

Abstract
Among the multiple cellular effects mediated by lysophosphatidic acid (LPA), the effect on cell proliferation has extensively been investigated. A recent study showed that LPA-mediated proliferation of colon cancer cells requires activation of β-catenin. However, the majority of colon cancer cells have deregulation of the Wnt/β-catenin pathway. This prompted us to hypothesize the presence of additional pathway(s) activated by LPA resulting in an increase in the proliferation of colon cancer cells. Krüppel-like factor 5 (KLF5) is a transcriptional factor highly expressed in the crypt compartment of the intestinal epithelium. In this work, we investigated a role of KLF5 in LPA-mediated proliferation. We show that LPA stimulated the expression levels of KLF5 mRNA and protein in colon cancer cells and this stimulation was mediated by LPA2 and LPA3. Silencing of KLF5 expression by small interfering RNA significantly attenuated LPA-mediated proliferation of SW480 and HCT116 cells. LPA-mediated KLF5 induction was partially blocked by inhibition of the mitogen-activated protein kinase kinase and protein kinase C-δ. Moreover, we observed that LPA regulates KLF5 expression via eukaryotic elongation factor 2 kinase (eEF2k). Inhibition of calmodulin or silencing of eEF2k blocked the stimulation in KLF5 expression. Knockdown of eEF2k specifically inhibited KLF5 induction by LPA but not by fetal bovine serum or phorbol 12-myristate 13-acetate. These results identify KLF5 as a target of LPA-mediated signaling and suggest a role of KLF5 in promoting proliferation of intestinal epithelia in response to LPA.
Lysophosphatidic acid (LPA)4 is a biologically active lysophospholipid that mediates a plethora of cellular effects, including cell survival, proliferation, migration, and induction of cytokines and growth factors. Signaling by LPA is mediated through LPA1, LPA2, and LPA3, which are members of a family of G protein-coupled receptors (1-3). LPA stimulates proliferation in many types of cells. Since the initial report by van Corven et al. (4), a growing body of work describes the effect of LPA on cell proliferation. LPA stimulates proliferation of many cell types, including fibroblasts, breast cancer cells, mesangial cells, vascular smooth muscle cells, neuronal cells and others (5-10). However, in some cases, LPA has a negative effect on cell growth. For example, LPA inhibits the proliferation of myeloma (5). In mesangial cells, LPA shows a positive effect to cell growth at micromolar concentrations but inhibits cell proliferation at a higher concentration (7). The mechanisms underlying the proliferative effect by LPA are diverse, and in many cases the mitogenic effects of LPA are mediated through multiple pathways that work independently. Activation of the extracellular signal-regulated kinases 1 and 2 (ERK1/2), protein kinase C (PKC), the phosphoinositide 3 kinase-Akt pathway, hydrolysis of phosphoinositide, Ca2+ mobilization, and activation of RhoA mediated by both pertussis toxin (PTX)-sensitive and -insensitive pathways have all been involved (5, 11-14).
The adenomatous polyposis coli (APC) gene is an important tumor suppressor for colorectal cancer and is mutated in 80% of colorectal cancer (15, 16). The Wnt/β-catenin pathway is activated upon binding of Wnt to a member of the Frizzled family of seven transmembrane receptors. In the absence of Wnt, the cytoplasmic level of β-catenin is kept low via a multiprotein complex consisting of APC, axin, and the glycogen synthase kinase 3β (GSK-3β). GSK-3β phosphorylates β-catenin, directing it to degradation by proteosome (17). When activated, β-catenin accumulates in the nucleus where it binds to the transcription factors, T-cell factor, and lymphoid enhancer-binding protein, leading to transcriptional activation of multiple target genes, such as c-myc and cyclin D (17, 18). Recently, work by Yang et al. (12) showed that LPA can activate the β-catenin pathway leading to nuclear translocation of β-catenin. This work defines another entry point to the Wnt/β-catenin pathway and adds a new dimension to the biological effects by LPA. However, the majority of colon cancer cells have mutations that result in inactivation of APC or activation of β-catenin and, hence, it is expected that β-catenin is constitutively activated in these cells. Therefore, it remains to be determined whether LPA can further activate the β-catenin pathway in these cells.
Krüppel is a zinc finger-containing transcription factor that is responsible for segmentation of the Drosophila melanogaster embryo (19-21). In mammals, there are at least 16 Krüppel-like factors (KLFs) that exhibit homology to Kruppel from Drosophila (22). In the intestine, two KLFs, KLF4 and KLF5, are highly expressed (23). KLF4 expression is enriched in differentiated enterocytes found in the upper villus region, whereas KLF5 is found mainly in the proliferating crypt cell population where it positively regulates cell proliferation (24-26). Despite the effect of LPA on proliferation of many types of cells, the mechanism of LPA-mediated proliferation is not fully elucidated and led us to hypothesize that KLF5 might be required for LPA-mediated signaling.
In this work, we report that LPA stimulates the expression level of KLF5 in colon cancer cells. LPA-mediated induction of KLF5 is observed in intestinal cells regardless of the mutational status of APC and KLF5 plays a crucial role in LPA-induced proliferation of colon cancer cells. Furthermore, we found that LPA induces KLF5 via pathways dependent on MEK1/2, PKC, and eukaryotic elongation factor 2 kinase (eEF2k).
EXPERIMENTAL PROCEDURES
Materials
1-Oleoyl LPA (18:1 LPA) and sphingosine 1-phosphate (S1P) were obtained from Avanti Polar Lipids. All antibodies were from Cell Signaling. LY290042, U0126, GF109203X, rotterlin, trifluoperazine, and rapamycin were from Calbiochem. All kinase inhibitors were added to the culture medium 10 min before the addition of LPA. PTX was added 14 h before LPA treatment. The concentrations used are as follow: GF109203X at 5 μM, rottlerin at 10 μM, LY294002 at 50 μM, SB203580 at 5 μM, U0126 at 10 μM, rapamycin at 100 nM, U73122 at 5 μM, AG1478 at 250 nM, PTX at 50 ng/ml, trifluoperazine at 3 μM, and calmidazolium at 6 μM. Stock solutions were prepared in dimethyl sulfoxide at a 1,000×concentration with respect to the working concentrations indicated above and dimethyl sulfoxide alone was added to control cells. All other chemicals were obtained from Sigma.
