

Abstract
A non-toxic, nanoparticulate polyelectrolyte complex (PEC) drug delivery system was formulated to maintain suitable physicochemical properties at physiological pH. Toxicity, binding, and internalization were evaluated in relevant microvascular endothelial cells. PEC were non-toxic, as indicated by cell proliferation studies and propidium iodide staining. Inhibitor studies revealed that PEC were bound, in part, via heparan sulfate proteoglycans and internalized through macropinocytosis. A novel, flow cytometric, Scatchard protocol was established and showed that PEC, in the absence of surface modification, bind cells non-specifically with positive cooperativity, as seen by graphical transformations.
Keywords: polyelectrolyte complexes, flow cytometry, Scatchard plots, nanoparticles, drug delivery
1. Introduction
A wide array of nanotechnologies is beginning to change the foundations of disease diagnosis, treatment, and prevention. Because molecules and structures inside cells operate at the nano- and micro-scale, the development of nanoparticulate approaches has become a key strategy for medical intervention at the cellular level. There are numerous nanoscale constructs, assemblies, architectures, and particulate systems being studied as targeted and drug delivery platforms. These include polymeric micelles, dendrimers, virus-derived capsid nanoparticles (NP), polyplexes, and liposomes[1–5]. These nanovehicles do not behave equivalently; their behavior within the biological microenvironment, stability, extracellular and cellular distribution vary with their chemical makeup, morphology and size[6].
NP systems, particularly polymer-based, can be formed by a variety of techniques, many of which utilize potentially toxic components and solvents. The most common fabrication methods involve polymerization reactions such as emulsion, dispersion, and inverse microemulsion polymerization of both biodegradable and non-biodegradable polymers. These techniques often induce polymerization using mineral oils and strong organic solvents, which may remain in the formulation along with other unreacted monomers, initiators, and surfactants. Such residues present safety issues in the final product[4, 7, 8]. Specifically, alkylcyanoacrylate nanotechnology suffers from toxic breakdown products[9].
One strategy to circumvent these processing limitations has involved the use of water-soluble, biodegradable, polymeric, polyelectrolyte NP. Biodegradable, polymeric polyelectrolytes degrade at a very slow rate into non-toxic products, and they do not alter normal cell function[10]. The polyelectrolyte system is aqueous, a major advantage for products to be used as drug delivery systems in humans. These nanoparticulate architectures, termed polyelectrolyte complex dispersions (PEC), result from strong electrostatic interactions between the charged microdomains of at least two oppositely charged polyelectrolytes[11]. The mixing of solutions of polyanions and polycations leads to the spontaneous formation of insoluble PEC under appropriate conditions. PEC formation is governed by the strength and location of ionic sites, polymer chain rigidity, precursor chemistries, pH, temperature, ionic strength, mixing intensity, and other controllable factors. Multi-component PEC, combined with Pluronic F-68 as a steric stabilizer, have been fabricated as a generic scaffold for incorporation of drugs and targeting moieties.
Cellular binding and internalization of PEC and NP by cells have not been subjected to detailed mechanistic studies. Frequently, PEC have been applied in gene delivery[12, 13], but there has been little focus on the process of PEC/cell interactions that occur before the expression of vector DNA. We have developed a fluorescent approach, by incorporation of a fluorophore, to describe the non-specific adsorptive mechanisms of nanoparticulate polyelectrolyte complex association in a human microvascular endothelial cell (HMVEC) model.
This cell type was chosen as a model because of its critical pathobiological significance. Vascular networks are lined with endothelial cells, which display unique surface markers for targeted delivery at sites of cancer and tissue repair[14]. Furthermore, the accessibility of endothelial cells from the blood stream is an important advantage over other target candidates such as tumor cells. Effective delivery of PEC is of paramount significance for the development of systems that target the vascular endothelium, the nutrient supply line for tumor and wound environments.
To improve polyelectrolyte drug delivery systems, it is critical to understand the mechanisms for and barriers to successful interactions with target cells. In the present study, fluorescent labeling and flow cytometry (FACS) permitted a multi-dimensional analysis of PEC/cell interactions with a stable non-radioactive marker. The mechanisms of PEC surface association were investigated by inhibitor studies. A novel, flow cytometric Scatchard analysis was used to define the cell-PEC interactions and the nature of internalization, components that have been rarely considered in studies to date.
2. Materials and Methods
2.1. PEC chemistries
All polyions were dissolved in distilled water before PEC fabrication. The anionic solution contained low molecular weight, low viscosity alginate, Mr=12 kDa (FMC Biopolymer, Drammen, Norway) and chondroitin sulfate, Mr=15 kDa, (Sigma Chemical Co., St. Louis, MO), both at 0.5 mg/ml. The cationic solution contained 0.5 mg/ml spermine tetrahydrochloride, Mr=0.348 kDa, (Sigma Chemical Co., St. Louis, MO), poly (methylene-co-guanidine) hydrochloride (PMCG), Mr=5 kDa (Scientific Polymer Products, Ontario, NY), calcium chloride (Sigma Chemical Co., St. Louis, MO), and 1% m/v Pluronic F-68 (Sigma Chemical Co., St. Louis, MO). All solutions were filtered through 0.22 μm nylon filters (Nalgene, Rochester, NY). With the exception of PMCG and Pluronic F-68, all components were derived from biological systems.
2.2. Polymer labeling
Fluorescein isothiocyanate (FITC, Sigma Chemical Co., St. Louis, MO) was covalently conjugated to terminal PMCG amines (pH 9.0) for 30 min under constant stirring at room temperature. The FITC-labeled product was purified by gel-permeation chromatography (Sephadex G-25, Amersham Biosciences, Piscataway, NJ) and eluted with distilled water.
2.3. PEC fabrication
A batch wise, non-stoichiometric process was used to create PEC. The solution of two anionic polyions (2 ml) was titrated into a cationic bath (20 ml), containing Pluronic F-68, with 20 kHz (maximum) frequency dispergation under conditions of mild mechanical stirring, defined as “one batch”. The system consisted of a needle (#26 gauge) connected to a 5 ml syringe, which was inserted into an ultrasonic, hollow, titanium probe with a 1.85 mm ID conical tip. The probe was connected to a transducer and power generator (Misonix, Farmingdale, NY). Anionic solution was slowly extruded via controlled air pressure (3 psig) at 1 ml/min. The complexes formed instantaneously. After preparation of PEC the 22 ml reaction suspension was transferred to 50 ml polypropylene ultracentrifuge tubes (Nalgene, Rochester, NY), the pH was measured (Fisher Accumet, Fairlawn, NJ), and the colloidal suspension was pelleted 3 times at 35000xg at 4°C for 10 minutes (Beckman, Model L5-50, Rotor Type 60 Ti). Subsequent to the first two centrifugations, the pellet was resuspended in 1 mM sodium acetate buffer, pH 4.2. Following the second wash, PECs were suspended in HMVEC complete growth media (Invitrogen/Gibco, Carlsbad, CA): MCDB 131 supplemented with 5% fetal calf serum (FCS), 2 mM L-Glutamine, 5 U/ml penicillin/streptomycin, 500 μg/ml hydrocortisone, 1 ng/ml epidermal growth factor. FITC-PMCG PEC were prepared by mixing 15.5 μg FITC-PMCG with the standard cationic bath. The addition of FITC-PMCG to the complex resulted in a colored PEC. Size and charge of PEC dispersions was determined by removing a small aliquot (1 ml) from any of the preparation steps and measurement by Malvern ZetaSizer Nano ZS (Malvern Instruments, Worcestershire, UK).
2.4. Transmission electron microscopy (TEM)
Reaction mixture preparations were analyzed for morphology and ZetaSizer validation by pipetting a 20 μl sample volume onto a dry, Formvar coated, 400 mesh copper grid (Electron Microscopy Sciences, Hatfield, PA). The volume was allowed to adsorb onto the grid surface for 30 s, after which the excess liquid was blotted carefully with filter paper. The specimen was then viewed with a Philips CM-12, 120 keV electron microscope equipped with a CCD camera. PEC diameter was evaluated by using point-to-point pixel based measurements for 1486 individual observations using software complementary to the TEM CCD camera (Advanced Microscopy Techniques, Danvers, MA).
