

Abstract
Reaction of trienes with α,β-unsaturated aldehydes produces bicyclic products via a tandem Diels-Alder/ene reaction. The adduct from tiglic aldehyde was converted into isoligularone by conversion to a furan followed by benzylic oxidation.
Keywords: Diels-Alder, ene reaction, oxidation
Introduction
In the context of developing new tandem radical reactions,1–3 we recently discovered a novel tandem reaction involving a Diels-Alder reaction followed by an ene reaction.4 Although the initial system studied (1 and 2) underwent the tandem reaction either at 80 °C in 56% yield or at 0 °C with catalysis by boron trifluoride etherate in 82% yield, less reactive aldehydes required Lewis acid catalysis.
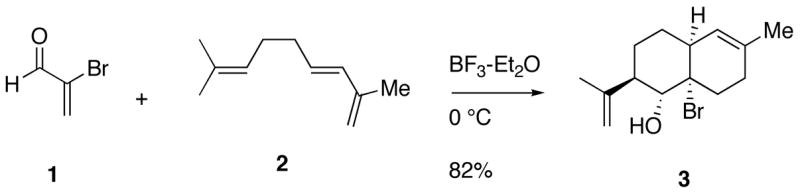
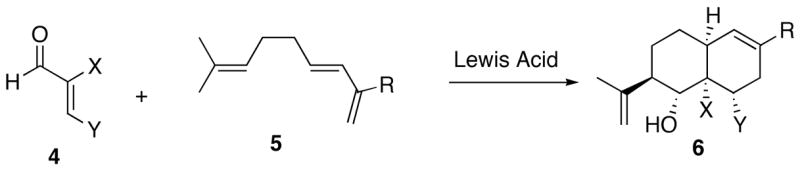
Because of the endo-selectivity of the Diels-Alder reaction, we anticipated a cis-fused decalin framework in product 3. NOESY NMR experiments indicated a trans-relationship between the isopropenyl group and the hydroxyl group but did not reveal the relative stereochemistry between the hydroxyl group and the ring juncture. An x-ray structure determination on a related adduct5 allowed us to assign retrospectively the relative stereochemistry of 3. In order to determine the scope of this interesting reaction, an array of dienophiles was treated with trienes. The results of our study showed that excellent stereochemical control could be achieved if X and Y in aldehyde 4 were both methyl groups or if X and Y were connected as part of a ring. Unsaturated ketones reacted with triene 5 to afford Diels-Alder adducts, but the subsequent ene reaction did not occur.
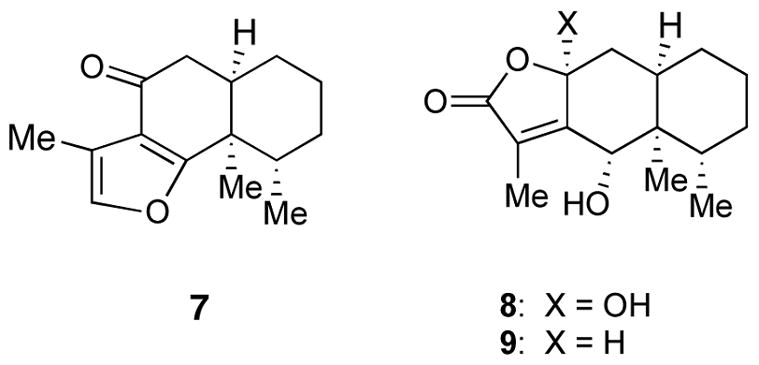
One application of this methodology would be to the synthesis of eremophilane sesquiterpenes.6 The sesquiterpene isoligularone (7),7 and the eremophilanolides 88 and 99 have been reported. Compound 7 has been synthesized by Yoshikoshi and by Tobinaga using novel Michael addition protocols.10 Since compounds bearing the 3-acylfuran subunit have shown useful antiviral and anticancer activity, we developed a direct synthesis of this natural product.