Cell Culture
SW480 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in 95% air, 5% CO2 atmosphere as previously described (27). For Caco-2 cells, RPMI 1640 was replaced with Dulbecco’s modified Eagle’s medium supplemented with 0.5% non-essential amino acids. HCT116 cells were maintained in McCoy’s 5A medium supplemented with 10% FBS, 100 μg/ml streptomycin, and 100 units/ml penicillin at 37 °C in 95% air, 5% CO2. IEC6 cells were obtained from the Emory Digestive Disease Research Development Center and cultured at 37 °C in 95% air, 5% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 5% FBS, 0.1 unit/ml insulin, 50 μg/ml streptomycin, and 50 units/ml penicillin. All the cells were serum starved 24 h before LPA treatment in their appropriate media without FBS.
Northern Blot Analysis
Total RNA from intestinal cells was isolated using TRIzol (Invitrogen). Thirty μg of total RNA for each sample was hybridized with [α-32P]dATP-labeled full-length mouse KLF5 cDNA probe (28) using ExpressHyb hybridization solution (BD Biosciences) at 68 °C with continuous shaking for 1 h. Washing was performed at 50 °C with 3 changes of washing solution. Phosphoprotein 36B4 (29) was used as a loading control to normalize the expression levels of KLF5 mRNA.
siRNA Transfection
Double-stranded siRNA oligonucleotides targeting LPA2, LPA3, PKCα, and eEF2k were from Dharmacon. siRNA targeting KLF5 or PKCδ was purchased from Invitrogen and Upstate, respectively. As a control, a scrambled 21-nucleotide RNA duplex was used. Cells seeded at 60% confluence on 60-mm culture plates were transfected with 40 nM siRNA using Lipofectamine 2000 (Invitrogen). Twenty-four h after transfection, cells were serum deprived for 16–24 h and then treated with LPA or carrier. The efficacy of gene silencing of LPA2 and LPA3 was determined by reverse transcriptase-PCR using a primer set specific for LPA2 and LPA3, respectively (30). Expression of KLF5, PKC, and eEF2k was determined by Western blot.
Luciferase Assay
The promoter of KLF5 was previously described (31). HCT116 cells were transfected with pGL3 harboring KLF5 promoter (32). A Renilla luciferase control vector was co-transfected to normalize the transfection efficiency. Cell lysis and reporter assay were performed 2 days after the transfection with the Dual Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Fluorescence-activated Cell Sorter Analysis
Cell cycle analysis was performed as previously described (33). Cells were rinsed twice in phosphate-buffered saline (PBS), treated with trypsin, and resuspended in their corresponding medium containing 10% FBS. Cells were then collected by centrifugation, washed with PBS, collected again by centrifugation, resuspended in 70% ethanol, and fixed at −20 °C overnight. Cells were pelleted once again by centrifugation and resuspended in a solution containing 50 μg/ml propidium iodide, 50 μg/ml RNase A, 0.1% Triton X-100, and 0.1 mM EDTA at room temperature for 30 min. Flow cytometry was performed on a FACSCalibur cytometer (BD Biosciences).
Cell Proliferation
Cells were seeded at 20,000 cells/well in a 24-well plate. Cells were serum starved 24 h and maintained in Dulbecco’s modified Eagle’s medium, 0.1% FBS supplemented with either 0.1% bovine serum albumin/PBS or 0.1–10 μM LPA in 0.1% bovine serum albumin/PBS for up 3 days. On the day of cell counting, cells were trypsinized and the number of cells was counted by using a hemacytometer.
Nuclear Extraction
SW480 and HCT cells were serum starved for 24 h followed by exposure to LPA. Nuclear proteins were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Pierce).
Western Immunoblot
Cells were rinsed three times with ice-cold PBS buffer, and lysed in lysis buffer composed of 10 mM Tris-Cl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1 mM phenylmethylsulfonyl fluoride, 10% glycerol, 2 mM sodium orthovanadate, 10 mM sodium fluoride, 20 mM sodium pyro-phosphate, 25 mM β-glycerophosphate, 1% Triton X-100, and protease inhibitors. The lysates were cleared by centrifugation at 14,000 × g at 4 °C for 10 min. Protein concentration was determined by the bicinchoninic acid assay (Sigma). The equal amount of lysate in 2× Laemmli sample buffer was resolved by 10% SDS-PAGE, and Western immunoblot analysis was performed as previously described (30). The amount of KLF5 protein was determined using polyclonal anti-KLF5 antibody generated against amino acids 106–122 of the mouse KLF5 (24). The expression level of KLF5 was normalized against the expression levels of β-actin. Densitometric analyses were performed using ImageJ (Scion Corp). Statistical significance was assessed by one-way analysis of variance using Origin software (OriginLab). Data are presented as the mean ± S.E.
RESULTS
LPA Induces KLF5 Expression
Previous studies have shown that LPA promotes proliferation of colon cancer cells, such as DLD1 and HCT116 (12, 34). Similarly, we found that LPA stimulated the rate of proliferation of human colon cancer Caco-2 and SW480 cells in a concentration-dependent manner (Fig. 1).
Figure. 1. LPA stimulates proliferation of colon cancer cells.
