruses and bacteria [4, 5]. The events that lead to the development of type 1 and type 2 immunity are generally thought to be distinct, and as each response matures, production of characteristic cytokines can down-regulate the reciprocal response, resulting in further polarization.
Typically, type 2 responses leading to resistance require the development of IL-4-producing effector CD4+ T cells, which then utilize autocrine IL-4 for their rapid expansion [3]. These Th2 cells mediate worm expulsion through their production of Th2 cytokines, with IL-4 and IL-13 being particularly important for the expulsion of gastrointestinal nematode parasites [1, 2]. The development and expansion of IL-4-producing T cells is preferentially dependent on B7 costimulatory molecules [6, 7], and in a number of infectious diseases, B7 blockade can actually deviate a Th2 response to an IFN-γ-dominant Th1 response [8, 9].
In the Th2 cell differentiation model just described, blockade of IL-4 production with B7 antagonists would be expected to also inhibit IL-13 production. However, other studies suggest that under certain circumstances IL-4 and IL-13 are regulated independently, and certain signaling pathways involving c-maf [10] and GATA-3 [11] can differentially regulate these two cytokines. Consistent with these findings, several in vivo studies have recently identified IL-4-independent elevations of IL-13 associated with an IFN-γ-dominant response [8, 12], although the cell surface or secreted molecules that induce the development of IL-13-expressing cells in this context remain uncertain. One candidate would be IL-18, which is up-regulated in many Th1 responses [13–15], can promote the development of IL-13-expressing cells in vitro [16, 17] and, when administered exogenously, can stimulate IL-13 production by memory Th1 cells in response to OVA peptide in vivo [18]. Furthermore, when IL-18 was combined with IL-2, there was a synergistic induction of IL-13 in both T and NK cells [17]. IL-18 can also induce IgE, IgG1 and Th2 cytokine production in murine experimental models [19, 20], and IL-18 transgenic mice produce higher levels of IL-4, IL-5, IL-13 and IFN-γ [21]. Taken together these studies indicate that IL-18 is a pleiotropic cytokine that may under some circumstances augment type 2 as well as type 1 immunity. Few studies, however, have examined the role of IL-18 in up-regulating Th2 cytokines physiologically during in vivo immune responses evoked by infectious agents.
The immune response to Trichuris muris is a particularly useful model for studies of immune regulation in vivo during infectious disease. This intestinal nematode parasite elicits a potent type 2 response in BALB/cmice that is dependent on the development of Th2 cells and leads to worm expulsion within several weeks after inoculation [3]. However, if B7 signaling is inhibited at the time of inoculation, an IFN-γ-dominant response associated with susceptibility develops. As expected, IL-4 production is inhibited in this response; however, elevations in IL-13 expression are sustained. If IFN-γ is also blocked, a host protective response is restored, which is IL-13-dependent [8]. The mechanisms of IL-13 regulation and the cell types producing IL-13 during this type 1 response remain unclear.
In this investigation, we examined the cell populations expressing IL-13 and the role of IL-12 and IL-18 in the development of the type 1 cytokine response that develops following B7 blockade, with a particular emphasis on the role of these cytokines in the stimulation of IL-13 expression leading to host protection after inhibition of IFN-γ. Our studies showed that CD4+ cells and NK cells are responsible for IL-13 elevations and that the IL-13 elevation and associated protection is CD4+ cell-dependent. Although IL-12 played a key role in the development of IFN-γ-producing T cells, its absence did not affect the development of IL-13-expressing cells. In contrast, IL-18 blockade abrogated increases in IL-13 and, furthermore, inhibited IL-13-mediated worm expulsion. These studies thus demonstrate that IL-18 but not IL-12 is necessary for the in vivo development of IL-13-producing cells that contribute to worm expulsion in the context of a type 1 immune response.
Results
DX5+ cells and CD4+ T cells are major, producers of IL-13 following B7 blockade in T. muris-infected mice
Previous studies have shown that blocking B7 ligand interactions increases mesenteric lymph node (MLN) IFN-γ gene expression levels and inhibits IL-4 but not IL-13 elevations following infection with the intestinal nematode parasite T. muris. Although this treatment blocked host protection, additional blockade of IFN-γ function induced IL-4-independent IL-13-mediated worm expulsion [8]. It remained uncertain what cell types produced the IL-13 that mediated resistance during this B7/IL-4-independent in vivo immune response. To address this, normal BALB/c mice (five per treatment group) were inoculated with T. muris eggs, and at days 0, 1 and 12 after inoculation, mice were administered 500 μg CTLA4-Ig or the control fusion protein L6. Mice were killed at day 21 after T. muris inoculation, MLN were collected and single-cell suspensions were prepared. Using magnetic bead cell sorting, four subpopulations were sequentially purified from T. muris-inoculated mice administered CTLA4-Ig or L6: CD4+, CD4−CD8+, CD4−CD8−DX5+ and CD4−CD8−DX5−. RNA was isolated from sorted and unsorted populations, and quantitative real-time fluorogenic RT-PCR was performed for IL-13, IL-4 and IFN-γ gene expression.
As shown in Fig. 1 and consistent with our previous studies [8], elevations in MLN IL-13 and IL-4 but not IFN-γ gene expression were detected on day 21 after T. muris inoculation. CTLA4-Ig administration blocked IL-4 and resulted in increased IFN-γ and IL-13 mRNA. Analysis of sorted cell populations revealed that the elevated IL-13 expression in T. muris-inoculated mice administered CTLA4-Ig was derived primarily from DX5+ cells and CD4+ cells. Of note, both sorted CD4+ and DX5+ cells expressed more IL-13 mRNA in T. muris-inoculated mice administered CTLA4-Ig compared to inoculated mice administered L6. Furthermore, following CTLA4-Ig administration, the sorted CD4+ T cells also expressed increased IFN-γ but decreased IL-4. Taken together, these results demonstrate that although B7 blockade inhibits IL-4 production and enhances IFN-γ production, IL-13 remains elevated and is expressed primarily by CD4+ and DX5+ cells.
Figure 1.
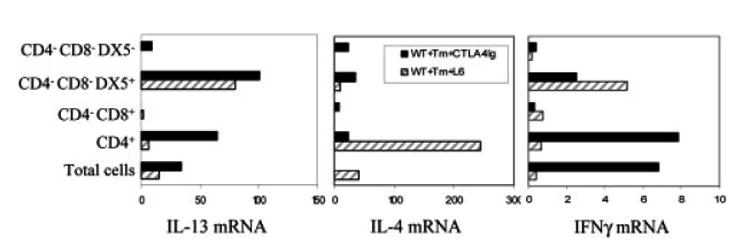
CD4+ and DX5+ cells are the major producers of IL-13 following B7 blockade in T. muris-immunized mice. CTLA4-Ig or L6 (500 μg) was administered at days 0, 1 and 12 after oral inoculation of BALB/c mice (five mice/group) with T. muris (Tm) eggs. At day 21, CD4+, CD4−CD8+ and CD4−CD8−DX5+ cell populations were purified from pooled MLN cells using anti-CD4, anti-CD8 or anti-DX5 magnetic beads. IL-13, IL-4 and IFN-γ mRNA expression were detected by quantitative fluorogenic real-time PCR. Data were individually normalized to rRNA, and data for treatment groups are expressed relative to the uninfected controls. Similar results were obtained in two additional experiments.