Results and Discussion
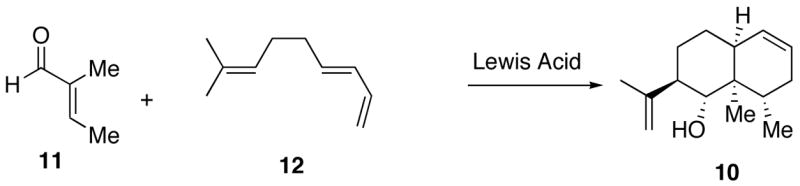
Our synthetic route begins with adduct 10. This was prepared in 77% yield as shown below from commercially available aldehyde 11 and triene11 12.
Hydroxyl-directed epoxidation of 10 with tert-butylhydroperoxide and molybdenum hexacarbonyl provided a separable mixture of two epoxides which could be reduced (H2, Pt/C) and oxidized with the Dess-Martin periodinane to yield 13 and 14 in 65% overall yield from 10 in a ratio of 60:40. Reaction of the major epoxide isomer with sodium hydroxide in ethanol at 25 °C followed by acidification afforded furan 15 in 82% yield. A small amount (5%) of enone 16 was also isolated. The minor epoxide isomer produced 15 and 16 in 20% and 60% yields, respectively. On the basis of the selectivities reported for the molybdenum hexacarbonyl-catalyzed epoxidation of (-)-isopulegol,12 we tentatively assign the structures of 13 and 14.

The conversion of furan 15 into isoligularone (7) required a benzylic oxidation. Although several methods have been advanced for this transformation,13 the application of most of these methods (CrO3, Pb(OAc)4, SeO2) to 15 led to decomposition of the furan subunit. Fortunately, the use of chromium hexacarbonyl and tert-butylhydroperoxide in boiling acetonitrile afforded a 32% yield of 7.14 The proton NMR and 13C NMR spectra for 7 matched the literature spectra15 for isoligularone.
The direct synthesis of 7 nicely demonstrates the utility of the tandem Diels-Alder/ene reaction sequence for the synthesis of natural products. This tandem sequence can create as many as five stereogenic centers in a single reaction. Applications of this chemistry to other natural product ring systems are in progress.
General Procedure for Tandem Diels-Alder/Ene reaction
To a solution of α,β-unsaturated aldehyde (1.1 mmol) in Et2O (3.5 mL) at 0 °C was added BF3·OEt2 (1.1 mmol) and the mixture was stirred for 10 min at 0 °C [in case of Et2AlCl, it was added to aldehyde at −78 °C and the mixture was stirred for 10 min at −78 °C]. To the resulting yellow solution was added via cannula a solution of triene (1 mmol) in Et2O (1 mL plus 0.5 mL rinse). After 1 h at 0 °C, the reaction mixture was warmed to rt and further stirred for 6 to 19 h (monitored by tlc). The reaction was quenched by the addition of saturated aqueous NH4Cl (3 mL) and the mixture was extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and was concentrated in vacuo. The residue was purified via silica gel flash chromatography using 5:1 hexanes: ethyl acetate to give the Diels-Alder/ene adduct.
8a-Bromo-2-isopropenyl-6-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1ol (3)
1H NMR (400 MHz, CDCl3) δ5.03 (s, 1H), 4.83 (bs, 1H), 4.81 (s, 1H), 3.28 (d, J = 10.4 Hz, 1H), 3.01 (bs, 1H), 2.86-2.82 (m, 1H), 2.45 (dt, J = 10.4, 4.0 Hz, 1H), 2.28-2.13 (m, 3H), 2.10-1.92 (m, 1H), 1.81-1.73 (m, 1H), 1.68 (s, 3H), 1.64 (s, 3H), 1.52-1.41 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 146.4, 135.0, 124.8, 112.8, 81.6, 69.0, 50.7, 45.8, 37.2, 29.8, 28.0, 27.0, 23.0, 19.6; Rf (hexane-EtOAc 5:1) = 0.44; HRMS m/e (EI) for C14H21BrO (M)+ calcd 284.0776, measured 284.0780.