2.5. PEC Size and Zeta Potential
Following colloidal preparations, washing and dispersion at varying pH, a 1 mL sample was removed for size and zeta potential measurement with the Malvern ZetaSizer Nano ZS (Malvern Instruments, Worcestershire, UK). Each measurement was performed in triplicate. The particle-sizing device uses non-invasive back scattering with photon correlation spectroscopy (PCS), which has a particle sensitivity in the range of 0.6 nm to 10 μm. Size-related measurements were reported as z-average mean, hydrodynamic diameter, and polydispersity index (PDI). The z-average mean is classically the parameter most comparable to diameters measured by transmission electron microscopy[15]. PDI is a dimensionless number that describes the heterogeneity of the sample, and it is scaled such that values less than 0.05 are rarely seen. The maximum value is 1.000. Values greater than 0.700 indicate a very broad size distribution and a lack of sample homogeneity.
2.6. Cell line and maintenance
The cell line used for these studies was an immortalized human microvascular endothelial cell line (HMVEC) generously provided by R. Swerlick from Emory University[16]. The cells were maintained in MCDB 131 medium supplemented with 5% fetal calf serum, 20 mM L-Glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 500 μg/ml hydrocortisone, 1 ng/ml epidermal growth factor (maintenance medium). During maintenance, the cells were cultured in T-flasks (Corning, Corning, NY) at 33°C, 5% CO2, and 95% relative humidity, which permitted expression of the introduced SV40 large T-antigen. After 3–4 days, cells were passaged by detachment with 0.25% trypsin/0.1% EDTA, counted by hemocytometer, and resuspended in fresh growth medium. All cell culture supplies were purchased from Invitrogen/Gibco, Carlsbad, CA.
2.7. Flow cytometric detection of PEC/cell interactions
Confluent HMVEC at approximately day 4 after plating were detached from 2 T-75 cm2 parent flasks and counted by hemocytometer. Aliquots equivalent to 5×105 cells were added to each well of 48-well plates (Nunc, Rochester, NY). HMVEC were permitted to attach overnight at 37°C (non-permissive conditions for the integrated large T-antigen), 5% CO2, and 95% relative humidity in complete MCDB131. Following a ~24 h incubation, experiments were performed by exposing HMVEC to PEC under various concentration and inhibitor schemes (detailed later). At the outset of exposure, the test media containing PEC were removed and the cultures washed thrice with 4°C phosphate buffered saline (PBS, Invitrogen/Gibco, Carlsbad, CA). Cells were then detached with 200 μl 0.25% trypsin/0.1% EDTA (2 min) or 5 min exposure to 5 mM EDTA in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Sigma Chemical Co., St. Louis MO), depending on the experiment. The majority of experiments were detached with EDTA, as trypsin was found to non-specifically affect PEC interactions. Detachment was terminated by the addition of FACS buffer (3% FCS in PBS). The detachment was followed by transfer of the cell suspension to 4 ml FACS tubes (BD Falcon, San Jose, CA) and centrifugation at 200xg for 10 min. The supernatant was removed, and cells were suspended in 200 μl FACS buffer. The tubes were kept on ice until arrival at the FACS location. Excitation of samples was performed with a 15 mW, 488 nm (λex= 485 nm) argon ion laser, and signals were detected with either λem=515–545 nm (FITC) or λem=564–606 nm (propidium iodide, PI) filters. Unless otherwise noted, acquisitions were performed using a 4-color FACSCalibur (BD Biosciences, Mountain View, CA).
A standard experiment included acquisition of HMVEC in the absence of PEC to establish a background. The samples exposed to PEC were then acquired. Side scatter (SSC), forward scatter (FSC), and FITC outputs were collected in list mode form and analyzed with FloJo (Treestar, Inc., Ashland, OR). The first round of acquisition yielded the total amount of cell-associated fluorescence (MFItotal), due to FITC PEC. The internalized fluorescence was determined by adding 120 μl 0.4 mg/ml trypan blue (TB, Mediatech, Herndon, VA) and re-acquiring the sample to give MFIinside. TB quenches extracellular FITC[17, 18] allowing the calculation of PEC cellular compartmentalization.
2.8. Binding and internalization kinetics
Fluorescent PEC were prepared as described above and suspended in 20 ml complete MCDB 131. Cell exposure, in 48-well plates, to 400 μl PEC was monitored for t=0, 5, 10, 15, 20, 30, 60, and 120 min. The 2 h incubation was followed by aspiration, washing at 4°C, detachment, centrifugation, and addition of FACS buffer. To account for FITC PMCG binding, 1.5 μg of the fluorescent polymer (FITC-PMCG:PMCG=1:10) was added to 20 ml complete MCDB131 and kinetics of association observed in an identical fashion. Flow cytometric measurements, minimally 10000 events, with and without TB were performed within 30 minutes.
2.9. PEC acute toxicity by propidium iodide (PI)
Toxicity of fluorescent PEC was detected by PI (Molecular Probes/Invitrogen, Eugene, OR) as described by Rasola, and Geuna[19]. Cells were exposed to PEC as described in time-course binding and internalization kinetics, detached after 2 h PEC exposure with 0.25% trypsin/0.1% EDTA, centrifuged and resuspended in 200 μl FACS buffer. PI (40 μl, 0.01 mg/ml) was added to each sample 1 min prior to FACS acquisition. For each sample, 10000 events were collected. The PI fluorescence was collected through a λem=564–606 nm filter after excitation of the fluorochrome at 488 nm (λex). The toxicity was represented as a percent of control cultures without PEC.
2.10. PEC effects on HMVEC-1 proliferation
One day before PEC exposure, cells were plated to 48 wells at a density of 2.5×103 cells/well. After an ~20 h attachment and acclimation period, cells were exposed to serial dilutions of PEC for 72 h without media change. Subsequent to the 72 h exposure, media for all wells was removed and HMVEC cell growth was measured using the Titer 96® Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA). Each well then received 300 μl fresh MCDB 131 and 20 μl of dye solution. Incubation was for 40 min at 37°C, 5% CO2, 95% relative humidity. Absorbance at 490 nm was determined by microplate reader (μQuant, Bio-Tek Instruments, Winooski, VT). All results were normalized to the control (no PEC).
2.11. Treatment of cells with various inhibitors
HMVEC were prepared as described as above. At the end of the treatments described below, cells were washed, detached and analyzed by flow cytometry as described. FITC-PMCG PEC were suspended in 20 ml of MCDB 131 complete medium. Wells received 400 μl PEC in medium or medium alone. In each case, at least one well was maintained under normal culture conditions. Inhibitors were applied for 30 min under standard incubation conditions (37°C, 5% CO2, and 95% relative humidity), unless otherwise indicated, followed by detachment with 5 mM EDTA. Flow cytometric measurements with and without TB were performed within 30 minutes. For each sample, 10000 events were collected. Inhibitors did not elicit any cellular toxicity as ascertained by FACS forward and side scatter.
To evaluate the effect of free glycosaminoglycans on PEC association[20, 21], cultures were pre-incubated 2h with 200 U/ml USP grade heparin (Celsus Laboratories, Cincinnati, OH) followed by a PBS wash, and then a mixture of 200 U/ml heparin and PEC in growth media.
Energy dependence of PEC-cell interaction was assessed by 2-deoxyglucose+sodium azide[22] or reduced temperature (4°C) [23]. For temperature effects, HMVEC were incubated at 4°C for 1 h in complete MCDB 131. The medium was then changed, PEC added, and incubation was continued for 30 min. 50 mM 2-deoxyglucose (Sigma Chemical Co., St. Louis, MO) and 0.05% v/v sodium azide (Sigma Chemical Co., St. Louis, MO) were prepared in complete medium and added to HMVEC for 1h, after which this mixture was replaced by PEC and 2-deoxyglucose+azide for a further 30 min.
The effect of actin on PEC association was evaluated by exposure of HMVEC to 10−8–10−5M cytochalasin D [24, 25] (Sigma Chemical Co., St. Louis, MO) in complete medium. After 2h, fresh medium with PEC/cytochalasin D treatment continued for 30 min.
To disrupt glycosaminoglycan synthesis, HMVEC that had been seeded in 48 well plates for 24h were washed and exposed to 500 μM α-xyl or β-xyl (Sigma Chemical Co., St. Louis, MO) overnight[26, 27] This treatment was followed by a further 30min exposure to PEC in the continued presence of α-xyl or β-xyl.
Trypsin sensitivity of PEC association with HMVEC was measured by detaching cells with 0.25% trypsin/0.1% EDTA as compared to 5 mM EDTA monolayer removal. PEC were added to cultures for 30 min. Subsequent to PBS washings, HMVEC were removed with either trypsin or EDTA.