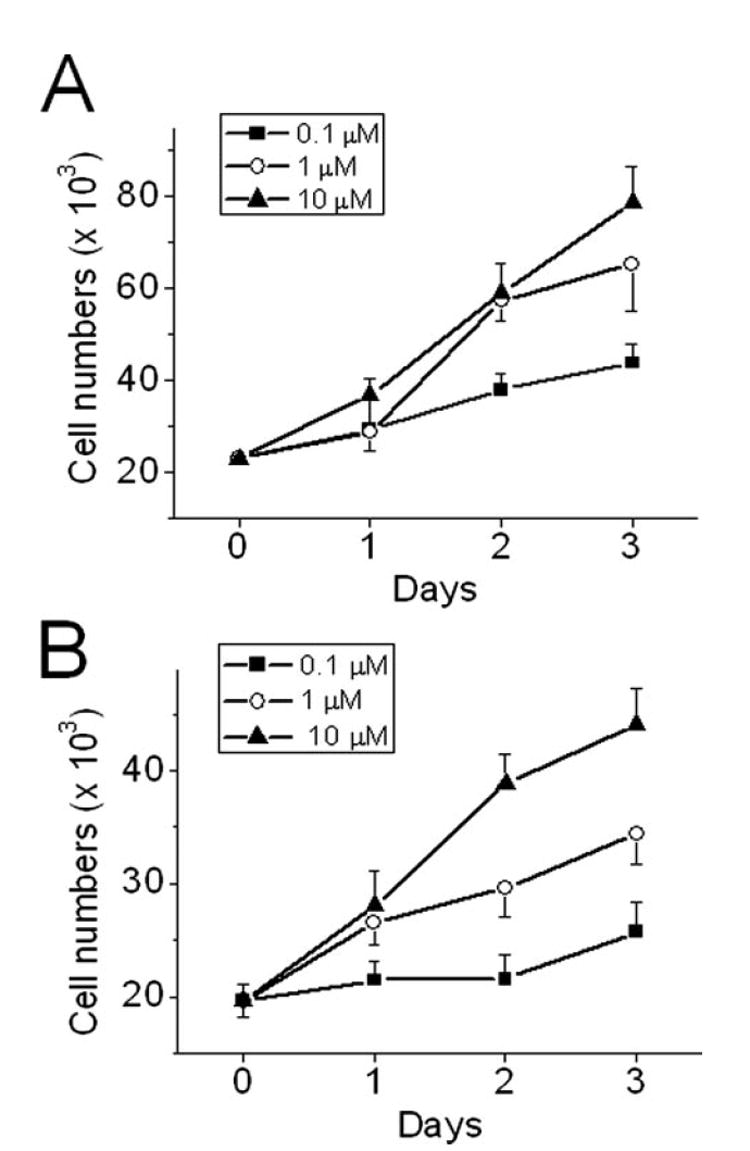
Caco-2 (A) and SW480 (B) cells cultured on 24-well plates in Dulbecco’s modified Eagle’s medium containing 0.1% FBS were treated with vehicle or various concentrations of LPA for up to 3 days. On each day, cells were trypsinized and the number of cells were counted by using a hemacytometer. Results presented are mean ± S.E. n = 6.
It has recently been reported that LPA-mediated signaling activates β-catenin in HCT116 and LS174T cells, which express wild type APC gene, and a decrease in β-catenin expression attenuates LPA-mediated proliferation of HCT116 cells (12). The APC gene is the most commonly mutated gene in colorectal cancer with the mutations resulting in deregulation of β-catenin activity (35). To determine whether LPA can also activate β-catenin in colon cancer cells with the mutated APC gene, we examined nuclear translocation of β-catenin in SW480 cells. As shown in Fig. 2, LPA treatment of serum-starved SW480 cells did not induce the nuclear translocation of β-catenin. In contrast, LPA treatment resulted in an increase in β-catenin expression in nuclear fractions in HCT116 cells, which have the wild type APC gene, consistent with a previous report (12). These data suggest that an alternative pathway(s) is responsible for the proliferation of SW480 and others, in which the APC gene is mutated.
Figure. 2. LPA activates β-catenin in HCT116 cells but not in SW480 cells.

SW480 and HCT116 cells were serum starved for 24 h followed by treatment with vehicle or 1 μM LPA for the indicated times. Nuclear translocation of β-catenin was determined by Western blot on nuclear extracts. Histone H1 was used as a control. Representatives of two to four independent experiments are shown.
KLF5 is a transcription factor highly expressed in epithelial cells in the proliferating compartment of the gastrointestinal tract (24), but whether KLF5 plays a role in LPA-mediated proliferation has not been studied. To test this, we first determined whether LPA induces KLF5 gene expression. Fig. 3A shows that LPA treatment led to an increase in the KLF5 mRNA level in Caco-2 and SW480 cells within 1–2 h of treatment, which was sustained for at least 24 h. The induction of KLF5 mRNA by LPA was not limited to cancer cells but also observed in the nontransformed rat intestinal cell line IEC-6 as well. We next determined the levels of the KLF5 protein. In SW480, Caco-2, and IEC6 cells LPA treatment resulted in a sustained increase in KLF5 protein level in these cells following 2–4 h treatment (Fig. 3B). It is noteworthy that the increase at 2 h was not always observed but the increase much more consistently occurred after 4 h of treatment. Interestingly, LPA also induced the expression level of KLF5 protein in HCT116 cells, in which LPA induced nuclear translocation of β-catenin (Fig. 2). The induction of KLF5 was maximal at 10 μM LPA, whereas submaximal activation was observed at 0.1–1.0 μM LPA (data not shown).
Figure. 3. LPA induces the expression levels of KLF5.