2-Isopropenyl-8,8a-dimethyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-ol (10)
1H NMR (300 MHz, CDCl3) δ 5.66-5.61 (m, 1H), 5.34-5.28 (m, 1H), 4.85 (d, J = 9Hz, 2H), 3.57 (d, J = 10.5Hz, 1H), 2.52-2.38 (m, 1H), 2.28-1.95 (m, 2H), 1.80-1.71 (m, 2H), 1.67 (s, 3H), 1.51-1.32 (m, 4H), 0.97 (s, 3H), 0.91 (d, J = 7.4Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 147.6, 130.8, 126.2, 113.1, 70.0, 49.2, 38.7, 32.3, 30.5, 26.9, 26.7, 19.2, 18.1, 15.4; Rf (hexane-EtOAc 5:1) = 0.51; HRMS m/e (EI) for C15H24O (M)+ calcd 220.1827, measured 220.1820. Mp 18.7-20.2 °C.
2-(1-Methyl-2-oxacyclopropyl)-8,8a-dimethyl-decahydronaphthalen-1-one (13/14)
To a solution of alcohol 10 (0.89 g, 4.1 mmol) and Mo(CO)6 (96 mg, 0.36 mmol) in 10 mL of benzene was added tert-butylhydroperoxide (0.84 mL, 5.5M solution in decane) at rt under argon. The reaction mixture was heated to reflux for 1h and then cooled to rt. To the mixtture was added 5 mL of 10% Na2S2O4(aq). It was extracted with ethyl acetate (3x20 mL). The organic layer was dried over MgSO4, filtered and was concentrated in vacuo. The residue was purified via silica gel flash chromatography using 5:1 hexanes:ethyl acetate to give epoxides (850mg, 80% yield).
To a solution of epoxide (850 mg, 3.6 mmol) in 10 mL of tetrahydrofuran was added Pt-C (1%, 200mg) and the flask was charged with H2 gas. The mixture was stirred at rt for 1 h and then was filtered through Celite. The filtrate was concentrated in vacuo (809mg, 94% yield).
To a solution of above crude compound (809 mg, 3.4 mmol) in 20 mL of CH2Cl2 was added the Dess-Martin periodinane (1.58 g, 3.7 mmol) at 0 °C. The mixture was allowed to warm to rt and stirred for 8 h. To the mixture was added 20 mL of saturated NaHCO3(aq) and 20mL of 10% Na2S2O3(aq). It was then extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered and was concentrated in vacuo. The residue was purified via silica gel flash chromatography using 5:1 hexanes:ethyl acetate to give epoxides 13 (418 mg, 52% yield) and 14 (280 mg, 35% yield).
Compound 13
1H NMR (300 MHz, CDCl3) δ 2.77 (d, J = 4.5Hz, 1H), 2.55 (d, J = 4.5Hz, 1H), 2.40-2.29 (m, 2H), 2.20-1.91(m, 5H), 1.49-1.33 (m, 5H), 1.30 (s, 3H), 1.25-1.18 (m, 1H), 1.12 (s, 3H), 0.88 (d, J = 6.8Hz, 3H) ; 13C NMR (75 MHz, CDCl3) δ 214.5, 57.1, 55.9, 53.1, 45.4, 39.4, 31.4, 30.6, 29.7, 26.7, 23.5, 20.5, 17.7, 14.8 ; Rf (hexane-EtOAc 5:1) = 0.43 ; HRMS m/e (EI) for C15H24O2 (M)+ calcd 236.1776 , measured 236.1771. Anal calcd for C15H24O2: C, 76.23; H, 10.24 Found C, 75.52, H, 10.89.