NK cells but not NK T cells in the DX5+ population are responsible for IL-13 elevation after CTLA4-Ig treatment in T. muris-infected mice
Previous studies have suggested that NK T cells produce Th2 cytokines [22]. To examine which cell subpopulation within the DX5+ population is the major source of IL-13, we electronically sorted DX5+ cells. To obtain sufficient cells for sorting, we used spleens. As shown in Fig. 2a, CTLA4-Ig treatment of T. muris-infected mice resulted in similarly increased levels of IL-13 mRNA in both spleens (p<0.05) and MLN. At day 21 post-T. muris inoculation, spleens were isolated from CTLA4-Ig-treated mice. CD4+ cells were depleted by magnetic bead-based cell sorting. The CD4− cells were stained with FITC-labeled anti-DX5 and PE-labeled anti-TCRαβ Ab. Cells were gated on DX5+TCRαβ+ and DX5+TCRαβ− populations for electronic cell sorting (Fig. 2b), and both populations were >96% pure. As shown in Fig. 2c, consistent with our previous studies with the MLN, CTLA4-Ig blocked IL-4 mRNA but increased IFN-γ (data not shown) and IL-13 mRNA in total spleen cells compared to the untreated group. Furthermore, direct comparison of sorted DX5+TCR− cells and DX5+TCR+ cells in the same experiment showed that the former, as well as being the major DX5+ cell population (see Fig. 2b), is the predominant source of IL-13 (Fig. 2c). Although DX5 is expressed on the majority of NK cells and NK T cells, it also can be expressed on basophils [23, 24]. In addition, both basophils and mast cells have been reported to be associated with protective Th2 responses [25] and to produce IL-4 and IL-13 when stimulated by FcεRI or with ionomycin [25, 26], suggesting these two cell populations may also be candidate cell types for these DX5+TCR− IL-13-expressing cells. To further identify the phenotype of the DX5+TCR− population, we examined expression of key cell surface markers used to distinguish these cell populations by flow cytometric analysis. As shown in Fig. 2d, the gated DX5+TCR− population expressed CD11b, CD122 (IL-2Rβ) and Ly49C but not c-kit or FcεRI, which is a phenotype characteristic of NK cells but not basophils or mast cells [27]. It should be noted that DX5+TCR− cells from MLN cells in CTLA4-Ig-treated T. muris-infected mice also expressed similar levels of these markers (data not shown). Thus, our results indicate that NK cells as well as CD4+ T cells are major producers of the IL-13 that contributes to resistance to T. muris following inhibition of B7 interactions and IFN-γ.
Figure 2.
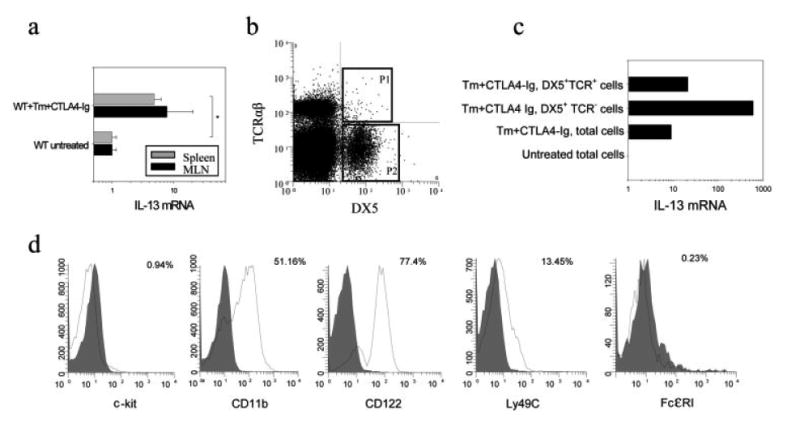
NK cells are a major non-T cell source of IL-13. BALB/c mice were inoculated orally with T. muris (Tm) eggs and treated with CTLA4-Ig as previously described. At day 21 after inoculation (a) MLN and spleens were assayed for IL-13 gene expression as described in Fig. 1, and (b) spleen cells from CTLA4-Ig-treated infected mice were depleted of CD4+ T cells by magnetic bead cell sorting and then stained with PE-labeled anti-mouse TCRαβ and FITC-labeled anti-mouse DX5 mAb. DX5+TCRαβ+ cells (gate P1) and DX5+TCRαβ− (gate P2) cells were purified by electronic cell sorting. (c) Isolated DX5+TCRαβ+ and DX5+TCRαβ− cells were analyzed for IL-13 gene expression. RNA from total spleen cells in untreated mice was used as a control. (d) DX5+TCRαβ− cells from spleens were analyzed using multi-color FACS staining. Single-color histograms represent the expression of c-kit, CD11b, IL-2Rβ, Ly49C and FcεRI on gated DX5+TCRαβ− cells (solid line). Corresponding isotype controls are shown in gray. Asterisk indicates a statistically significant difference (*p<0.05) between the indicated groups.
Depletion of CD4+ T cells abrogates the IL-13-dependent worm expulsion
To examine whether CD4+ Tcells are required for the IL-13-mediated protective immune responses, T. muris-inoculated mice administered both CTLA4-Ig and anti-IFN-γ were given depleting anti-CD4 antibody at days 0, 7 and 14, as previously described [28]. As shown in Fig. 3a, effective worm expulsion was inhibited by anti-CD4 treatment (p<0.01). These results indicate that CD4+ T cells are not only required for the IL-4-dependent protection observed after T. muris infection but are also required for the IL-13-mediated IL-4-independent protective response that occurs following blockade of both B7 and IFN-γ.
Figure 3.
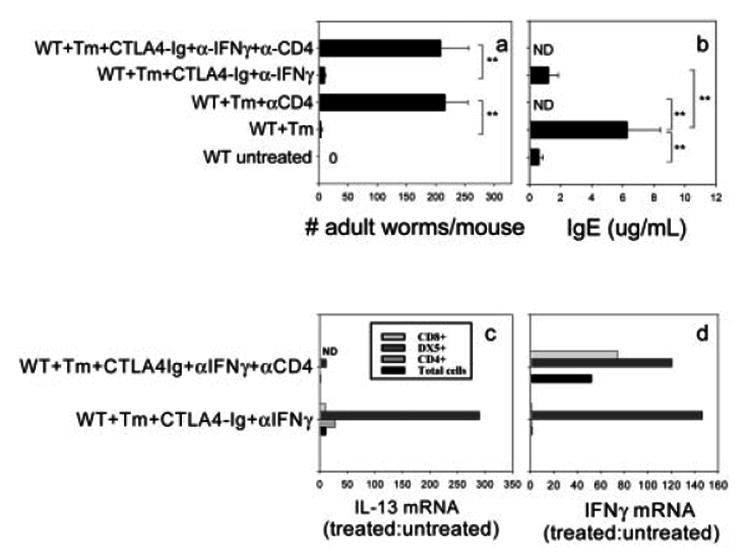
IL-4-independent IL-13 up-regulation and the associated protection against T. muris after blockade of both B7 and IFN-γ is CD4+ cell-dependent. BALB/c mice were administered CTLA4-Ig or L6 after inoculation with T. muris (Tm) eggs as previously described. At days 0, 7 and 14, 1 mg anti-IFN-γ mAb or the isotype control (GL113) and anti-CD4 mAb were administered to specific treatment groups. At day 21, (a) susceptibility was determined by assessment of worm burden. (b) Serum IgE levels were determined by ELISA. The means and SE from five individual BALB/c mice are shown for each group (ND: not detectable). (c, d) At day 21, CD4+, CD4−CD8+ and CD4−CD8−NK+ cells were purified from pooled MLN cells. RNA was isolated from these cell populations. Expression of IL-13 (c) and IFN-γ (d) mRNA was detected by real-time PCR. Data were individually normalized to rRNA, and treatment groups are expressed relative to the uninfected control. Similar results were obtained in two independent experiments. Asterisks indicate a statistically significant difference (**p<0.01) between the indicated groups.