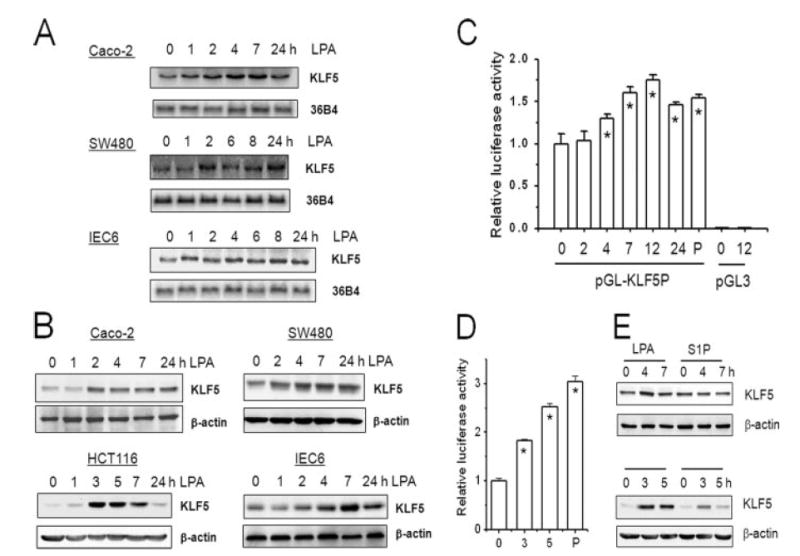
A, serum-starved Caco-2, SW480, and IEC6 cells were treated with 10 μM LPA for 0–24 h. Total RNA was isolated and the expression of KLF5 mRNA was detected by Northern blot hybridization using mouse KLF5 cDNA as probe. The amount of KLF5 mRNA was normalized to the expression levels of phosphoprotein 36B4 as a loading control (n = 3). B, serum-starved Caco-2, SW480, HCT116, and IEC6 cells were treated with 10 μM LPA for 0–24 h. 30 μg of whole cell lysates were resolved by SDS-PAGE and the amount of KLF5 protein was determined by Western blot using polyclonal anti-KLF5 antibody. Membranes were stripped and probed with an antibody against β-actin as a loading control. Representative Western blots from four separate experiments are shown. C, SW480cells were transiently transfected with pGL3 harboring KLF5 promoter, pGL-KLF5P, or pGL3. A Renilla luciferase control vector was co-transfected to normalize the transfection efficiency. 24 h post-transfection, cells were serum deprived for 24 h followed by treatment with LPA for the indicated times. As a control, PMA (P) was used as a positive agonist of the KLF5 promoter. Cell lysis and reporter assay were performed with Dual Luciferase Reporter Assay System. Three separate independent experiments were performed with at least 3 samples per condition. Results presented are mean ± S.E. *, p < 0.01 compared with untreated control. D, KLF5 promoter activity in HCT cells was determined as described above. *, p < 0.001. E, SW480 (upper panel) and HCT116 (lower panel) cells were treated with 10μM LPA or 1μM S1P. The amount of KLF5 protein was determined by Western blot as described earlier. Representatives of three independent experiments are shown.
To further substantiate the increase in KLF5 expression by LPA, a luciferase reporter driven by a KLF5 promoter was transiently transfected in SW480 or HCT116 cells for the reporter gene assay (31). Exposure of SW480 cells to LPA for 2 h had a minimal effect on promoter activity (Fig. 3C), as we observed that a similar treatment resulted in an inconsistent increase in KLF5 protein level (Fig. 3B). In contrast, there was a significant increase at 4 h, which further increased after 7 h of treatment. Similarly, LPA treatment of HCT116 stimulated the luciferase activity by 85% at 3 h and 152% at 5 h (Fig. 3D). As a control, 4 h incubation with 100 nM phorbol 12-myristate 13-acetate (PMA), a known agonist of KLF5 expression (26), resulted in an increase in the KLF5 promoter activity in both cell lines (Fig. 3D-E).
A previous report showed that another lysophospholipid S1P induces KLF5 expression (36). Hence, we next compared the induction of KLF5 expression by LPA and S1P. As shown in Fig. 3E, S1P did not show a significant effect on the expression level of KLF5 in SW480 cells (upper panel). On the other hand, we observed an increase in KLF5 expression in HCT116 cells (lower panel) treated with S1P, but the effect of S1P was relatively smaller and was short lived compared with LPA.
Induction of KLF5 Is Important for LPA-mediated Proliferation
Previous findings that KLF5 positively regulates cell proliferation led us to hypothesize that KLF5 may be a mediator of LPA-mediated cell proliferation (26, 37-39). Because many colon cancer cells such as Caco-2 and SW480 have high basal levels of KLF5 expression, we examined the role of KLF5 via silencing of the KLF5 gene using siRNA. Fig. 4A shows that KLF5 siRNA decreased KLF5 expression by ~90% compared with control siRNA-transfected SW480 cells (Fig. 4A). Despite the knockdown, LPA treatment led to an increase in KLF5 expression in siKLF5-treated cells. Silencing of the KLF5 gene resulted in a significant decrease in LPA-induced proliferation of SW480 cells (Fig. 4B). Similarly, knockdown of KLF5 decreased the rate of LPA-mediated proliferation of HCT116 cells (Fig. 4C). However, LPA treatment resulted in a significant increase in cell numbers in siKLF5-treated cells compared with untreated cells, which may be attributed to the small induction of KLF5 observed in these cells (Fig. 4A).
Figure. 4. Knockdown of KLF5 by siRNA attenuates LPA-mediated cell proliferation.

A, SW480 cells were transfected with siRNA specific for KLF5 (siKLF5) or control siRNA (siCont). The levels of KLF5 proteins were determined by Western blot. B and C, the effects of KLF5 knockdown on the rate of proliferation of SW480 (B) and HCT116 (C) cells were determined. Cells transfected with control or KLF5 siRNA were treated with 10 μM LPA for 3 days. Numbers of cells were counted by using a hemacytometer. Three independent experiments were performed with triplicate samples. Results presented are mean ± S.E. D, serum-starved SW480 cells transfected with control of KLF5 siRNA were treated with 10 μM LPA for 72 h. Cells were trypsinized, collected, and subjected to flow cytometric analysis. Experiments were repeated twice with quadruple samples. *, p < 0.0001; **, p < 0.05 compared with control.
To further understand the role of KLF5, we performed cell-cycle analysis on cells treated with siKLF5 or control siRNA. Following LPA treatment for 72 h, the population of cells in S phase is almost 2-fold greater in control cells compared with siKLF5-treated cells (15.55 versus 8.29%). These data indicate that the induction of KLF5 is important for LPA-mediated proliferation of colon cancer cells and it stimulates cell proliferation by promoting G1/S transition.
LPA2 and LPA3 Receptor Mediate LPA-induced KLF5 Induction
We previously reported that colon cancer cells, such as SW480 and HCT116, overexpress LPA2 (30). At the same time, LPA1 expression was significantly decreased in these cells compared with normal colon epithelial cells (30). We next sought to determine which LPA receptors, LPA2 or LPA3, are responsible for the induction of KLF5. We omitted LPA1 based on the previous finding that the level of LPA1 in these cells is low (30). LPA2 and LPA3 expression in HCT116 cells was blocked using siRNA specific for LPA2 or LPA3. Silencing of either LPA2 or LPA3 markedly decreased activation of ERK 1 and 2 in these cells. Similarly, the induction of KLF5 by LPA was abrogated by knockdown of either LPA2 or LPA3 and when combined a greater inhibition was achieved (Fig. 5).