2.12. Physical counting of PEC
Particle concentration, in terms of PEC/ml, was determined by flow cytometry (FACS Aria, BD Biosciences). The FACSAria was fitted with SSC, FITC, and FSC detection lasers. In particular, the FSC laser was adapted with a photomultiplier tube (PMT) to magnify the signal generated from the PEC. Firefli™ 200 nm fluorescent green (λex=468 nm/λem=530 nm) polystyrene microspheres (Duke Scientific Corporation, Palo Alto, CA) of known concentration were processed and detected using FSC PMT and SSC lasers. The number of events recorded over fixed period of time (60s) was used to develop a standard calibration curve. This calibration curve was then used to evaluate PEC recorded events and thus concentration over four logs of arbitrary dilution for one batch of fluorescent PEC. The populations of PEC were analyzed for fluorescence by FACSDiva software (BD Biosciences, Mountain View, CA) to normalize fluorescence units in terms of PEC/ml. Free and bound particle concentrations were defined based on ordinary linear interpolation and proportionality. Routine PEC concentrations for direct association and inhibitor experiments were 1.54×109 PEC/ml. In each FACS analysis, media and cell fluorescent backgrounds were established.
2.13. Scatchard plots
Scatchard titrations were prepared by exposing HMVEC to serial dilutions of PEC for 3 h under normal culture conditions. One day prior to analysis, 5×105 cells/well were seeded and incubated as previously described. Selected plates were incubated at 4°C to block the endocyotosis of added PEC[23]. The exposure was followed by cell detachment and acquisition of MFI by FACSAria (BD Biosciences, Mountain View, CA). For each sample, 10000 events were collected by list-mode data that consisted of side scatter, forward scatter, and fluorescence emission centered at 530 nm (FITC). Bound MFI was determined using FACSDiva software. Free, unbound MFI was evaluated by material balance. These MFI values were then converted to PEC concentration. Scatchard plots were prepared by plotting bound PEC/free PEC versus bound PEC as described classically[28].
2.14. Confocal microscopy
One day prior to PEC fabrication, confluent HMVEC at approximately day 4 of culture, with a viability of ≥90% or greater by TB exclusion, were detached from one T-75 cm2 parent flask, diluted, and seeded into 8-well microscope slides (Lab-Tek™ II Chamber Slide System, Electron Microscopy Sciences, Hatfield, PA) at 5000 cell/well. Cells were allowed to attach overnight at 37°C, 5% CO2, and 95% relative humidity in standard medium. FITC-PMCG labeled PEC were prepared and suspended in 20 ml MCDB 131 (1.54 × 109 PEC/ml) complete medium in preparation for microscopic studies. After the 24 h incubation, FITC-PMCG PEC (400 μl/chamber) were added. Cells were allowed to interact with PEC for up to 24 h, followed by aspiration and three washing steps with PBS at 4°C. The cultures were then fixed with cold methanol (200 μl) at −20°C for 30 min. The nuclei were stained with 150μl of TOPRO-3 (200 ng/ml; Molecular Probes/Invitrogen, Eugene, OR) for 10 min. The stain was then removed along with the chamber walls and the preparation was mounted with a glass coverslip. Images were collected with a 3-track confocal microscope (Zeiss LSM-510, Thornwood, NY) using the appropriate fluorescence filters: λex=488 nm (FITC) and λem=633 nm (TOPRO-3). Z-sectioning was applied to determine if PEC localized in the same optical plane as cells. By varying the distance between the objective pinhole and the specimen over fixed increments, a ‘z-series’ was generated that dissects and collects images through the specimen.
2.15. Statistical analysis
Statistical analysis was performed using JMP-IN 5.1 (SAS, Cary, NC). Reaction mixture formulations and final preparations in cell growth medium were compared by two-sample t-test to evaluate the differences between sizes, zeta potentials, and PDI. One-way ANOVA was used to compare cell proliferation data with controls (PEC-free exposures) while one-tail t-tests were applied to analyze statistical deviations of inhibitor responses from controls. All statistical tests were performed at p<0.05 (95% confidence level). Results are displayed as average ± standard error for at least 3 replicates.
3. Results and Discussion
3.1. PEC physicochemistry
As detailed previously[12], the non-stoichiometric titration of the anions chondroitin sulfate and sodium alginate into a multi-component cation bath, containing Pluronic F-68, PMCG, spermine, and CaCl2, caused spontaneous assembly of cationic nanoparticulate architectures in water. Fabrication was aided by a 20 kHz frequency ultrasound input to the anionic stream. These suspensions exhibited a colloidal, Tyndall effect. PEC, as prepared in their native state, had a measured pH of 4.2, due to excess cations present in the reaction mixture. The complexation led to a core-shell morphology, with the cations dominating the corona surface and a neutralized inner phase of polyanions. Hydrodynamic diameter and zeta potential of PEC were assessed by PCS, while morphological properties were evaluated with TEM. PEC in their reaction mixture were positively charged (34.7 mV) with a mean diameter of 164.6 nm and a polydispersity index of 0.199 (Figure 1A–C). The significant zeta potential, ≥ | ±30 mV |, was indicative of a stable colloidal suspension as has been pragmatically defined[29, 30]. TEM images (Figure 1D) showed that the complexes were spherical in nature, and they had a mean diameter of 141.5 nm, averaged over several separate micrographs and observations (n=1486). The correlation factor between PCS and TEM size estimates was 0.86. This ratio was consistent with measurements in many other polymeric nanoparticle systems[31, 32].
Figure 1.
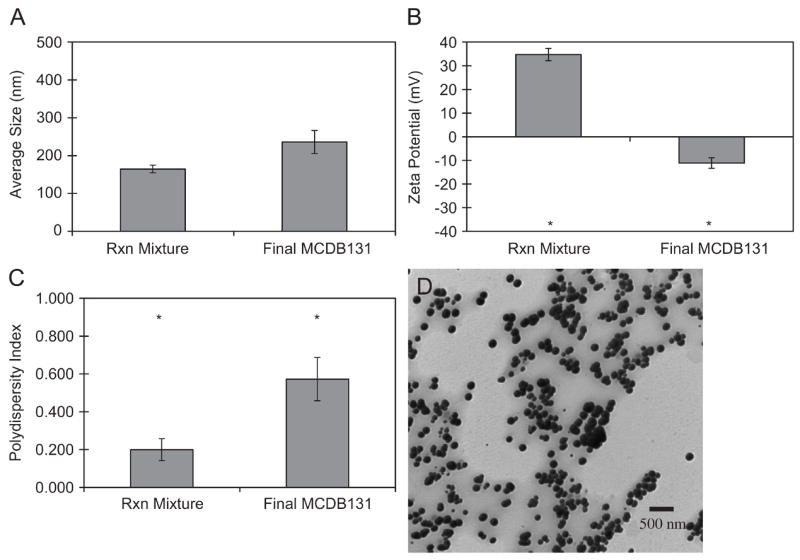
Physicochemical properties of the initial PEC reaction (Rxn) mixture and final, washed preparations in culture media for cellular studies. (A), (B), and (C) correspond to hydrodynamic diameter, zeta potential, and polydispersity index, respectively, measured by PCS. The average PEC diameter was 164.6 nm±10.2 nm and 235.9 nm±30.5 nm (mean±standard error, n=3) before and after washing, respectively; (D) is a representative TEM image. Asterisks indicate means that differ statistically by two-sample t-test at the 95% confidence interval.
PEC isolation and resuspension in endothelial cell growth medium led to an increase in size and PDI and a decrease in zeta potential (Figure 1A–C). Two-sample t-tests did not reveal a statistical difference in size, but there was a significant difference in surface charge and PDI (p<0.05). Hydrodynamic diameter was not significantly different from 200 nm, an empirical benchmark diameter for efficient cellular uptake and systemic delivery of nanoparticulate material[8, 33]. The change in PDI and zeta potential was likely due to the presence of serum particles and proteins, which have intrinsic light scattering properties detected by PCS. Alterations in surface charge were attributed to changes in the extent of ionization of surface groups[34] and/or the protrusion of ionized, core hydroxyl groups[35]. The preservation of hydrodynamic diameter may be related to the presence and passive entrapment of Pluronic F-68, which was added to provide steric stabilization. When adsorbed to the PEC surface, Pluronic F-68 could create osmotic and entropic barriers to particle-particle interactions and discourage adhesion of electronegatively charged serum molecules, resulting in maintenance of structural integrity[36, 37].