To examine whether B cell antibody class switching and secretion occurred following administration of anti-CD4 Ab, serum IgE was assessed after T. muris inoculation. In Fig. 3b, increases in serum IgE were completely blocked (p<0.01) and IgE levels undetectable following anti-CD4 treatment of T. muris-inoculated mice whether CTLA4-Ig and/or anti-IFN-γ Ab were administered. Furthermore, serum IgE levels were comparable to untreated controls in T. muris-inoculated mice administered CTLA4-Ig and anti-IFN-γ. These findings indicate that the anti-CD4 treatment inhibited Ig class switching to IgE, confirming that the anti-CD4 Ab administration was effectively blocking T helper cell activity.
Increases in IL-13 production remain CD4+ T cell-dependent
Our results showing that CD4+ T cell depletion abrogates IL-13-mediated resistance suggested that CD4 depletion may abrogate IL-13 production by NK cells in addition to directly eliminating CD4+ T cells. To address this possibility, anti-CD4 mAb was used to deplete CD4+ T cells in vivo in T. muris-inoculated mice administered CTLA4-Ig and anti-IFN-γ Ab. On day 21 after inoculation, MLN were removed, and CD4+, CD4−CD8+ and CD4−CD8−DX5+ populations were sequentially purified. As shown in Fig. 3c and d, consistent with our previous data, blocking both B7 and IFN-γ inhibited IL-4 (data not shown) and IFN-γ gene expression but increased IL-13 gene expression in total MLN cells post-infection. Additional treatment with anti-CD4 markedly decreased IL-13 gene expression in unsorted MLN cells. Analysis of sorted CD4−CD8−DX5+ cells showed significantly increased levels of IL-13 mRNA in T. muris-inoculated mice administered both CTLA4-Ig and anti-IFN-γ Ab. Anti-CD4 Ab treatment dramatically reduced elevations in IL-13 mRNA by these cells. CD4 depletion did not abrogate IFN-γ mRNA expression by these CD4−CD8−DX5+ cells. These findings thus demonstrate that IL-13 gene expression by NK cells is CD4+ T cell-dependent.
Blocking both B7 ligand interactions and IFN-γ function is not associated with increased susceptibility to T. muris in IL-12p40–/– mice
Blocking B7 interactions during the T. muris immune response results in sustained IL-13 production, although IL-4 is inhibited and IFN-γ is increased. The sustained elevations in IL-13 may be induced through similar mechanisms such as IL-12 or IL-18 that promote the Th1 response. In fact, several studies have suggested that IL-13 may be induced by IL-12 and IL-18 or IL-18 alone [29, 30]. To examine whether the IL-13-mediated host protective response after blockade of both B7 and IFN-γ is impaired in the absence of IL-12, IL-12p40–/– and WT BALB/c mice (five per treatment group) were inoculated with T. muris eggs and administered either CTLA4-Ig or CTLA4-Ig with anti-IFN-γ Ab. At day 21, adult worm survival and fecundity were determined. As shown in Fig. 4a and as previously published [8], CTLA4-Ig administration to T. muris-inoculated WT mice blocked worm expulsion (p<0.01), which was restored following anti-IFN-γ Ab administration (p<0.01). In marked contrast, CTLA4-Ig administration to T. muris-inoculated IL-12p40–/– mice did not block the host protective response (p<0.01). The host resistance was sustained after additional inhibition of IFN-γ. These studies indicate that IL-12 does not stimulate the IL-13-dependent host protective response observed following combined blockade of B7 and IFN-γ.
Figure 4.
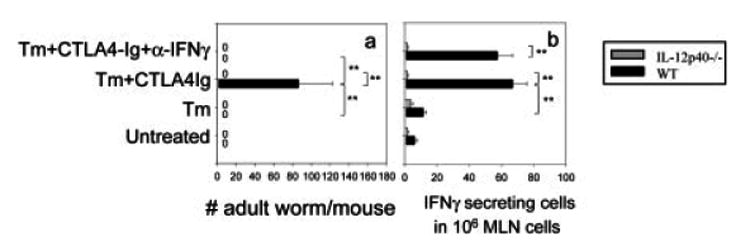
IL-13-mediated protection to T. muris after blockade of both B7 and IFN-γ is not dependent on IL-12. IL-12p40–/– and WT BALB/c mice (five mice/group) were administered CTLA4-Ig or L6 after T. muris (Tm) inoculation. Anti-IFN-γ mAb was used to block IFN-γ function in vivo. At day 21 post-immunization, (a) worm burdens were assessed by counting total numbers of adult worms in the gut, and (b) MLN cells were collected and IFN-γ-producing cells detected by ELISPOT. The means and SE from five individual mice are shown for each group. Similar results were obtained in two additional experiments. Asterisks indicate a statistically significant difference (**p<0.01) between the indicated groups.
Our finding that blocking B7 alone inhibited worm expulsion in T. muris-inoculated WT mice but not in inoculated IL-12p40–/– mice suggested that IL-12-induced IFN-γ production contributed to the susceptibility observed following B7 blockade. To examine whether elevations in IFN-γ were IL-12-dependent, IFN-γ ELISPOT assays on MLN cell suspensions were performed at day 21 after T. muris inoculation. As shown in Fig. 4b, in T. muris-inoculated WT mice, CTLA4-Ig administration enhanced IFN-γ production by MLN cells (p<0.01). Sustained increases of IFN-γ production in anti-IFN-γ c mAb-treated mice were due to lack of anti-IFN-γ mAb in the in vitro culture when ELISPOT was performed. Elevations in IFN-γ that occurred following B7 blockade were blocked in IL-12p40–/– mice (p<0.01). These findings indicate that IFN-γ-mediated host susceptibility induced following B7 blockade is IL-12-dependent and, most importantly, that protective immunity that occurs following combined B7 and IFN-γ blockade is IL-12-independent.
IL-13 protein remains high in T. muris-infected IL-12p40–/– mice
The observation that IL-12p40–/– mice can generate intact protection following B7 blockade and IFN-γ neutralization suggested that IL-4-independent IL-13 production does not require IL-12. To test this directly, MLN cells obtained from mice at day 21 after T. muris inoculation were restimulated in vitro with anti-CD3 for 72 h, after which supernatants were assayed by ELISA for IL-13 protein. As shown in Fig. 5a, elevations in IL-13 levels in T. muris-inoculated IL-12p40–/– mice given CTLA4-Ig + control Ab or CTLA4-Ig + anti-IFN-γ were not statistically significantly reduced compared to similarly treated WT mice. This finding was confirmed by analysis of MLN cytokine gene expression using quantitative real-time fluorogenic RT-PCR, where again IL-13 elevations were not inhibited in T. muris-inoculated IL-12p40–/– mice administered CTLA4-Ig or both CTLA4-Ig and anti-IFN-γ (Fig. 5b). To confirm that elevations in IL-4 remain blocked in IL-12p40–/– mice administered CTLA4-Ig, IL-4 mRNA levels and serum IgE levels were assessed in the same experiment. After T. muris inoculation, IL-4 gene expression (p<0.01) and serum IgE levels (p<0.05) were similarly increased in WT and IL-12p40–/– mice. CTLA4-Ig similarly blocked IL-4 (p<0.01) and serum IgE elevations (p<0.05) in both WT and IL-12p40–/– mice (Fig. 5c, d), indicating that increases in IL-13 remain IL-4-independent in IL-12p40–/– mice. These results confirmed that IL-13 elevations observed in T. muris-inoculated mice administered CTLA4-Ig and anti-IFN-γ are IL-12-independent and, when considered with the findings presented in Fig. 4, demonstrate that IL-13-mediated protective immunity is IL-12-independent.