Compound 14
1H NMR (300 MHz, CDCl3) δ 2.71-2.62 (m, 1H), 2.60 (d, J = 4.5Hz, 1H), 2.49 (d, J = 4.5Hz, 1H), 2.41-2.28 (m, 2H), 2.21-1.81(m, 5H), 1.50-1.40 (m, 5H) 1.37 (s, 3H), 1.16 (s, 3H), 0.88 (d, J = 6.8Hz, 3H) ; 13C NMR (75 MHz, CDCl3) δ 214.5, 56.9, 55.1, 53.5, 51.9, 45.8, 39.4, 32.6, 30.1, 26.0, 25.4, 21.4, 18.1, 14.1; Rf (hexane-Et2O 5:1) = 0.37. Anal calcd for C15H24O2: C, 76.23; H, 10.24 Found C, 75.98, H, 10.62.
3,9,9a-Trimethyl-4,5,5a,6,7,8,9,9a-octahydro-naphtho[1,2-b]furan (15)
To a solution of compounds 13 and 14 (0.24 g, 1 mmol) in 5 mL of ethanol was added NaOH (4 mg, 0.1 mmol) at rt. The solution was stirred for 8 h at rt. To the solution was added 2 mL of 10% HCl (aq) and it was extracted with ethyl ether (2x10 mL). The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified via silica gel flash chromatography using 2:1 hexanes:ethyl acetate to give compound 15 (124 mg, 57% yield) and 16 (63 mg, 27% yield).
Compound 15
1H NMR (400 MHz, CDCl3) δ 7.04 (d, J = 1.2Hz, 1H), 2.41-2.26 (m, 2H), 2.09-2.04 (m, 1H), 1.91 (d, J = 1.2Hz, 3H), 1.85-1.25 (m, 9H), 1.19 (s, 3H), 0.96 (d, J = 6.8Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.1, 137.1, 119.5, 116.4, 42.5, 38.9, 36.6, 31.2, 29.9, 27.8, 25.5, 21.5, 20.7, 17.8, 8.4 ; Rf (hexane-EtOAc 2:1) = 0.81 ; HRMS m/e (EI) for C15H22O (M)+ calcd 218.1670 , measured 218.1665. The boiling point of 15 was 189-194 °C/40 mm Hg.
Compound 16
1H NMR (400 MHz, CDCl3) δ 4.17 (s, 2H), 2.95-2.82 (m, 1H), 2.19-1.95 (m, 4H), 1.85 (s, 3H), 1.70-1.25 (m, 8H), 1.09 (s, 3H), 0.67 (d, J = 6.8Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 203.9, 142.0, 131.1, 62.4, 47.6, 40.2, 33.7, 30.1, 29.2, 28.4, 25.2, 24.0, 18.8, 10.4; Rf (hexane-EtOAc 2:1) = 0.48
Isoligularone (7)
To a solution of compound 15 (73 mg, 0.33 mmol) in 2 mL of acetonitrile was added Cr(CO)6 (31mg, 0.17mmol) and tert-butylhydroperoxide (0.073 mL, 5.5M solution in decane) and then heated to reflux for 7 h. The mixture was cooled to rt and then diluted with ethyl ether (10 mL). It was washed with H2O, sat NaHCO3(aq) and brine. The organic layer was dried over MgSO4, filtered and concentrated in vacuo. The residue was purified via preparative TLC using 5:1 hexanes:ethyl acetate to give compound 7 (24 mg, 32% yield).
1H NMR (400 MHz, CDCl3) δ 7.07 (d, J = 1.2Hz, 1H), 2.85 (m, 1H), 2.25-2.20 (m, 2H), 2.19 (d, J = 1.2Hz, 3H), 1.61-1.26 (m, 8H), 1.30 (s, 3H), 0.92 (d, J = 6.8Hz, 3H); 13C NMR (100 MHz, CDCl3) δ196.3, 174.8, 139.3, 119.2, 118.7, 42.3, 41.4, 39.9, 35.0, 30.1, 26.3, 20.6, 17.3, 16.2, 9.4 ; Rf (hexane-EtOAc 5:1) = 0.33 ; HRMS m/e (EI) for C15H20O2 (M)+ calcd 232.1463 , measured 232.1466.