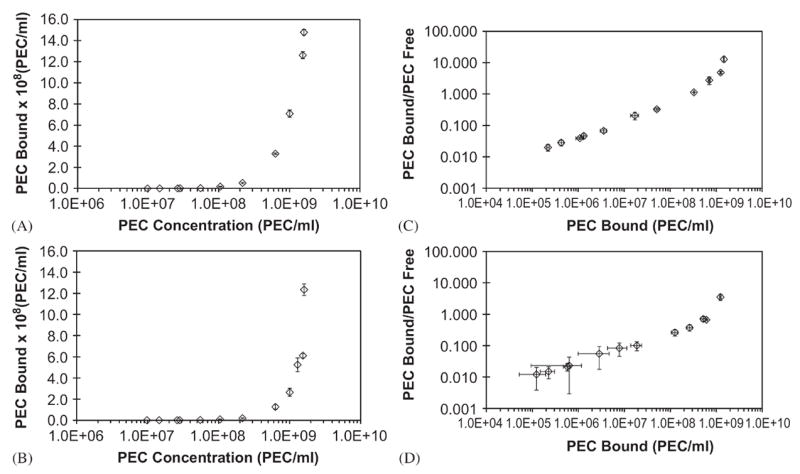
3.2. Determination of PEC number by flow cytometry
Flow cytometry was used to measure the concentration of PEC for subsequent Scatchard analysis and cytotoxicity assays. This method was far less labor-intensive and more accurate than electron microscope-based analysis. Serial dilutions of NIST-traceable fluorescent nanospheres (stock=2.3 × 1012 beads/ml, 200 nm nominal diameter) were prepared, diluted and processed using a FACSAria cytometer to calibrate the system for free versus cell-associated PEC. Plots of forward scatter versus side scatter showed no overlapping populations between 200 nm beads, FITC-labeled PEC, and HMVEC (Figure 2A–C). The ratio of events per second to the time elapsed was plotted against bead concentration (Figure 2D). The resultant, analogous PEC data showed a linear response from 9.80×106 to 1.54×109 PEC/ml (Figure 2E–F). Results were confirmed with PCS (data not shown), although with much lower sensitivity. Concentrations >1.5×109 PEC/ml or >2.×109 bead/ml could not be accurately measured, either due to saturation of the detector or aggregation due to a decrease in particle interstitial space.
Figure 2.
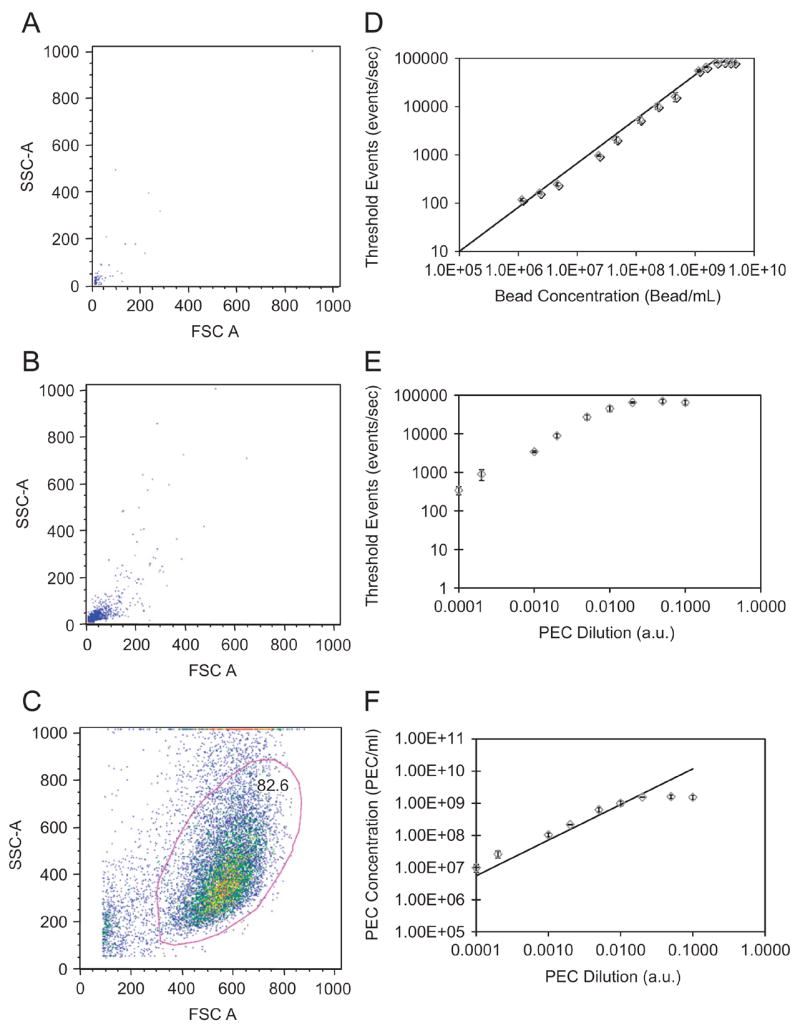
Evaluation of PEC concentrations by flow cytometry. FACS analyses of (A) NIST traceable fluorescent green beads and (B) multi-component PEC suspended in HMVEC growth media; (C) gating of HMVECs showed little or no overlap between cellular and PEC gates, while PEC and 200 nm bead share similar forward (FSC) and side (SSC) scattering. (D) calibration plot (detected events v. bead concentration) generated for various dilutions of NIST beads. (E) measured detected events versus arbitrary PEC dilutions per batch; (F) the calculated concentration of PEC based on the bead standardization. Error bars are the standard error for n=3.
3.3. PEC cytotoxicity
Cytotoxicity of multi-component PEC was tested by MTS reduction and propidium iodide (PI) internalization in confluent HMVEC, markers of cell metabolism and membrane integrity, respectively. Concentrations from 4×102 to 6×104 PECs/cell did not cause significant changes in MTS reduction as verified by one-way ANOVA and Dunnet’s Test (Figure 3A). No change in viability was detected when HMVEC were exposed to 1.5×109 PEC/ml for 2 h, detached, stained with PI and analyzed by FACS (Figure 3B). PI, which is excluded from viable cells, provided a higher–throughput, FACS-based approach to analyze toxicity after PEC exposure as compared to methods such as trypan blue dye exclusion. In the present study, trypan blue was used as a means to discriminate externally bound and internalized fluorescent signals. The limited cytotoxicity of the PEC system made it a suitable candidate for drug delivery to endothelial cells.
Figure 3.
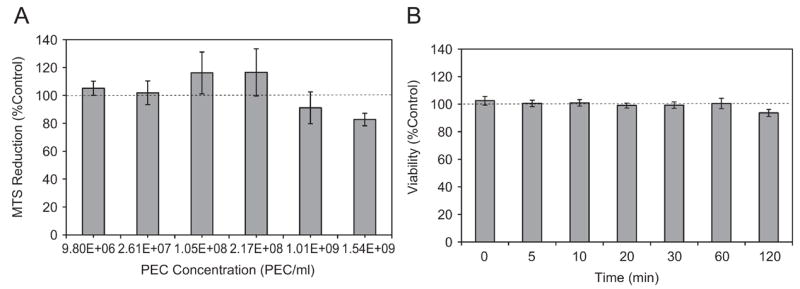
PEC cytotoxicity profiles for multi-component PEC. (A) MTS reduction after 72 h incubation over various serial dilutions; (B) propidium iodide staining at a fixed concentration of 1.54×109 PEC/ml. One-way ANOVA showed no significant difference as a function of dose or time, while Dunnet’s test showed that each mean was statistically the same as the control (no PEC), p <0.05. Data are means of at least 3 experiments (mean ± s.e.).
3.4. Suppression and quenching of extracellular fluorescence by TB
Binding and internalization of FITC-PMCG PEC was distinguished by quenching extracellular fluorescence with 0.4 mg/ml TB[17, 18]. Figure 4A shows a complete shift in FITC distribution by FACS after addition of TB to a 200 μl suspension of 1.5×109 PEC/ml in a 1:1 volumetric ratio. TB treatment of FITC-PMCG PEC resulted in a 98% suppression in the median fluorescence index (MFI), from median values of 6142 a.u. (TB+) to 130 a.u. (TB−), as illustrated in Figure 4B. Thus, TB was used to characterize compartmentalization (surface versus inside) of multi-component PEC by a simple material balance of MFI:
Figure 4.