Figure 5.
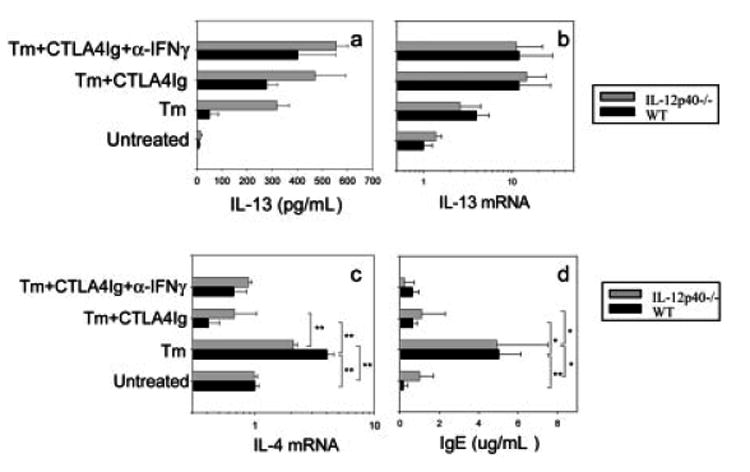
Increases in IL-13 are comparable in T. muris-inoculated IL-12p40–/– and WT mice following B7 and IFN-γ blockade. IL-12p40–/– and WT BALB/c mice (five mice/group) were immunized with T. muris (Tm) and treated with CTLA4-Ig or L6 and anti-IFN-γ or GL113 (isotype control). At day 21 post-inoculation, (a) MLN cells were restimulated with anti-CD3 (0.625 μg/mL) for 72 h and IL-13 supernatant levels detected by ELISA; (b, c) IL-13 and IL-4 gene expression were determined by quantitative fluorogenic real-time PCR; and (d) total serum IgE levels were determined by ELISA. The means and SE from five individual BALB/c mice are shown for each group. Similar results were obtained in two additional experiments. Asterisks indicate a statistically significant difference (*p<0.05, **p<0.01) between the indicated groups.
Blocking IL-18 function abrogates increased IL-13 and the associated host protective response
Previous studies have shown that IL-18 can induce NK cells or T cells to produce IL-13 [16, 17]. We were interested in examining whether IL-18 also plays a role in the in vivo IL-13-mediated resistance to T. muris that occurs following inhibition of B7 and IFN-γ. To address this possibility, we used IL-18 binding protein (IL-18BP) [31] to block IL-18 function in vivo. WT BALB/c mice were inoculated with T. muris eggs. CTLA4-Ig and anti-IFN-γ mAb were administered as already described. In addition IL-18BP-Fc was given to inhibit in vivo IL-18 function. At day 21 after T. muris inoculation, adult worm burden was determined. As shown in Fig. 6a, although T. muris-inoculated mice effectively expulsed worms when administered CTLA4-Ig and anti-IFN-γ Ab, additional administration of IL-18BP-Fc yielded a significantly increased worm burden (p<0.01), suggesting that IL-18 is required for generating host protection in the T. muris-infected mice when B7 and IFN-γ function is inhibited.
Figure 6.
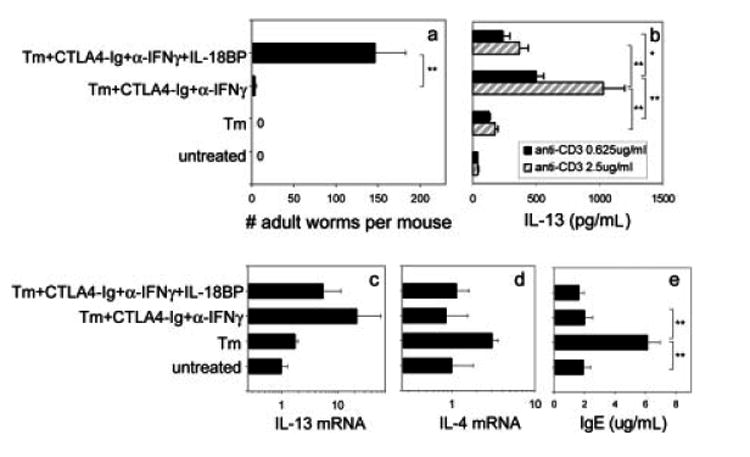
IL-18BP-Fc treatment abrogates IL-13 production and associated protection in T. muris-inoculated WT mice after B7 and IFN-γ blockade. BALB/c mice (five mice/group) were immunized with T. muris (Tm) and treated with CTLA4-Ig and anti-IFN-γ. IL- 18BP-Fc was administered weekly by i.v. injection to appropriate treatment groups. At day 21, (a) worm burden was determined. (b) MLN cells were restimulated with anti-CD3, and IL-13 production in the supernatants was determined by ELISA. (c, d) IL-13 and IL-4 mRNA expression were determined by real-time quantitative PCR. (e) Sera were collected from the mice and total serum IgE levels determined by ELISA. The means and SE from five individual BALB/c mice are shown for each group. Asterisks indicate a statistically significant difference (*p<0.05, **p<0.01) between the indicated groups. Similar results were obtained in two experiments.
To address directly whether IL-18 induces IL-13 production in this system, MLN cells from T. muris-inoculated mice were restimulated with anti-CD3 at varying concentrations for 72 h, and IL-13 production in the supernatants after restimulation was determined by ELISA. As shown in Fig. 6b, consistent with our previous results, a pronounced increase in IL-13 was detected in T. muris-inoculated mice given CTLA4-Ig plus anti-IFN-γ compared with T. muris-inoculated mice given control Ab (p<0.01 at both 0.625 μg/mL and 2.5 μg/mL anti-CD3). However, IL-18BP-Fc treatment significantly decreased IL-13 production by MLN cells in this group (p<0.01 at 2.5 μg/mL and p<0.05 at 0.625 μg/mL anti-CD3), indicating that IL-18 is required for IL-13 production in T. muris-infected mice given CTLA4-Ig plus anti-IFN-γ. To confirm this result and test the possibility that IL-4 may be increased after IL-18 blockade [13], MLN were collected for cytokine mRNA expression (Fig. 6c, d). Quantitative real-time fluorogenic RT-PCR results showed that: 1) T. muris inoculation increased IL-4 mRNA; 2) combined CTLA4-Ig and anti-IFN-γ administration blocked IL-4 mRNA but increased IL-13 mRNA; 3) IL-18BP-Fc treatment markedly decreased IL-13 mRNA but had little effect on IL-4 mRNA; 4) IFN-γ mRNA remained low in all treatment groups (all <1; data not shown). Total serum IgE levels were also detected by ELISA to specifically determine whether IL-4-dependent increases in serum Ig were inhibited in the different treatment groups administered CTLA4-Ig. As shown in Fig. 6e, pronounced increases in serum IgE (p<0.01) after T. muris inoculation were markedly reduced in CTLA4-Ig-and anti-IFN-γ-treated groups (p<0.01). Blocking IL-18 with IL-18BP-Fc had little effect on serum IgE levels. These findings indicate that IL-4 was not produced at sufficient levels to mediate IgE class switching and secretion following CTLA4-Ig administration whether or not IL-18BP-Fc was also given. Taken together with results from MLN IL-4 mRNA determinations, our findings indicate that IL-18 did not affect IL-4 function after B7 and IFN-γ blockade.