Figure. 5. LPA2 and LPA3 induce KLF5 expression.

HCT116 cells were treated with siRNA specific for LPA2, LPA3, or both. As a control, cells were transfected with control siRNA. Cells were then treated with LPA and phosphorylation of ERK1/2 (A) and the expression level of KLF5 (B) was determined by Western blot. Representatives of three separate experiments are shown.
Induction of KLF5 Is Dependent on PKCδ and MEK1/2
A myriad of signaling pathways regulates LPA-mediated cell proliferation. Earlier studies have indicated involvement of ERK1/2, the phosphoinositide 3-kinase-Akt cascade, and in some cases the p38 group of protein kinases. To determine the signal transduction pathway involved in the induction of KLF5, we used pharmaceutical inhibitors specific against these protein kinases. The specific Gαi/o inhibitor PTX, the phosphoinositide 3-kinase inhibitor LY290042, the p38 inhibitor SB203580, the phospholipase Cβ inhibitor U37122, and the epidermal growth factor (EGF) receptor inhibitor AG1478 did not block the induction of KLF5 in SW480 and HCT116 cells (Fig. 6A and B). On the other hand, the MEK1/2 inhibitor U0126 was able to partially block the effect of LPA on KLF5 protein expression in both SW480 and HCT116 cells (Fig. 6C and D).
Figure. 6. LPA-mediated induction of KLF5 is dependent onPKCand MEK1/2.

SW480 (A and C) and HCT116 (B and D) cells were pretreated with PTX for 14 h or other inhibitors for 10 min before treatment with vehicle or LPA. The concentration of the inhibitors are described under “Experimental Procedures.” The expression level of KLF5 was determined by Western blot. Membranes were stripped and probed with an antibody against β-actin as a loading control. Representative Western blots from three separate experiments are shown. E, HCT116 cells transiently co-transfected with pGL-KLF5P and Renilla luciferase control vector were serum-starved for 24 h and treated with LPA in the presence or absence of U0126 (U), GF109203X (GF), or both (U+GF). Results presented are mean ± S.E. Luciferase assays were repeated twice with triplicate samples. *, p < 0.005 compared with untreated cells. **, p < 0.02; ***, p < 0.08.
Previous studies have shown that LPA regulates PKC (12, 40). Moreover, LPA induces interleukin-8 secretion via a PKCδ-dependent mechanism (41, 42). We, therefore, examined whether PKC is involved in the activation of KLF5. SW480 cells were treated with LPA in the presence or absence of the broad-spectrum PKC inhibitor GF109203X or the PKCδ-specific inhibitor rottlerin. GF109203X showed a modest inhibitory effect in both SW480 and HCT116 cells (Fig. 6C and D). Interestingly, when the cells were pretreated with both U0126 and GF109203X, the inhibitory effect on KLF5 was substantially greater than with each inhibitor in both cell lines. The effects of the inhibitors were confirmed by the KLF5 promoter reporter assay, which shows a significant decrease in the luciferase activity in the presence of U0126, GF109203X, or both (Fig. 6E). This indicates that the effects of these inhibitors are additive. In addition to GF109203X, 10 μM rottlerin drastically blocked the induction of KLF5 expression by LPA in SW480 cells and HCT116 cells (Fig. 7A). The expression of KLF5 was partly recovered at 7 and 5 h in SW480 and HCT116, respectively, but we did not observe such a recovery when 20 μM rottlerin was used (data not shown). Consistently, the inhibitory effect of rottlerin was observed in Caco-2 cells as well.
Figure. 7. LPA-mediated induction of KLF5 is sensitive to rottlerin.

A, SW480, HCT116, and Caco-2 cells were treated with LPA or vehicle in the presence or absence of rottlerin. The expression level of KLF5 was determined by Western blot. Membranes were stripped and probed with an antibody against β-actin as a loading control. B, HCT116 cells were transfected with control, PKCα, or PKCδ siRNA. Transfected cells were treated with 1 μM LPA or vehicle and Western blotting was performed to determine KLF5 protein expression. Specific knockdown of PKCα or PKCδ protein by siRNA treatment is shown below. C, HCT116 cells were treated with LPA in the absence or presence of Gö6976 and KLF5 protein was determined. Representatives of three separate experiments are shown.
To confirm the specificity of rottlerin, PKCδ and PKCα expression was decreased by means of siRNA. The siRNA constructs specifically silenced the expression of either PKCδ or PKCα without affecting the other isoform. However, as shown in Fig. 7B, knockdown of PKCδ (~85%) in HCT116 cells resulted in an ~50% decrease in KLF5 expression by LPA, whereas knockdown of PKCα showed no effect. A similar result was observed in SW480 cells (data not shown). The PKCα/β inhibitor Gö6976 (Fig. 7C) did not affect the induction of KLF5 protein expression by LPA, confirming the lack of an effect by PKCα siRNA.
Induction of KLF5 Is eEF2k-dependent
Knockdown of PKCδ as shown above confirms a role of PKCδ in regulation of KLF5 expression. However, the extent of the effect by knockdown of PKCδ expression was comparatively smaller than the effect by rottlerin. Although the remaining ~15% of PKCδ protein may be responsible for the regulation of KLF5 expression, rottlerin may be acting on an another target. Rottlerin binds to eEF2k, also known as calmodulin (CaM)-and Ca2+ -dependent kinase III, in addition to PKCδ (43, 44). The activity of eEF2k is dependent on CaM and Ca2+ and phosphorylation of eEF2k leads to the inactivation of eEF2k and subsequent dephosphorylation of eEF2 (45). Hence, we next examined the potential involvement of eEF2k by using an inhibitor to CaM, trifluoperazine. As shown in Fig. 8, trifluoperazine, a phenothiazine, abrogated the induction of KLF5 expression in SW480 and HCT116 cells. For an unknown reason trifluoperazine raised the basal KLF5 expression level in HCT116 cells, but when the basal level was taken into account, it is evident that trifluoperazine effectively abrogated the induction of KLF5 by LPA (Fig. 8B). Because trifluoperazine is known to block calcium transporting proteins (46), we also tested another CaM antagonist, calmidazolium chloride. Similarly to trifluoroperazine, calmidazolium effectively blocked LPA-mediated induction of KLF5 expression in both SW480 and HCT116 cells (Fig. 8).