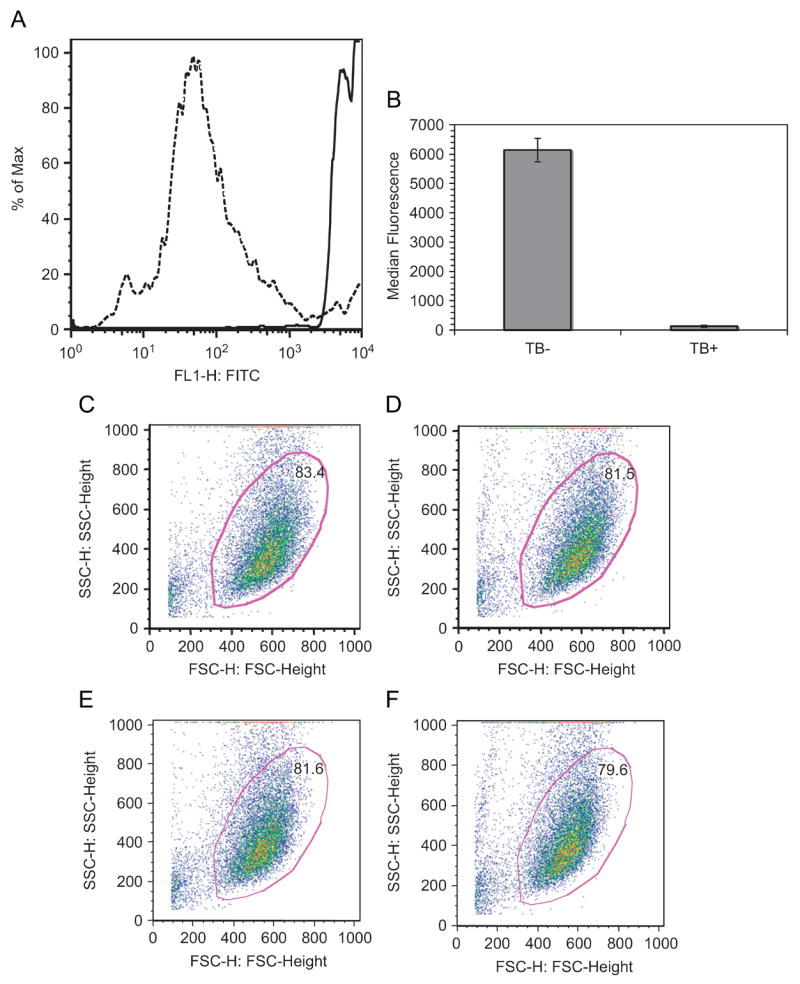
Suppression of FITC via addition of trypan blue. (A) Addition of 0.4 mg/ml trypan blue abolished the fluorescence of free PEC in suspension. Lines indicate the distributions of fluorescent events before (solid) and after (dashed) exposure to trypan blue, respectively. (B) median fluorescent signals (means ± standard error) for three independent experiments and PEC preparations. The use of trypan blue did not affect the viable, gated cell population in cultures alone (C), with trypan blue (E), incubated with PEC for 2 h (D), and PEC 2 h with trypan blue (F).
One of the main advantages of using flow cytometry to define PEC interactions in cellular environments is that the two scattering lasers are markers for cell size (forward scatter) and internal morphology (side scatter). Because dead cells differ in size compared to viable cultures, lower forward scatter values were considered non-viable populations. Independent of trypan blue exposure, live cellular gating remained constant (Figures 4C–F). Cell only, initial acquisitions, and trypan blue quenched populations comprised from 79–84% of the total events.
3.5. Kinetics of PEC binding and uptake: tryptic degradation of interactions
We employed FACS to track the binding and uptake of 1.54×109 FITC-containing PEC by HMVEC, in serum-containing medium at intervals up to 2 h. After exposure, cells were detached with 5 mM EDTA in 1X HBSS (pH=7.6), and the FACS signals were expressed as MFI. Despite the reversed zeta potential measured in MCDB131, PEC binding (Figure 5A) and uptake (Figure 5B) were rapid and reached saturation. The shift in fluorescence began immediately after addition of PEC and washing (t=0). FITC-PMCG PEC that were incubated with HMVEC for 2 h were examined by confocal laser scanning microscopy, which showed clear evidence of perinuclear accumulation of PEC (Figure 5D; green fluorescence). Z-sectioning indicated that perinuclear PEC were in the same plane as the cytoplasm. Cellular morphology was unchanged except for the association of refractile PEC with individual, intact cells (data not shown). PEC association was also observed in many other cell types: mouse fibroblasts (CRL-10225 and NIH3T3), CHO, CT26 colon carcinoma, primary human endothelium, macrophages, and hepatocytes (data not shown). The binding and internalization was not due to dissociation of FITC or FITC PMCG from the PEC. Over 2 h, the cumulative release in HMVEC growth media (5% FCS) was 1.75% of the initial amount incorporated, while after 6 h only 4.62% was liberated. Cellular surface proteins were involved in PEC binding and uptake, as trypsin decreased the total amount of surface bound (60%) and internalized (40%) PEC, relative to values in cells detached by EDTA (Figure 5C). Divalent cations did not appear to play in role in PEC/cell interaction.
Figure 5.
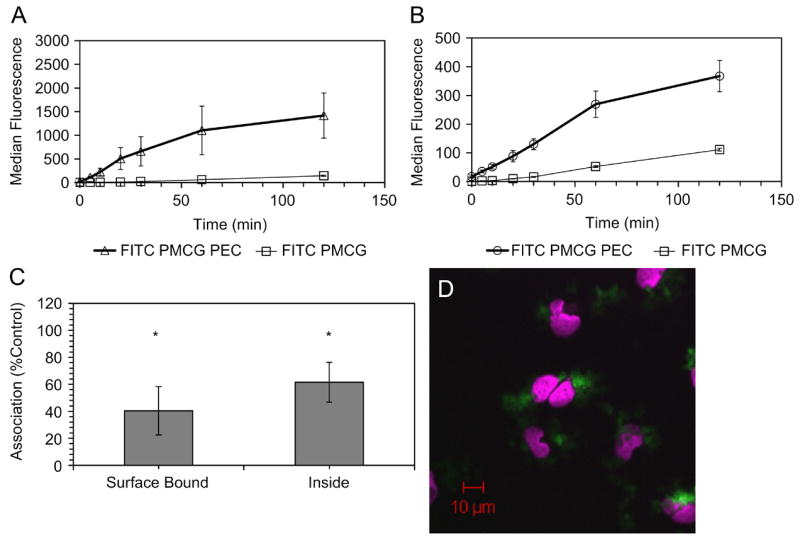
PEC-HMVEC association kinetics and trypsin sensitivity for HMVEC. Kinetics of rapid binding (A) and internalization (B) for 2 h PEC or free FITC PMCG exposure measured by FACS showed saturability(n=3). Binding and uptake at 30 min (C) were also sensitive to tryptic detachment (p<0.05, n=3). A one-sample t-test compared bound and internalized fluorescence compared to EDTA treated (control). Confocal laser scanning microscopy imaging (D) of PECs (green) incubated with HMVEC for 2 h showed a perinuclear accumulation. Nuclei were stained with TOPRO-3 (violet).
The kinetics of FITC-PMCG PEC binding (Figure 5A) and internalization (Figure 5B) was compared to 0.075 μg/ml of free fluorescent FITC-PMCG in HMVEC for 2h. For fluorescent PEC, the binding and internalization showed a saturable profile, with first order kinetics up to 1h. Free FITC-PMCG exhibited first-order linear binding and internalization kinetics for 2 h, indicative of a non-saturable phenomenon at this concentration, and overall binding and uptake was greatly reduced as compared to the NP formulation. The mass and charge of the PEC and free FITC-PMCG differed substantially. Although the bulk PEC surface charge was negative, microdomains of positive charge may contribute to the attachment of PEC to cells through guandinium-rich PMCG, which possibly induced electrostatic adsorption of PEC via bidentate hydrogen bonding to distal, electron-rich polyanionic residues located on the cell surface[38, 39].