Acknowledgments
We thank the National Institutes of Health (grant P01 ES12020) and the Office of Dietary Supplements for partial financial support through the Center for Research on Botanical Dietary Supplements at Iowa State University.Experimental
References
- 1.Kraus GA, Kim J. Tandem Diels-Alder Reaction/Radical Cyclizations for the Rapid Construction of Bridged Ring Systems. Tetrahedron Lett. 2004;45:1457–1459. [Google Scholar]
- 2.Kraus GA, Kim I. A direct route to isoflavan quinones. The Synthesis of Colutequinones A and B. Tetrahedron. 2003;59:7935. [Google Scholar]
- 3.For excellent reviews on tandem reactions, see: Parsons PJ, Penkett CS, Shell AJ. Tandem Reactions in Organic Synthesis. Chem Rev. 1996;96:195. doi: 10.1021/cr950023+.Winkler JD. Tandem Diels-Alder Cycloadditions in Organic Synthesis. Chem Rev. 1996;96:167–76. doi: 10.1021/cr950029z.
- 4.Kraus GA, Kim J. Tandem Diels-Alder/Ene Reactions. Org Lett. 2004;6:3115–17. doi: 10.1021/ol048847d. [DOI] [PubMed] [Google Scholar]
- 5.The x-ray structure was determined on the adduct of 2 with 2,5-dihydrothiophene carboxaldehyde.
- 6.Fraga BM. Natural sesquiterpenoids. Natural Prod Reports. 2004;21:669–693. doi: 10.1039/b407376m. [DOI] [PubMed] [Google Scholar]
- 7.Sato T, Tada M, Takahashi T, Horibe I, Ishii H, Iwata T, Kuriyama K, Tamura Y, Tori K. Chem Lett. 1977:1191–4. [Google Scholar]
- 8.Sugama K, Hayashi K, Mitsuhashi H. Eremophilenolides from Petasites japonicus. Phytochemistry. 1985;24:1531–5. [Google Scholar]
- 9.Ishii H, Tozyo T, Minato H. J Chem Soc (C) 1966:1545. [Google Scholar]
- 10.Koike T, Takeuchi N, Ohta T, Tobinaga S. Total synthesis of racemic ligularone and isoligularone. Chem Pharm Bull. 1999;47:897–899. [Google Scholar]; Miyashita M, Kumazawa T, Yoshikoshi A. 1-Nitro-1-(phenylthio)propene as a new nitro olefin reagent for 3-methylfuran annulation and its application to the synthesis of some furanoterpenoids. J Org Chem. 1980;45:2945–50. [Google Scholar]
- 11.Renslo AR, Danheiser RL. Synthesis of Substituted Pyridines via Regiocontrolled [4 + 2] Cycloadditions of Oximinosulfonates. J Org Chem. 1998;63:7840–7850. [Google Scholar]
- 12.Kim JH, Lim HJ, Cheon SH. A facile synthesis of (6S,1’S)-(+)-hernandulcin and (6S,1’R)-(+)-epihernandulcin. Tetrahedron. 2003;59:7501–7507. [Google Scholar]
- 13.Ohta Y, Doe M, Morimoto Y, Kinoshita T. Regiospecific synthesis of 2-substituted furanonaphthoquinones. J Heterocyc Chem. 2000;37:731–734. [Google Scholar]; Corey EJ, Xiang YB. A method for the off-ring functionalization of carbon-substituted furans. Synthesis of kahweol from cafestol. Tetrahedron Lett. 1987;28:5403–6. [Google Scholar]
- 14.Pearson AJ, Chen YS, Han GR, Hsu SY, Ray T. A new method for the oxidation of alkenes to enones. An efficient synthesis of δ5-7-oxo steroids. J Chem Soc Perkin Trans. 1985;1:267–73. [Google Scholar]
- 15.Tada M, Sato T, Takahashi T, Tori K, Horibe I, Kuriyama K. Conformational equilibriums of eremophilanes, naturally occurring cis-decalin derivatives, studied by NMR and CD spectroscopy. J Chem Soc Perkin. 1981;1:2695–2701. [Google Scholar]