Discussion
Our previous studies demonstrated that blocking both B7 and IFN-γ function induces IL-13-mediated worm expulsion following T. muris inoculation [8]. The results reported herein extend these studies by providing novel insights concerning the regulation of IL-13-mediated host protective responses during an IFN-γ-dominant response to T. muris. They demonstrate that in this immune environment: 1) the major sources of IL-13 are CD4+ Tcells and NK cells; 2) IL-13 production and IL-13-mediated protection is CD4+ cell-dependent; 3) although IL-12 induces the IFN-γ response that mediates susceptibility, it is not required for IL-13 up-regulation; 4) IL-18 mediates up-regulation of IL-13 expression and the associated worm expulsion independently of IL-4 and B7 costimulation.
IL-13, originally described as a T cell-derived anti-inflammatory cytokine, has multiple biological activities. Like IL-4, IL-13 up-regulates CD23, CD71 and MHC class II expression on B cells and monocytes [32], elicits B cell proliferation and induces IgG4 and IgE production by human B cells [32], and promotes Th2 cell differentiation [33]. In addition, IL-13 plays a dominant role to IL-4 in mediating worm expulsion to Nippostrongylus brasiliensis [34], exerts protective functions both in the chronic stages of some Leishmaniasis major infections [35] and in Listeria monocytogenes infection in the context of a Th1 cytokine response [36], and acts as the key mediator in the pathogenesis of allergic asthma [37]. Studies of T. muris showing that IL-13 mediates host protection in IL-4–/– BALB/c mice suggest an IL-4-independent role for IL-13 in resistance to T. muris [38]. Our previous findings support this concept and further suggest that IL-13 can mediate resistance to T. muris in the absence of both IL-4 signaling and B7 costimulation [8]. These studies indicated either a novel B7-independent pathway of CD4+ T cell differentiation leading to IL-13 but not IL-4 expression or an alternative cell source that can differentiate to produce IL-13 if B7 is blocked and/or IL-4 is neutralized.
Although IL-13 is mainly produced by T cells [39, 40], several sources of IL-13 have been suggested, including human and mouse NK cells [16, 17], NKT cells [41], basophils [25, 26] and mast cells [42]. To specifically address the question of which cell populations produce IL-13 during this IL-4/B7-independent in vivo immune response to T. muris, we performed cell sorting and quantitative RT-PCR. Our findings that CD4+ T cells are the principal source of IL-13 are consistent with previous studies showing that Th2 cells are major producers of IL-13 [43]. However, IL-4 was blocked, as demonstrated by inhibition of both IL-4 mRNA and serum IgE elevations, when B7 costimulation was abrogated. These studies thus demonstrate differential regulation of IL-4 and IL-13 in T cells by costimulatory molecules during the in vivo immune response, consistent with previous studies indicating that these Th2 cytokines can be differentially regulated by certain transcription factors [10, 11]. Our studies further indicate that DX5+ cells are also a major source of IL-13.
DX5 has been shown to be expressed by a number of cell types, including NK cells, NK T cells, basophils and some T cells [23, 24]. Mast cells and basophils can be distinguished by the expression of the high-affinity IgE receptor (FcεRI), which is absent from murine NK cells, and anti-c-kit mAb specifically identifies mast cells [44, 45]. NK cells express IL-2Rβ and CD11b, and subsets express Ly49C. Furthermore, NK cells can be distinguished from T cells by the absence of TCR expression. Taken together, our results indicate that the IL-13-producing DX5+ cells are NK cells, since they expressed IL-2Rβ, CD11b and Ly49C but not FcεRI, c-kit or TCR. It has consistently been reported that NK cells are an important source of IL-13 in vitro and in vivo [17, 46, 47]. In particular, our findings demonstrate that NK cells express IL-13 as well as IFN-γ in vivo during infectious disease. Thus, even in the absence of IL-4 and B7 signaling (pathways usually required for the development of a Th2 response), a potent IL-13-mediated response, derived from both CD4+ T cells and NK cells, can develop sufficiently to mediate worm expulsion when IFN-γ function is inhibited. It will be useful in future studies to examine the mechanisms that drive IL-13 as well as IFN-γ production by T cells following B7 blockade.
As with other nematode parasites, such as N. brasiliensis and Heligmosomoides polygyrus, CD4+ T cells have been shown to be required for the induction and expression of Th2 immunity to T. muris [48]. We were interested in examining whether the IL-13-mediated protective response that develops in the absence of B7 and IFN-γ is still dependent on CD4+ T cells. To address this question, we used anti-CD4 mAb, which has been shown to deplete CD4+ T cells successfully in vivo [28]. Since our data showed that this response can develop in the absence of B7/IL-4 and that IL-13 can be produced by non-CD4+ T cells (NK cells), we expected that depletion of CD4+ T cells would at most only partially affect host protection and IL-13 production in MLN cells. However, surprisingly, depletion of CD4+ T cells not only completely blocked IL-13 production by both total MLN cells and sorted NK cells but also blocked worm expulsion, indicating that IL-13 production and its associated resistance is still CD4+ T cell-dependent. How CD4+ T cells control the capability of NK cells to produce IL-13 is not clear. It is possible that CD4+ T cells may either release some soluble mediators such as cytokines or interact with NK cells directly to trigger the latter to produce IL-13. Previous in vitro studies also demonstrated that depletion of CD3+ T cells decreased IL-13 production by NK cells from IFNγ–/– mice, suggesting that T cell-NK cell interactions might be important for optimal IL-13 production in the absence of IFN-γ [16]. Our findings suggest that similar interactions occur between CD4+ T cells and NK cells in vivo.
Since IL-13 plays a critical role in inducing a protective response to T. muris after B7 and IFN-γ blockade, understanding the mechanisms that up-regulate IL-13 expression and function in this immune environment is significant. Typically, IL-4 is the critical differentiation factor for Th2 cell development [49], and IL-13-producing cells are thought to develop in conjunction with the production of autocrine IL-4 [50]. However, in our system, it seems unlikely that IL-4 triggers IL-13 production, since B7 blockade inhibited IL-4 expression and IL-4-dependent elevations in IgE. Instead, sustained IL-13 elevation was associated with increased IFN-γ and susceptibility, suggesting that IL-13 was up-regulated by mechanisms similar to those that promote the Th1 response. Several studies have indeed shown that other cytokines, in particular IL-12 and IL-18, can drive the development of IL-13-producing cells [29]. We therefore examined whether IL-12 is required for IL-13 production by using IL-12p40–/– mice. Our results showed that the lack of IL-12 had no effects on IL-13 elevations and associated worm expulsion, although IFN-γ up-regulation was inhibited. The effect of IL-12 on IFN-γ is consistent with recent studies indicating that during T. muris infection, IFN-γ upregulation and the associated Th1 response is dependent on TLR4 signaling, which includes a MyD88-mediated pathway known to induce IL-12 [51]. Previous studies showed that although IL-18 alone up-regulates IFN-γ production and the Th1 response [52], in most cases this IFN-γ-promoting capability synergizes with IL-12 [53]. Our findings suggest that IL-12 is primarily responsible for IFN-γ up-regulation and are consistent with previous studies showing that T. muris infection of BL/6 IL-12p40–/– mice resulted in markedly reduced IFN-γ production even when IL-18 secretion remained elevated [13]. In addition, we further showed that CTLA4-Ig could successfully block IL-4 expression and increases of serum IgE levels in IL-12p40–/– mice, indicating that the absence of IL-12 does not lessen the requirement of B7 costimulation for the development of IL-4-producing cells. Taken together, our studies thus indicate that IL-12 induces IFN-γ elevations during this response but is not required for concomitant increases in IL-13.