Figure. 8. Induction of KLF5 by LPA is blocked by CaM inhibitors.
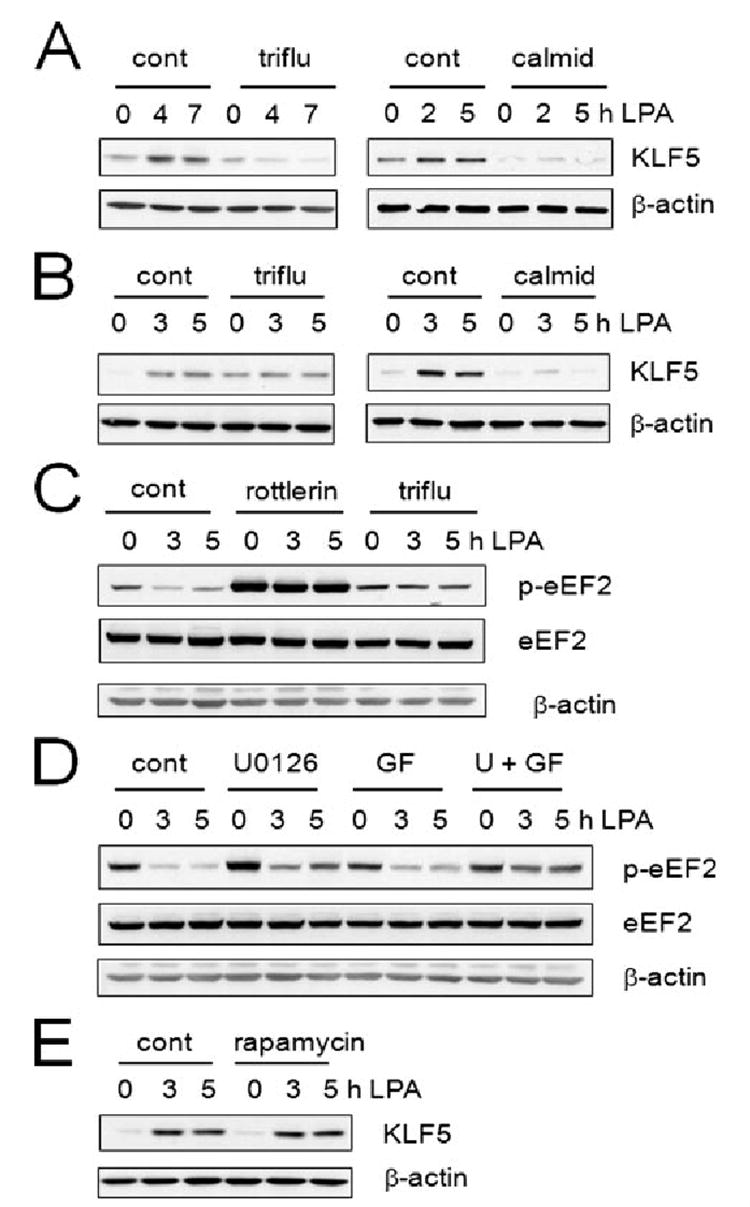
Effect of the CaM inhibitor trifluoperazine was determined. SW480 (A) or HCT116 (B) cells were treated with LPA in the presence of 3 μM trifluoperazine or 6 μM calmidazolium. C, the effects of trifluoperazine or rottlerin on phosphorylation levels of eEF2 in HCT116 cells treated with LPA were determined by Western blot. D, the effects of trifluoperazine or rottlerin on phosphorylation levels of eEF2 in HCT116 cells are shown. E, the effect of rapamycin on LPA-mediated induction of KLF5 was determined.
Next, we examined whether LPA treatment regulates phosphorylation levels of eEF2. Fig. 8C shows that LPA treatment resulted in a significant decrease in the level of eEF2 phosphorylation, which facilitates translation of mRNA. The presence of rottlerin completely abrogated the effect of LPA on eEF2 phosphorylation. The preincubation with rottlerin increased the basal phosphorylation level of eEF2k without affecting the protein expression. The reason for this effect is not known but rottlerin conceivably inhibits the basal activity of eEK2k. Our data in Fig. 6 showed that the induction of KLF5 is sensitive to inhibition by U0126. To explore the contribution of MEK1/2 on regulation of eEF2k, the phosphorylation level of eEF2 was determined in the presence of U0126 (Fig. 8D). U0126 significantly blocked the activation of eEF2 by LPA. On the other hand, GF109203X showed a marginal effect on eEF2 phosphorylation compared with control.
eEF2k is a known downstream target of the rapamycin-sensitive pathway involving the mammalian target of rapamycin (mTOR). However, we found that LPA-mediated induction of KLF5 was not blocked by rapamycin (Fig. 8E), suggesting that KLF5 induction is either mediated by the rapamycin-insensitive mTOR pathway or independent of mTOR.
The role of eEF2k was further substantiated by silencing of eEF2k expression by siRNA. siRNA targeting of eEF2k decreased the expression level of eEF2k by ~55% compared with control siRNA-transfected or untransfected cells (Fig. 9). More importantly, LPA-mediated KLF5 induction was decreased in cells treated with eEF2k siRNA. However, the decrease in KLF5 induction by eEF2k knockdown was not complete but was about 25%. On the contrary, the induction of KLF5 by PMA or FBS was unaffected by the knockdown of eEF2k, indicating that the regulation of KLF5 via eEF2k is specific to LPA.
Figure. 9. eEF2k is involved in induction of KLF5 by LPA.