Although the experimental design did not mimic an in vivo flow environment, PEC can interact with charged groups in microvascular endothelial beds. Incorporation of near infrared fluorescent derivatives of PMCG into PEC has enabled the real-time fluorescent imaging of PEC distribution in mice. Preliminary evidence has revealed that PEC interact extensively with the lung, liver, and spleen after several forms of intravenous administration (data not shown). This phenomenon may be due to the extensive elecropositivity embedded in the particle architecture from PMCG and spermine,
3.6. Probing endocytic mechanisms
Thermodynamic and metabolic energy requirements for binding and internalization were probed by incubating cells at reduced temperature or with glycolysis inhibitors, respectively. FACS analysis showed that both treatments produced significant decreases in binding and uptake (Figure 6A). Sodium azide/2-deoxyglucose treatment brought about a significant decrease in bound (−55%) and internalized (−70%) PEC through a mechanism mediated by depletion of ATP with secondary effects on membrane potential (Figure 6A, gray)[22, 39]. This response provided further evidence that PEC attachment and endocytosis was an active process that began with adsorptive fusion with the cell membrane. However, some residual attachment and internalization was resistant to metabolic inhibitors, indicating a process other than endocytosis. Transduction or direct PEC entry after diffusion of complexes to the cell surface, may contribute to this portion of the internalization after diffusion of complexes to the cell surface[40].
Figure 6.
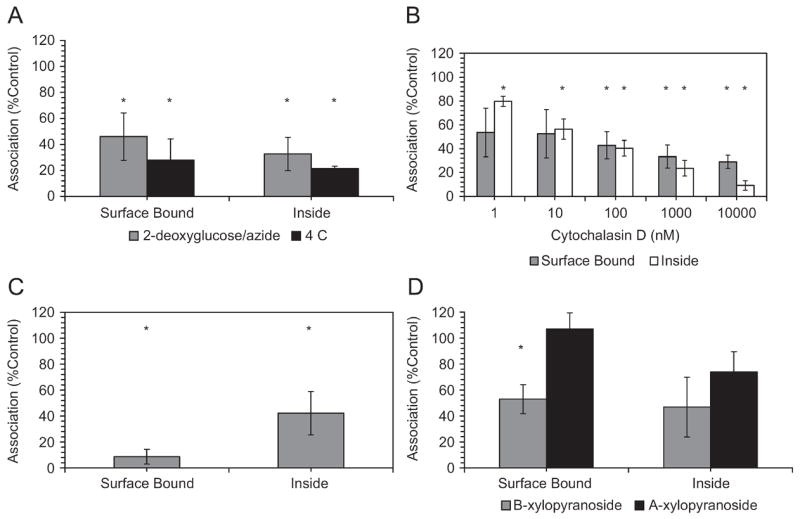
Inhibition of binding (surface bound) and internalization (inside) mechanisms is modulated by multiple mechanisms. (A) Energy-mediated PEC internalization and binding. Dependence on metabolic and thermodynamic was evaluated by incubation with 2-deoxyglucose/sodium azide (gray) and reduced temperature (black), respectively. Statistically significant differences were seen at the 95% confidence interval for surface binding and uptake (n=3). (B) Actin assembly facilitates the binding and internalization of PEC. Preincubation of HMVEC with 10 μM cytochalasin D for 2 h significantly inhibited particle uptake, but binding inhibition did not reach significance until higher doses were applied (one-sample t-test p<0.05). Data are the average of 3 replicates with error bars corresponding to standard error. (C) shows the competitive inhibition of attachment and binding by incubation of cells with PEC and 200 U/ml heparin while (D) displays the role of HSPG in PEC binding. HMVEC were 24 h pre-incubated with either 4-nitrophenyl-α-xylopyranoside (α-xyl, black) or 4-nitrophenyl-β-xylopyranoside (β-xyl, gray), where the α-isomer does not inhibit HSPG biosynthesis. Only the surface binding differed statistically from the control (p<0.05, n=3)
Reduced temperature is a classical endocytosis and active transport inhibitor, where only passive PEC membrane fusion occurs[23], and decreased temperature reduces the motility of surface protein groups and the lateral diffusion of PEC to the cell boundary layer. Internalization of PEC decreased by 80% at 4°C (Figure 6A, black). The apparent residual uptake may have been due to strong interactions of PMCG with the plasma membrane, as observed with arginine-rich peptides[24].
The role of the actin cytoskeleton in PEC binding and endocytosis was studied by addition of increasing concentrations of cytochalasin D, including a level (10 μM) that is reported to block phagocytosis and initial stages of macropinocytosis through microfilament disruption[24]. Although drug treatment reduced both surface binding and uptake at all concentrations (Figure 6C), only PEC internalization showed significant reduction at 1 nM and 10 nM (−28% and −42%, respectively, p<0.05). Higher concentrations resulted in significant decreases in both of the compartments. Consistent with other studies[24, 41], inhibition of PEC uptake by cytochalasin D suggested a role for macropinocytosis and non-clathrin, non-caveolae dependent endocytosis in the trafficking of PEC to the cell interior. Macropinocytosis is lipid raft-mediated, with macropinosomes often being larger than 1 μm, and it is dependent on cytoskeletal rearrangements[42]. Clathrin- and caveolae-dependent endocytosis rely on clathrin coated pits (~120 nm) or caveolae invaginations (~60 nm), respectively[43]. The PEC system applied in this study had hydrodynamic diameters consistently around 200 nm, making them too large for these processes[44].
The major charged surface component on many cell types are HSPG in the form of syndecans and glypicans[45]. The contributions of cell-surface HSPG to PEC binding and internalization was suggested by trypsin sensitivity treatment [46]. The role of HSPG in PEC association was defined by two approaches: (a) competitive inhibition of PEC binding by adding the anionic glycosaminoglycan heparin to the media prior to and during PEC exposure; (b) perturbed biosynthesis of HSPG by preincubation of HMVEC with α-xyl or β-xyl, where only β-xyl disrupts HSPG glycosaminoglycan chain elongation[26]. The HMVEC pre-treatments were followed by 30 min exposure to PEC.
Heparin treatment completely abolished PEC cellular interactions (Figure 6C), consistent with previous data using lipo- and polyplexes[47]. Surface binding was reduced by 93% (p<0.05), while internalization was diminished by 58% (p<0.05). Some PEC may have escaped heparin displacement by rapid cell entry. Heparin, because of its highly anionic nature, likely bound to and caused the neutralization of positively charged groups on the PEC corona, thus masking prospective cell binding sites. The charge neutralization may have also destabilized the complexes or caused PEC swelling[21], preventing cell binding and entry[20].
Incubation with a HSPG biosynthesis inhibitor, β-xyl, supported the hypothesis that membrane-associated HSPG partly mediated the attachment and internalization of PEC (Figure 6D). HSPG depletion led to a 47% reduction in surface PEC binding compared to the control (Figure 6D, gray). As expected, exposure to the inactive α-xyl isomer led to no significant deviation in binding and uptake as compared to the control (Figure 6D, black). Internalization was not statistically reduced for either xylopyranoside, although β-xyl did produce an obvious decrease, suggesting a possible role of HSPG in endocytosis. The xyloside-resistant cell surface component may have been non-specific and electrostatic, but it was more likely related to low turnover rate of HSPG. It is possible that other sulfated glycoproteins or glycolipids may account for PEC attachment. The HSPG-dependent aspect of PEC-cell association further supported the role of macropinocytosis.
Because HSPG, in the form of syndecans and glypicans, are involved in adhesion, migration, and cytoskeletal organization[45], it is likely that the inhibition of HSPG binding is coupled to actin microfilament-mediated uptake. These binding and uptake observations were consistent with a process that involves the association of cell surface HSPG with the actin cytoskeleton. HSPG cytoplasmic tails and actin filaments interact within the cytoplasm through linker proteins. Binding to adhesion receptors can provide the initial step for particle engulfment via membrane ruffling driven by networks of cortical actin fibers[38].
3.7. Non-Specific Binding Verified by Scatchard Plots
Using FITC-labeled PEC, we developed a FACS-based Scatchard equilibrium experiment for the detection and characterization of binding. The FACS acquisition was performed on a FACSAria system fitted with a photomultiplier tube on the forward scattering laser for more sensitive detection of cell bound PEC. The conventional binding curves are shown in Figure 7A–B. Titrations were prepared by exposing HMVEC under non-toxic conditions (1.54×109 PEC/ml-9.80×106 PEC/ml) for 3 h at 37°C and 4°C. Upper concentration limits were defined by particle aggregation. Thus, the calculated ratio of PEC concentration to cell density ranged from 32000:1 to 200:1, while the proportion of PEC bound varied from 30000:1 to 5:1. The incubation time was derived from the time course of binding (Figure 5A–B). The approach allowed the Scatchard transformation (bound/free versus bound), and it also provided a demonstration of non-specific, adsorptive binding at 37°C and 4°C (Figures 7C–D). These data are the first such FACS-based Scatchard representations of cell interactions for a polymeric drug delivery system.