Given that IL-18 induces IL-13 production in both NK cells and T cells in synergy with IL-2 [54] and that in vivo administration of IL-18 increases serum IgE levels and splenocyte production of Th2 cytokines [20], IL-18 seemed a likely candidate for driving IL-13 production during this IL-4/B7-independent protective immune response to T. muris. To address this, we used IL-18BP-Fc, which has been shown to completely block LPS-induced IFN-γ production in vivo [31]. Our findings showed that blocking IL-18 in vivo abrogated IL-13-dependent host protection following B7 and IFN-γ blockade, indicating that IL-18 induces IL-13 production under these conditions. Previous findings showed that IL-18 promotes NK cells/NK T cells to produce IL-13 in synergy with IL-12 [29, 30] and that IL-12 induces IL-18Rα expression on naive T cells, Th1 cells and B cells [55], suggesting that IL-12 may be a key player required for IL-18-mediated IL-13 up-regulation. However, our results indicate that this is unlikely, because the absence of IL-12 did not decrease either IL-13 production by MLN cells or IL-13-dependent worm expulsion. Previous studies also suggested that IL-18 could induce IL-4 and associated IgE and IgG1 production in vivo [19, 20]. In the IL-4-dominant response to T. muris, IL-18 has further been shown to down-regulate IL-13 production [13]. In contrast, our studies demonstrate that IL-18 did not induce IL-4 or associated Ig isotypes in vivo following B7 blockade and that under these conditions it could markedly up-regulate IL-13 production and IL-13-mediated resistance. We speculate that this may be due to different effects of IL-18 on Th1 vs. Th2 cells. In an IL-4-dominant Th2 setting, IL-18 may act as a suppressing factor to inhibit Th2 cell function, including IL-13 production; in a Th1 setting (in the absence of IL-4), IL-18 may act as a promoting factor to enhance IL-13 production by Th1 cells. Consistent with our findings, recent studies have shown that IL-18 can induce memory Th1 cells to produce IL-13 in an airway inflammation model [18]. It should be noted that as IL-18–/– BALB/c mice become readily available, it will be of interest to compare our results using the IL-18 antagonist in WT mice with constitutively IL-18-deficient mice. Taken together, our results demonstrate that in this in vivo model system, IL-18 is required for IL-12-independent elevations in IL-13 production and associated IL-13-dependent host protection against T. muris. Although we expect APC to be a likely source, it will be important in future studies to identify the cells that produce IL-12 and especially IL-18.
In summary, this study provides novel insights concerning the in vivo regulation of IL-13 during an immune response that can culminate in worm expulsion. Our findings indicate that in the absence of IL-4 elevations and B7 costimulation, CD4+ T cells and NK cells are predominant sources of IL-13, which mediates protection following IFN-γ blockade. Furthermore, during a Th1 response, IL-18 promotes IL-13 elevations at sufficient levels to mediate a host protective response if IFN-γ is neutralized. Our studies further showed that blockade of dominant immune responses in vivo can reveal underlying responses that may also be important in resistance during infectious disease, an indication of redundancy in the in vivo immune response that may have particular significance for specific genetic backgrounds including deficiencies in molecules that mediate the immune response.
Materials and methods
Mice
Female 8- to 12-wk-old BALB/c female mice (National Cancer Institute, Frederick, MD) were used for all studies of wild-type (WT) mice and as controls for the BALB/c IL-12p40-deficient mice. IL-12p40-deficient mice (IL-12p40–/–) on a BALB/c background were purchased from the JAX Laboratory (Bar Harbor, Maine). The experiments herein were conducted according to the principles set forth in the Guide for the Care and Use of Laboratory Animals, Institute of Animal Resources, National Research Council, Department of Health, Education and Welfare (National Institutes of Health) 78–23.
Parasite, worm burdens and fecundity
Infective Trichuris muris eggs originally from Dr. Richard Grencis (University of Manchester, Manchester, UK) and Dr. David Artis (University of Pennsylvania, Philadelphia, PA) were cultured and maintained as previously described [8]. Mice were orally infected on day 0 with 250–500 T. muris eggs. On day 21 post-infection, the cecum and adjacent colon were removed to determine worm burdens [8].
Reagents
Rat anti-mouse IFN-γ (XMG-6) and its control rat IgG1 anti-β-galactosidase (GL113) Ab were given i.v. at a concentration of 1 mg at weekly intervals starting on the day of inoculation of T. muris eggs. Murine CTLA4-Ig, the chimeric fusion protein used to block B7 ligand interactions [8], and its control, L6, were administered at a dose of 500 μg. IL-18BP-Fc, generously provided by Amgen (Thousand Oaks, CA) and its control Ab, human IgG1, were administered i.v. at a concentration of 5 mg/kg weekly after inoculation. In several experiments, on the same days, CD4+ T cells were depleted in vivo by i.v. administration of 1 mg anti-CD4 mAb (clone GK1.5, purified from ascites). This dose has previously been shown to effectively deplete CD4+ T cells in vivo [28]. All i.v. treatments were given through the orbital plexus in the eye socket.
Flow cytometric analysis
MLN and spleen cell suspensions were collected from treated mice and incubated with FITC- or allophycocyanin (APC)-conjugated anti-DX5; PE- or FITC-conjugated anti-TCRαβ; and APC- or PE-conjugated anti-FcεRI, anti-c-kit, anti-CD11b, anti-CD122 or anti-Ly49C Ab. Fc receptor was blocked with Fc block (BD Pharmingen, San Diego, CA) in all experiments. After washes, cell were fixed with 1% paraformaldehyde (Fisher) and analyzed by flow cytometry with a FACSCalibur (Becton Dickinson) for three-color staining. All antibodies were from BD Pharmingen except anti-FcεRI and anti-CD122 which were from e Bioscience (San Diego, CA).
Cell sorting
Single-cell suspensions were prepared from MLN and spleens as previously described [28]. Cells were washed and then resuspended in RPMI 1640 medium supplemented with 10% FCS, 2 mM l-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (all from GIBCO BRL, NY). For magnetic beads sorting, MLN cells were labeled with anti-CD4 beads and passed through LS+ columns (Miltenyi Biotec), followed by CD8 positive selection. FACS analysis showed that the CD4+ population was >98% pure. The CD8+ population was >95% pure. The CD4−CD8− population was >95% pure in all sorts described. For NK cell sorting, the CD4−CD8− cells were mixed with anti-DX5 beads and passed through MS+ columns two times. The purity of NK+ cells was 44–50%. Anti-mouse CD4, CD8 and DX5 beads were all purchased from Miltenyi Biotec. For electronic cell sorting, CD4+ cells were first depleted using CD4 microbeads. The CD4− cells were stained with FITC-anti-DX5 vs. PE-anti-TCRαβ Ab. DX5+TCRαβ+ and DX5+TCRαβ− populations were sorted electronically using a FACSVantage cell sorter with high-speed upgrade (Becton Dickinson). The cell purities were >96%.
Cytokine gene expression by RT-PCR
Total RNA was extracted from purified cell populations by using the RNA Isolation Kit (Stratagene, Cedar Creek, TX), specially developed for isolating small RNA quantities and then reverse transcribed as previously described [56]. Real-time PCR kits (Applied Biosystems, Foster City, CA) specific for individual cytokines or ribosomal RNA were used to quantitate differences in gene expression. Ribosomal RNA was used as an endogenous internal standard. The Applied Biosystems 7700 sequence detector (Applied Biosystems) was used for amplification of target mRNA. Quantification of differences between treatment groups was calculated according to the manufacturer's instructions.