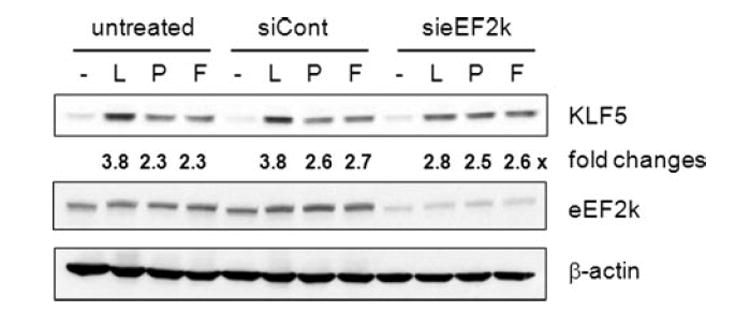
HCT116 cells were transfected with control or eEF2k siRNA. Cells were serum starved and treated for 3 h with 10 μM LPA, 100 nM PMA, or 15% FBS. Cells were lysed and the expression levels of KLF5, eEF2k, and β-actin were determined by Western blot. Representatives of three separate experiments are shown.
DISCUSSION
In adult intestine, the stem cell located at the base of the crypt continuously divide to generate epithelial cells, which migrate toward the tip of adjacent villi where the cells become differentiated and assume absorptive functions. To date, only a limited number of genes have been implicated in regulating the growth and differentiation of the intestinal epithelial cells. KLF5 is one such gene that plays a significant role in controlling cell proliferation in the intestine. KLF5 is predominantly expressed in the crypt compartment of intestine where cells rapidly replicate (23). A body of study has shown that KLF5 is a mediator of cell proliferation. KLF5 stimulates cell growth of NIH3T3 and IEC6 cells (26, 37). In NIH3T3 cells, KLF5 promotes and activates the cyclin B1/Cdc complex during mitosis and promotes transforming properties of oncogenic H-Ras (38, 39). KLF5 expression is regulated by serum and phorbol ester (26). In addition, KLF5 is also regulated by transcriptional regulators and nuclear receptors, such as retinoic acid receptor-α, NF-κB, and peroxisome proliferator-activated receptor γ (47). In this study, we identified KLF5 as a downstream target of LPA-mediated signaling. The induction of KLF5 by LPA was observed in all the colon cancer cells studied. Silencing of KLF5 expression substantially inhibited the proliferative effect of LPA, indicating that KLF5 plays a functional role in LPA-mediated proliferation. Last, we provided evidence that LPA induces KLF5 through signaling cascades that depend on MEK1/2, PKCδ, and eEF2k.
LPA stimulates proliferation of many types of cells, including fibroblasts, breast cancer cells, mesangial cells, vascular smooth muscle cells, neuronal cells, and others (5-10). The pro-proliferative effects of LPA on intestinal cells have previously been demonstrated. LPA enhances the metastatic potential of the DLD1 cell by promoting cell proliferation and migration (34). The proliferation of DLD1 cells is mediated by both LPA1 and LPA2, but only LPA1 is capable of enhancing migration. On the other hand, both LPA2 and LPA3, but not LPA1, promote the proliferation in HCT116 and LS174T cells via inactivation of GSK-3β and nuclear translocation of β-catenin (12). In this work, we show that both LPA2 and LPA3 stimulate the expression of KLF5. We did not investigate the role of LPA1 in induction of KLF5 under an assumption that any contribution by LPA1 will be minor based on the low expression level of LPA1. It appears that the roles of LPA2 and LPA3 in regulation of KLF5 expression are redundant as knockdown of either LPA2 or LPA3 each attenuated the induction of KLF5 expression by more than 50%. In keeping with redundancy of these LPA receptors, knockdown of LPA2 or LPA3 equally had a drastic effect on LPA-mediated proliferation of HCT116 cells (12).
The APC gene is the most commonly mutated gene in colorectal cancer with the mutations resulting in loss of the regulation of β-catenin activity (35). Unlike HCT116 and LS174T cells, the APC gene is mutated in SW480 and Caco-2 cells and we found that LPA did not stimulate the nuclear localization of β-catenin in SW480 cells. Our data showed that LPA specifically enhanced the expression of KLF5 in SW480 and Caco-2 cells and knockdown of KLF5 expression by siRNA significantly attenuated LPA-mediated proliferation. However, the induction of KLF5 is not limited to cells with mutated APC gene. In both nontransformed intestinal IEC6 and HCT116 colon cancer cells with wild type β-catenin, LPA stimulated KLF5 expression. Therefore, our data should not be taken to suggest that the LPA-β-catenin pathway and the LPA-KLF5 pathway are mutually exclusive. It is possible that KLF5 induction is dependent on β-catenin translocation. However, the following evidence does not favor this possibility. First, LPA does not induce translocation of β-catenin in SW480 cells in which the APC gene is mutated. Second, although both KLF5 and β-catenin can be stimulated in response to Wnt-1, the activation of KLF-5 by Wnt-1 takes places via a non-canonical Wnt pathway and is independent of β-catenin (32). Third, inhibition of GSK-3 did not alter the magnitude of KLF5 induction.5
KLF5 expression can be stimulated by a number of stimuli, including PMA and FBS (26, 32). In addition, a previous study has shown that in H-Ras-transformed cells, KLF5 is up-regulated via H-Ras-activated MAPK (38). In the present study, LPA-mediated induction of KLF5 was not sensitive to the presence of PTX, LY294002, and U73122. The lack of an effect by these inhibitors is similar to the activation of β-catenin by LPA (12), although the activation of β-catenin and KLF5 appears to be independent as discussed earlier. Studies have shown that LPA transactivates the EGF receptor in various cell types and that EGF activates KLF5 mRNA expression in primary esophageal cells (48-50). However, the presence of the EGF receptor inhibitor, AG1478, did not block the induction of KLF5 expression by LPA (Fig. 6), indicating that transactivation of the EGF receptor by LPA does not play a significant role in LPA-mediated induction of KLF5. LPA has been shown to activate MAPKs (51, 52), but inhibition of p38 MAPK had no effect on induction of KLF5, whereas blocking MEK1/2 with U0126 partially attenuated KLF5 induction. In addition, GF109203X moderately blocked LPA-mediated induction of KLF5. Multiple PKCs have been shown to regulate the Raf-MEK-Erk cascade (53), and we observed that GF109203X significantly attenuated LPA-mediated phosphorylation of ERK1/2 in SW480 cells (data not shown). However, the presence of U0126 completely abrogated phosphorylation of ERK1/2 and yet the effects of GF109203X and U0126 on KLF5 expression were additive in this study, as evidenced by both Western blot and the promoter activity (Figs. 7 and 8). Therefore, the effect of GF109203X is unlikely ERK1/2-dependent.