Figure 7.
Binding isotherms for PEC on HMVEC at 37°C and 4°C. 3h incubations were performed at 37°C, (A) and (C), or at 4°C, (B) and (D). Binding was estimated by FACS (n=3). (A) and (B) show dose-dependent binding curves; (C) and (D) represented Scatchard plot analysis. The ratios of bound and free PEC for each concentration were determined based on the median fluorescence index of each dose in the absence of cells, follwed by correlation to calibration curves.
Both binding isotherms, at 37°C and 4°C (Figure 7A–B), formed the lower half of the standard S-curve saturation behavior seen in steady-state experiments at these temperatures. However, the curves never approached saturation. Similar behavior has been observed in human serum albumin binding to drugs such as paclitaxel[48, 49]. The Scatchard transformations (Figure 7C–D) showed an inverse relationship for the apparent binding constant due to extensive cooperativity. When specific, high-affinity interactions are present, the initial positive slope reaches a maximum, followed by a monotonic decrease as the total ligand increases in concentration[50, 51]. The binding isotherms lacked an inflection point without saturation. Within the practical concentration limits, PEC binding was largely determined by charge neutralization between the anionic cell surface and cationic microdomains of the PEC.
The binding curves had positive slopes, independent of temperature, under conditions where PEC bind to a heterogeneous group of receptors with cooperativity. The amorphous nature of PEC likely led to conformational flexibility and adaptability, further facilitating cell association. Thus, the data did not lend themselves to the analysis by Scatchard plots in terms of calculation of binding parameters[52]. The positive cooperativity would suggest that the binding of one PEC to any receptor on the cell surface created an energetically favorable site for another PEC.
Mathematically, the total concentration of bound PEC (Bound) to a single class of receptors can be defined by the Michaelis-Menten equation below:
where LFree, Rtotal, and Kd represent the concentration of free PEC (ligand), total receptor concentration, and equilibrium binding constant, respectively. In the case of the unmodified PEC, the free ligand was at concentrations much lower than the Kd, leading to a linear relationship between bound and free PEC with a slope of Rtotal/Kd. Breakdown in mass action laws may also be a contributing factor to PEC binding: receptor heterogeneity, cooperativity, and irreversible binding.
4. Conclusion
This study describes PEC interactions with endothelial cells in vitro through the use of applications ideally suited for nanoscale systems: PCS and FACS. The incorporation of a fluorescent polymer constituent allowed the use of flow cytometry to evaluate cellular interactions, while trypan blue permitted the delineation between surface associated and internalized PEC. This facilitated the exploration of (non-specific) PEC cellular compartmentalization without cell lysis, a method commonly used in other studies[53–56]. Binding and internalization studies showed that the associative phenomena were sensitive to proteolysis, exogenous heparin, metabolic energy, cytoskeletal integrity, and, at least in part, interactions with HSPG. No data exist to date on the specific mechanisms of PEC uptake, although liposomes have been found to be endocytosed[41, 57]. Scatchard plots are largely ignored for polymeric drug delivery systems, but characterizations of the type of binding occurring in biological systems is critical for the development of target-specific nanovehicles. Due to its favorable physical attributes and rapid extra- and intracellular accumulation, the system presented in this study provides a platform that can be readily modified for a targeted delivery approach. The presentation of outer-shell amine groups allows the conjugation of ligands for targeted accumulation in pathologic tissue, while inner core carboxyl chemical linkages are available for the incorporation of therapeutic payloads. Meeting requirements such as low cytoxicity and significant uptake into a target cell line, this system provides a promising candidate for further development and study.
Acknowledgments
We acknowledge support of National Institutes of Health Grants 1R01EB002825 (J.M.D. and A.P) and 1RO1AI50213-01 (W.N.K), support from the Department of Veterans Affairs (J.M.D.), and American Cancer Society RSG 102299 (W.N.K.). In addition we would like to thank the Vanderbilt Institute for Nanoscale Science and Engineering (VINSE) for use of the Malvern ZetaSizer Nano ZS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kramer M, Stumbe JF, Grimm G, Kaufmann B, Kruger U, Weber M, et al. Dendritic polyamines: Simple access to new materials with defined treelike structures for application in nonviral gene delivery. Chembiochem. 2004;5:1081–87. doi: 10.1002/cbic.200300905. [DOI] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Drug delivery systems: Entering the mainstream. Science. 2004;303:1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 3.Haag R, Vogtle F. Highly branched macromolecules at the interface of chemistry, biology, physics, and medicine. Angew Chem-Int Edit. 2004;43:272–73. doi: 10.1002/anie.200320082. [DOI] [PubMed] [Google Scholar]
- 4.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: Theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 5.Verderone G, van Craynest N, Boussif O, Santaella C, Bischoff R, Kolbe HVJ, et al. Lipopolycationic telomers for gene transfer: Synthesis and evaluation of their in vitro transfection efficiency. J Med Chem. 2000;43:1367–79. doi: 10.1021/jm9911579. [DOI] [PubMed] [Google Scholar]
- 6.Chithrani BD, Ghazani AA, Chan WCW. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–68. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 7.Vauthier-Holtzscherer C, Benabbou S, Spenlehauer G, Veillard M, Couvreur P. Methodology for the preparation of of ultra-dispersed polymer systems. STP Pharma Sci. 1991;2:109–16. [Google Scholar]
- 8.Panyam P, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55:329–47. doi: 10.1016/s0169-409x(02)00228-4. [DOI] [PubMed] [Google Scholar]
- 9.Cruz T, Gaspar R, Donato A, Lopes C. Interaction between polyalkylcyanoacrylate nanoparticles and peritoneal macrophages: MTT metabolism, NBT reduction, and NO production. Pharm Res. 1997;14:73–79. doi: 10.1023/a:1012059501947. [DOI] [PubMed] [Google Scholar]
- 10.Matsusue Y, Hanafusa S, Yamamuro T, Shikinami Y, Ikada Y. Tissue reaction of bioabsorbable ultra-high strength poly(L-lactide) rod - a long-term study in rabbits. Clin Orthop Rel Res. 1995;317:246–53. [PubMed] [Google Scholar]
- 11.Schatz C, Lucas JM, Viton C, Domard A, Pichot C, Delair T. Formation and properties of positively charged colloids based on polyelectrolyte complexes of biopolymers. Langmuir. 2004;20:7766–78. doi: 10.1021/la049460m. [DOI] [PubMed] [Google Scholar]
- 12.Carlesso G, Kozlov E, Prokop A, Unutmaz D, Davidson JM. Nanoparticulate system for efficient gene transfer into refractory cell targets. Biomacromolecules. 2005;6:1185–92. doi: 10.1021/bm0492531. [DOI] [PubMed] [Google Scholar]
- 13.Fisher KD, Ulbrich K, Subr V, Ward CM, Mautner V, Blakey D, et al. A versatile system for receptor-mediated gene delivery permits increased entry of DNA into target cells, enhanced delivery to the nucleus and elevated rates of transgene expression. Gene Ther. 2000;7:1337–43. doi: 10.1038/sj.gt.3301230. [DOI] [PubMed] [Google Scholar]
- 14.Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, et al. Tumor regression by targeted gene delivery to the neovasculature. Science. 2002;296:2404–07. doi: 10.1126/science.1070200. [DOI] [PubMed] [Google Scholar]
- 15.Ito T, Sun L, Bevan MA, Crooks RM. Comparison of nanoparticle size and electrophoretic mobility measurements using a carbon-nanotube-based coulter counter, dynamic light scattering, transmission electron microscopy, and phase analysis light scattering. Langmuir. 2004;20:6940–45. doi: 10.1021/la049524t. [DOI] [PubMed] [Google Scholar]