Cell cultures
The MLN cells were plated at 3 × 106 cells per well in 24-well plates that were coated with various concentrations of anti-CD3 mAb (Pharmingen). The cells were cultured at 37°C in an atmosphere of 5% CO2. Cell-free supernatant fluids were harvested from these cell cultures at 72 h and stored at –70°C for cytokine production analysis.
Quantification of IL-13
ELISA assays were performed on supernatants from cultured cells using a commercial kit according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
ELISPOT
ELISPOT assays were used as previously described [56]. Briefly, single-cell lymph node suspensions were prepared in RPMI 1640 containing 10% heat-inactivated FCS, 100 U/mL penicillin, 100 μg/mL streptomycin and 2 mM l-glutamine (GIBCO BRL). The cells (0.5 × 106) were seeded into each well of an Immulon IV 96-well microtiter plate (Microtiter, Chantilly, VA) coated with anti-IFN-γ (clone R46A2; Pharmingen). After 12 h culture, the plate was washed several times with PBS, followed by washes with PBS/Tween-20. Secondary biotinylated anti-IFN-γ Ab (clone XMG1.2; Pharmingen) was diluted in PBS/0.05% Tween/5% FCS, added at 100 μL/well and incubated overnight at 4°C. Plates were then washed, and a 1/2000 dilution of streptavidin-alkaline phosphatase (AKP) (Jackson ImmunoResearch, West Grove, PA) was added. Plates were developed and results counted as described [56].
Quantification of serum Ig
Serum IgE levels were quantified by ELISA [57].
Statistical analysis
Statistical differences (p<0.05) between groups were assessed using ANOVA and Fisher's protected least significant difference (LSD) test for pairwise comparisons. The software program SigmaStat (Jandel, San Rafael, CA) was used for all statistical analyses.
Acknowledgments
This work was supported by the National Institutes of Health Grant RO1AI47478 and partially supported by USDA CRIS 1265–32000–064–00D. The opinions or assertions contained within are the private views of the authors and should not be construed as official or necessarily reflecting the views of Uniformed Services University of the Health Sciences, the Department of Defense or the Department of Agriculture.
Abbreviations
- IL-18BP
interleukin 18 binding protein
- MLN
mesenteric lymph node
References
- 1.Wynn TA. IL-13 effector functions. Annu Rev Immunol. 2003;21:425–456. doi: 10.1146/annurev.immunol.21.120601.141142. [DOI] [PubMed] [Google Scholar]
- 2.Liu Z, Liu Q, Pesce J, Anthony RM, Lamb E, Whitmire J, Hamed H, et al. Requirements for the development of IL-4-producing Tcells during intestinal nematode infections: what it takes to make a Th2 cell in vivo. Immunol Rev. 2004;201:57–74. doi: 10.1111/j.0105-2896.2004.00186.x. [DOI] [PubMed] [Google Scholar]
- 3.Gause WC, Urban JF, Stadecker MJ. The immune response to parasitic helminths: insights from murine models. Trends Immunol. 2003;24:269–277. doi: 10.1016/s1471-4906(03)00101-7. [DOI] [PubMed] [Google Scholar]
- 4.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 5.Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 6.Lu P, Zhou X, Chen SJ, Moorman M, Morris SC, Finkelman FD, Linsley P, et al. CTLA-4 ligands are required in an in vivo interleukin 4 response to a gastrointestinal nematode parasite. J Exp Med. 1994;180:693–698. doi: 10.1084/jem.180.2.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenwald R, Lu P, Zhou XD, Nguyen H, Chen SJ, Perrin PJ, Madden KB, et al. Effects of blocking B7-1 and B7-2 interactions during a type 2 in vivo immune response. J Immunol. 1997;158:4088–4096. [PubMed] [Google Scholar]
- 8.Urban J, Fang H, Liu Q, Ekkens MJ, Chen SJ, Nguyen D, Mitro V, et al. IL-13-mediated worm expulsion is B7 independent and IFN-gamma sensitive. J Immunol. 2000;164:4250–4256. doi: 10.4049/jimmunol.164.8.4250. [DOI] [PubMed] [Google Scholar]
- 9.Corry DB, Reiner SL, Linsley PS, Locksley RM. Differential effects of blockade of CD28-B7 on the development of Th1 or Th2 effector cells in experimental Leishmaniasis. J Immunol. 1994;153:4142–4148. [PubMed] [Google Scholar]
- 10.Kim JI, Ho IC, Grusby MJ, Glimcher LH. The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity. 1999;10:745–751. doi: 10.1016/s1074-7613(00)80073-4. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, et al. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 12.Babaloo Z, Kaye PM, Eslami MB. Interleukin-13 in Iranian patients with visceral leishmaniasis: relationship to other Th2 and Th1 cytokines. Trans R Soc Trop Med Hyg. 2001;95:85–88. doi: 10.1016/s0035-9203(01)90344-x. [DOI] [PubMed] [Google Scholar]
- 13.Helmby H, Takeda K, Akira S, Grencis RK. Interleukin (IL)-18 promotes the development of chronic gastrointestinal helminth infection by downregulating IL-13. J Exp Med. 2001;194:355–364. doi: 10.1084/jem.194.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akira S. The role of IL-18 in innate immunity. Curr Opin Immunol. 2000;12:59–63. doi: 10.1016/s0952-7915(99)00051-5. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi K, Yoshimoto T, Tsutsui H, Okamura H. Interleukin-18 regulates both Th1 and Th2 responses. Annu Rev Immunol. 2001;19:423–474. doi: 10.1146/annurev.immunol.19.1.423. [DOI] [PubMed] [Google Scholar]
- 16.Hoshino T, Winkler-Pickett RT, Mason AT, Ortaldo JR, Young HA. IL-13 production by NK cells: IL-13-producing NK and T cells are present in vivo in the absence of IFN-gamma. J Immunol. 1999;162:51–59. [PubMed] [Google Scholar]
- 17.Hoshino T, Wiltrout RH, Young HA. IL-18 is a potent coinducer of IL-13 in NK and T cells: a new potential role for IL-18 in modulating the immune response. J Immunol. 1999;162:5070–5077. [PubMed] [Google Scholar]
- 18.Sugimoto T, Ishikawa Y, Yoshimoto T, Hayashi N, Fujimoto J, Nakanishi K. Interleukin 18 acts on memory T helper cells type 1 to induce airway inflammation and hyperresponsiveness in a naive host mouse. J Exp Med. 2004;199:535–545. doi: 10.1084/jem.20031368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshimoto T, Mizutani H, Tsutsui H, Noben-Trauth N, Yamanaka K, Tanaka M, Izumi S, et al. IL-18 induction of IgE: dependence on CD4+ T cells, IL-4 and STAT6. Nat Immunol. 2000;1:132–137. doi: 10.1038/77811. [DOI] [PubMed] [Google Scholar]
- 20.Wild JS, Sigounas A, Sur N, Siddiqui MS, Alam R, Kurimoto M, Sur S. IFN-gamma-inducing factor (IL-18) increases allergic sensitization, serum IgE, Th2 cytokines, and airway eosinophilia in a mouse model of allergic asthma. J Immunol. 2000;164:2701–2710. doi: 10.4049/jimmunol.164.5.2701. [DOI] [PubMed] [Google Scholar]
- 21.Hoshino T, Kawase Y, Okamoto M, Yokota K, Yoshino K, Yamamura K, Miyazaki J, et al. Cutting edge: IL-18-transgenic mice: in vivo evidence of a broad role for IL-18 in modulating immune function. J Immunol. 2001;166:7014–7018. doi: 10.4049/jimmunol.166.12.7014. [DOI] [PubMed] [Google Scholar]