- 16.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, et al. HMEC-1 -establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–90. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 17.Jevprasesphant R, Penny J, Attwood D, D’Emanuele A. Transport of dendrimer nanocarriers through epithelial cells via the transcellular route. J Control Release. 2004;97:259–67. doi: 10.1016/j.jconrel.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin-and caveolae-mediated endocytosis. Biochem J. 2004;377:159–69. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasola A, Geuna M. A flow cytometry assay simultaneously detects independent apoptotic parameters. Cytometry. 2001;45:151–57. doi: 10.1002/1097-0320(20011001)45:2<151::aid-cyto1157>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 20.Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci USA. 1996;93:12349–54. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiethoff CM, Smith JG, Koe GS, Middaugh CR. The potential role of proteoglycans in cationic lipid-mediated gene delivery - Studies of the interaction of cationic lipid-DNA complexes with model glycosaminoglycans. J Biol Chem. 2001;276:32806–13. doi: 10.1074/jbc.M007940200. [DOI] [PubMed] [Google Scholar]
- 22.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly(D,L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20:212–20. doi: 10.1023/a:1022219003551. [DOI] [PubMed] [Google Scholar]
- 23.Kessner S, Krause A, Rothe U, Bendas G. Investigation of the cellular uptake of E-Selectin-targeted immunoliposomes by activated human endothelial cells. Biochim Biophys Acta-Biomembr. 2001;1514:177–90. doi: 10.1016/s0005-2736(01)00368-6. [DOI] [PubMed] [Google Scholar]
- 24.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–22. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. 2002;277:2437–43. doi: 10.1074/jbc.M110017200. [DOI] [PubMed] [Google Scholar]
- 26.Belting M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem Sci. 2003;28:145–51. doi: 10.1016/S0968-0004(03)00031-8. [DOI] [PubMed] [Google Scholar]
- 27.Belting M, Persson S, Fransson LA. Proteoglycan involvement in polyamine uptake. Biochem J. 1999;338:317–23. [PMC free article] [PubMed] [Google Scholar]
- 28.Scatchard G. The attractions of proteins for small molecules and ions. Ann NY Acad Sci. 1949;51:660–72. [Google Scholar]
- 29.Chern CS, Lee CK, Chang CJ. Electrostatic interactions between amphoteric latex particles and proteins. Colloid Polym Sci. 2004;283:257–64. [Google Scholar]
- 30.Sugrue S. Predicting and controlling colloid suspension stability using electrophoretic mobility and particle size measurements. Am Lab. 1992;24:64–71. [Google Scholar]
- 31.Du JZ, Tang YP, Lewis AL, Armes SP. pH-sensitive vesicles based on a biocompatible zwitterionic diblock copolymer. J Am Chem Soc. 2005;127:17982–83. doi: 10.1021/ja056514l. [DOI] [PubMed] [Google Scholar]
- 32.Taylor S, Qu LW, Kitaygorodskiy A, Teske J, Latour RA, Sun YP. Synthesis and characterization of peptide-functionalized polymeric nanoparticles. Biomacromolecules. 2004;5:245–48. doi: 10.1021/bm034326m. [DOI] [PubMed] [Google Scholar]
- 33.Wood KC, Little SR, Langer R, Hammond PT. A family of hierarchically self-assembling linear-dendritic hybrid polymers for highly efficient targeted gene delivery. Angew Chem-Int Edit. 2005;44:6704–08. doi: 10.1002/anie.200502152. [DOI] [PubMed] [Google Scholar]
- 34.Ogawa K, Sato S, Kokufuta E. Formation of intra- and interparticle polyelectrolyte complexes between cationic nanogel and strong polyanion. Langmuir. 2005;21:4830–36. doi: 10.1021/la047071m. [DOI] [PubMed] [Google Scholar]
- 35.Tong WJ, Gao CY, Mohwald H. Stable weak polyelectrolyte microcapsules with pH-responsive permeability. Macromolecules. 2006;39:335–40. [Google Scholar]
- 36.Csaba N, Caamano P, Sanchez A, Dominguez F, Alonso MJ. PLGA: poloxamer and PLGA: poloxamine blend nanoparticies: New carriers for gene delivery. Biomacromolecules. 2005;6:271–78. doi: 10.1021/bm049577p. [DOI] [PubMed] [Google Scholar]
- 37.Shenoy D, Little S, Langer R, Amiji M. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive system for tumor-targeted delivery of hydrophobic drugs: Part 2. In vivo distribution and tumor localization studies. Pharm Res. 2005;22:2107–14. doi: 10.1007/s11095-005-8343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopatz I, Remy JS, Behr JP. A model for non-viral gene delivery: through syndecan adhesion molecules and powered by actin. J Gene Med. 2004;6:769–76. doi: 10.1002/jgm.558. [DOI] [PubMed] [Google Scholar]
- 39.Rothbard JB, Jessop TC, Wender PA. Adaptive translocation: the role of hydrogen bonding and membrane potential in the uptake of guanidinium-rich transporters into cells. Adv Drug Deliv Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Zaro JL, Shen WC. Quantitative comparison of membrane transduction and endocytosis of oligopeptides. Biochem Biophys Res Commun. 2003;307:241–47. doi: 10.1016/s0006-291x(03)01167-7. [DOI] [PubMed] [Google Scholar]
- 41.Huth US, Schubert R, Peschka-Suss R. Investigating the uptake and intracellular fate of pH-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110:490–504. doi: 10.1016/j.jconrel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 42.Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–28. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 43.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 44.Rupper A, Cardelli J. Regulation of phagocytosis and endo-phagosomal trafficking pathways in Dictyostelium discoideum. Biochim Biophys Acta-Gen Subj. 2001;1525:205–16. doi: 10.1016/s0304-4165(01)00106-4. [DOI] [PubMed] [Google Scholar]
- 45.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–77. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- 46.Gill PJ, Silbert CK, Silbert JE. Effects Of Heparan-Sulfate Removal On Attachment And Reattachment Of Fibroblasts And Endothelial-Cells. Biochemistry. 1986;25:405–10. doi: 10.1021/bi00350a020. [DOI] [PubMed] [Google Scholar]
- 47.Ruponen M, Ronkko S, Honkakoski P, Pelkonen J, Tammi M, Urtti A. Extracellular glycosaminoglycans modify cellular trafficking of lipoplexes and polyplexes. J Biol Chem. 2001;276:33875–80. doi: 10.1074/jbc.M011553200. [DOI] [PubMed] [Google Scholar]
- 48.Brodersen R, Honore B, Larsen FG. Serum albumin - A non-saturable carrier. Acta Pharmacol et Toxicol. 1984;54:129–33. doi: 10.1111/j.1600-0773.1984.tb01906.x. [DOI] [PubMed] [Google Scholar]
- 49.Paal K, Muller J, Hegedus L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268:2187–91. doi: 10.1046/j.1432-1327.2001.02107.x. [DOI] [PubMed] [Google Scholar]
- 50.Clegg LS, Lindup WE. Scatchard plots with a positive slope - dependence upon ligand concentration. J Pharm Pharmacol. 1984;36:776–78. doi: 10.1111/j.2042-7158.1984.tb04873.x. [DOI] [PubMed] [Google Scholar]
- 51.Thaler CD, Cardullo RA. The initial molecular interaction between mouse sperm and the zona pellucida is a complex binding event. J Biol Chem. 1996;271:23289–97. doi: 10.1074/jbc.271.38.23289. [DOI] [PubMed] [Google Scholar]
- 52.Judis J. Binding of codeine, morphine, and methadone to human serum proteins. J Pharm Sci. 1977;66:802–06. doi: 10.1002/jps.2600660615. [DOI] [PubMed] [Google Scholar]
- 53.Behrens I, Pena AIV, Alonso MJ, Kissel T. Comparative uptake studies of bioadhesive and non-bioadhesive nanoparticles in human intestinal cell lines and rats: The effect of mucus on particle adsorption and transport. Pharm Res. 2002;19:1185–93. doi: 10.1023/a:1019854327540. [DOI] [PubMed] [Google Scholar]
- 54.Panyam J, Dali MA, Sahoo SK, Ma WX, Chakravarthi SS, Amidon GL, et al. Polymer degradation and in vitro release of a model protein from poly(D,L-lactide-co-glycolide) nano-and microparticles. J Control Release. 2003;92:173–87. doi: 10.1016/s0168-3659(03)00328-6. [DOI] [PubMed] [Google Scholar]
- 55.Panyam J, Sahoo SK, Prabha S, Bargar T, Labhasetwar V. Fluorescence and electron microscopy probes for cellular and tissue uptake of poly(D,L-lactide-co-glycolide) nanoparticles. Int J Pharm. 2003;262:1–11. doi: 10.1016/s0378-5173(03)00295-3. [DOI] [PubMed] [Google Scholar]
- 56.Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26:2713–22. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 57.Simoes S, Slepushkin V, Pires P, Gaspar R, de Lima MCP, Duzgunes N. Human serum albumin enhances DNA transfection by lipoplexes and confers resistance to inhibition by serum. Biochim Biophys Acta-Biomembr. 2000;1463:459–69. doi: 10.1016/s0005-2736(99)00238-2. [DOI] [PubMed] [Google Scholar]