- 22.Akbari O, Stock P, Meyer E, Kronenberg M, Sidobre S, Nakayama T, Taniguchi M, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 23.Min B, Prout M, Hu-Li J, Zhu J, Jankovic D, Morgan ES, Urban JF, Jr, et al. Basophils produce IL-4 and accumulate in tissues after infection with a Th2-inducing parasite. J Exp Med. 2004;200:507–517. doi: 10.1084/jem.20040590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voehringer D, Shinkai K, Locksley RM. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity. 2004;20:267–277. doi: 10.1016/s1074-7613(04)00026-3. [DOI] [PubMed] [Google Scholar]
- 25.Kawakami T, Galli SJ. Regulation of mast-cell and basophil function and survival by IgE. Nat Rev Immunol. 2002;2:773–786. doi: 10.1038/nri914. [DOI] [PubMed] [Google Scholar]
- 26.Gessner A, Mohrs K, Mohrs M. Mast cells, basophils, and eosinophils acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid cytokine production. J Immunol. 2005;174:1063–1072. doi: 10.4049/jimmunol.174.2.1063. [DOI] [PubMed] [Google Scholar]
- 27.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. 2005;6:600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 28.Liu Z, Liu Q, Pesce J, Whitmire J, Ekkens MJ, Foster A, VanNoy J, et al. Nippostrongylus brasiliensis can induce B7-independent antigen-specific development of IL-4-producing Tcells from naive CD4 Tcells in vivo. J Immunol. 2002;169:6959–6968. doi: 10.4049/jimmunol.169.12.6959. [DOI] [PubMed] [Google Scholar]
- 29.Chakir H, Lemay AM, Webb JR. Cytokine expression by murine DX5+ cells in response to IL-12, IL-18, or the combination of IL-12 and IL-18. Cell Immunol. 2001;212:71–81. doi: 10.1006/cimm.2001.1844. [DOI] [PubMed] [Google Scholar]
- 30.Lauwerys BR, Garot N, Renauld JC, Houssiau FA. Cytokine production and killer activity of NK/T-NK cells derived with IL-2, IL-15, or the combination of IL-12 and IL-18. J Immunol. 2000;165:1847–1853. doi: 10.4049/jimmunol.165.4.1847. [DOI] [PubMed] [Google Scholar]
- 31.Faggioni R, Cattley RC, Guo J, Flores S, Brown H, Qi M, Yin S, et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of Fas/Fas ligand-mediated models of liver disease in mice. J Immunol. 2001;167:5913–5920. doi: 10.4049/jimmunol.167.10.5913. [DOI] [PubMed] [Google Scholar]
- 32.Punnonen J, Aversa G, Cocks BG, McKenzie AN, Menon S, Zurawski G, de Waal MR, de Vries JE. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci USA. 1993;90:3730–3734. doi: 10.1073/pnas.90.8.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, Murray R, et al. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–432. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 34.Urban JF, Jr, Noben-Trauth N, Donaldson DD, Madden KB, Morris SC, Collins M, Finkelman FD. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- 35.Mohrs M, Ledermann B, Kohler G, Dorfmuller A, Gessner A, Brombacher F. Differences between IL-4- and IL-4 receptor alpha-deficient mice in chronic leishmaniasis reveal a protective role for IL-13 receptor signaling. J Immunol. 1999;162:7302–7308. [PubMed] [Google Scholar]
- 36.Flesch IE, Wandersee A, Kaufmann SH. Effects of IL-13 on murine listeriosis. Int Immunol. 1997;9:467–474. doi: 10.1093/intimm/9.4.467. [DOI] [PubMed] [Google Scholar]
- 37.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [see comments] [DOI] [PubMed] [Google Scholar]
- 38.Bancroft AJ, McKenzie AN, Grencis RK. A critical role for IL-13 in resistance to intestinal nematode infection. J Immunol. 1998;160:3453–3461. [PubMed] [Google Scholar]
- 39.Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, Hogan SP, et al. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J Exp Med. 2002;195:1433–1444. doi: 10.1084/jem.20020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 41.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, et al. Nonclassical CD1d-restricted NK Tcells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burd PR, Thompson WC, Max EE, Mills FC. Activated mast cells produce interleukin 13. J Exp Med. 1995;181:1373–1380. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minty A, Asselin S, Bensussan A, Shire D, Vita N, Vyakarnam A, Wijdenes J, et al. The related cytokines interleukin-13 and interleukin-4 are distinguished by differential production and differential effects on T lymphocytes. Eur Cytokine Netw. 1997;8:203–213. [PubMed] [Google Scholar]
- 44.Kronenberg M, Gapin L. The unconventional lifestyle of NKT cells. Nat Rev Immunol. 2002;2:557–568. doi: 10.1038/nri854. [DOI] [PubMed] [Google Scholar]
- 45.Yoshimoto T, Tsutsui H, Tominaga K, Hoshino K, Okamura H, Akira S, Paul WE, Nakanishi K. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci USA. 1999;96:13962–13966. doi: 10.1073/pnas.96.24.13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Katsumoto T, Kimura M, Yamashita M, Hosokawa H, Hashimoto K, Hasegawa A, Omori M, et al. STAT6-dependent differentiation and production of IL-5 and IL-13 in murine NK2 cells. J Immunol. 2004;173:4967–4975. doi: 10.4049/jimmunol.173.8.4967. [DOI] [PubMed] [Google Scholar]
- 47.McDermott JR, Humphreys NE, Forman SP, Donaldson DD, Grencis RK. Intraepithelial NK cell-derived IL-13 induces intestinal pathology associated with nematode infection. J Immunol. 2005;175:3207–3213. doi: 10.4049/jimmunol.175.5.3207. [DOI] [PubMed] [Google Scholar]
- 48.Grencis RK. Cytokine regulation of resistance and susceptibility to intestinal nematode infection - from host to parasite. Vet Parasitol. 2001;100:45–50. doi: 10.1016/s0304-4017(01)00482-4. [DOI] [PubMed] [Google Scholar]
- 49.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 50.Noben-Trauth N, Hu-Li J, Paul WE. IL-4 secreted from individual naive CD4+ Tcells acts in an autocrine manner to induce Th2 differentiation. Eur J Immunol. 2002;32:1428–1433. doi: 10.1002/1521-4141(200205)32:5<1428::AID-IMMU1428>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 51.Helmby H, Grencis RK. Essential role for TLR4 and MyD88 in the development of chronic intestinal nematode infection. Eur J Immunol. 2003;33:2974–2979. doi: 10.1002/eji.200324264. [DOI] [PubMed] [Google Scholar]
- 52.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 53.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 54.Yoshimoto T, Min B, Sugimoto T, Hayashi N, Ishikawa Y, Sasaki Y, Hata H, et al. Nonredundant roles for CD1d-restricted natural killer T cells and conventional CD4+ T cells in the induction of immunoglobulin E antibodies in response to interleukin 18 treatment of mice. J Exp Med. 2003;197:997–1005. doi: 10.1084/jem.20021701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. IL-12 up-regulates IL-18 receptor expression on Tcells, Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 56.Liu Z, Liu Q, Hamed H, Anthony RM, Foster A, Finkelman FD, Urban JF, Jr, Gause WC. IL-2 and autocrine IL-4 drive the in vivo development of antigen-specific Th2 Tcells elicited by nematode parasites. J Immunol. 2005;174:2242–2249. doi: 10.4049/jimmunol.174.4.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Greenwald RJ, Urban JF, Ekkens MJ, Chen S, Nguyen D, Fang H, Finkelman FD, et al. B7-2 is required for the progression but not the initiation of the type 2 immune response to a gastrointestinal nematode parasite. J Immunol. 1999;162:4133–4139. [PubMed] [Google Scholar]