We assumed that the effect of rottlerin was solely mediated via PKCδ based on the broad usage of rottlerin as a specific inhibitor of PKCδ and a previous study that LPA-mediated secretion of interleukin-8 was blocked by a dominant negative form of PKCδ (54). However, the partial of effect by silencing of PKCδ expression suggested the presence of an additional target(s) by rottlerin. Despite its wide usage as a PKCδ-specific inhibitor, rottlerin also binds eEF2k with a similar affinity (5.3 μM for eEF2k versus 3–6 μM for PKCδ) (43). The potential role of eEF2k in LPA-mediated induction of KLF5 is evidence by the inhibition of LPA-mediated KLF5 induction by CaM inhibitors, trifluoroperazine and calmidazolium. However, a caution on the results from the CaM inhibitors should be noted. Trifluoroperazine is known to block calcium transporting proteins at a higher dose (55). A recent study (56) has shown that the commonly used membrane-permeable CaM inhibitors, such as W-7, trifluoroperazine, and calmidazolium, all bind significantly to the inner leaflet of the plasma membrane at approximately the same concentrations at which they are used to inhibit CaM in cells. Thus, the common assumption that using two chemically different inhibitors (e.g. trifluoroperazine and W-7) provides evidence that they are acting specifically on CaM is not warranted (56). In addition, previous reports showed that eEF2k can be regulated via a Ca2+/CaM-independent pathway (57, 58). Despite this limitation, LPA decreased the phosphorylation level of eEF2 in both HCT116 and SW480 cells, which is assumed to stimulate translation of mRNA (Fig. 8). A further evidence for a role of eEF2k in regulation of KLF5 protein expression comes from the knockdown of eEF2k expression that attenuated the induction of KLF5 by LPA.
eEF2k is an unusual kinase. The sequence of its catalytic domain differs substantially from that of the main Ser-Thr-Tyr kinase superfamily (59). eEF2K is a Ca2+/CaM-dependent protein kinase that phosphorylates and inactivates eEF2, an elongation factor that facilitates translocation of peptidyl t-RNA from the ribosomal A site to P site (60). It has been shown that several protein kinases can phosphorylate eEF2k and the activity of eEF2k is altered by phosphorylation. These protein kinases include p70S6k, p90RSK, and stress-activated protein kinase/p38δ (45, 61). Phosphorylation at Ser-366 by p70S6k (regulated via mTOR) and p90RSK (activated by ERK) inhibits the enzyme activity of eEF2k (45). On the other hand, the activation of AMP-activated protein kinase by myocardial ischemia stimulated the phosphorylation level of eEF2k and its activity independent of the mTOR pathway (62). Activation of eEF2k phosphorylates eEF2, temporarily inhibiting translation (60). Surprisingly, mice lacking eEF2k do not exhibit any phenotypic defect, indicating that eEF2k is not essential for development, behavior, or reproduction (62, 63). Because eEF2k functions in regulation of the translation process, it seems plausible that all translational processes affecting KLF5 protein expression are gated through eEF2k and eEF2. However, it appears that eEF2k does not govern all translation of KLF5 mRNA based on our observation that knockdown of eEF2k specifically decreased LPA-mediated but not PMA- or FBS-mediated induction of KLF5.
The study using KLF5+/− mouse has revealed diverse roles in regulation of cellular differentiation and tissue development. KLF5−/− homozygotes die before embryonic day 8.5, but KLF5+/− mice are apparently normal and survive until adulthood (64). However, the KLF5+/− mice have misshapen intestinal villi, consistent with in vitro studies that KLF5 positively regulates cell proliferation in the intestine. In addition, KLF5+/− mice show reduced cardiac hypertrophy and fibrosis in response to external stress, suggesting that KLF5 plays a critical role in cardiovascular remodeling (64). Previous studies have shown that LPA stimulates protein synthesis and hypertropic cell growth in cultured neonate rat cardiac myocytes (65, 66). Although the present study is not aimed at the effect of LPA in cardiomyocytes, it remains an intriguing possibility that KLF5 may be a mediator of LPA-induced effects in the cardiovascular system.
Phospholipase A2 family members play a key role in the metabolism of membrane lipids into lysophospholipids, which are used to generate arachidonic acids and prostaglandins (67). Deletion of cytosolic phospholipase A2 results in a drastic reduction in tumor number in APCMin/+ mice, suggesting a link between lipid metabolism and intestinal tumor formation (68). However, a number of questions remain unanswered in regard to the role of LPA in the tumorigenesis of colon cancer. A recent study has shown that Caco-2 cells are capable of producing LPA and other phospholipids (69), but whether LPA is produced in the gastrointestinal tract in vivo and whether LPA concentration is elevated in colorectal cancer as in ovarian or other gynecological cancers is not yet known.
In summary, we have identified KLF5 as an effector of LPA-mediated signaling. KLF5 plays an important role in proliferation of colon cancer cells. LPA stimulates proliferation of colon cancer cells via induction of KLF5 and we have shown that the induction of KLF5 is mediated via MEK and PKCδ-dependent pathways. Our results also demonstrate for the first time that LPA regulates protein expression via regulation of eEF2k.
Acknowledgments
We are grateful to Dr. J. Lingrel for the mouse KLF5 cDNA. We thank Mandayam Nandan for helpful discussion on anti-KLF5 antibody.
Footnotes
This work was supported in part by National Institutes of Health Grant DK071597, the Emory University Research Committee, and a Pilot & Feasibility Grant DK64399 from the Digestive Diseases Research Development Center.
The abbreviations used are: LPA, lysophosphatidic acid; KLF, Kru¨ ppel-like factor; S1P, sphingosine 1-phosphate; eEF2k, eukaryotic elongation factor 2 kinase; PKC, protein kinase C; MAPK, mitogen-activate protein kinase; MEK, mitogen-activate protein kinase kinase; ERK, extracellular signal-regulated kinase; PTX, pertussis toxin; siRNA, small interfering RNA; APC, adenomatous polyposis coli; PMA, phorbol 12-myristate 13-acetate; GSK-3_, glycogen synthase kinase 3_; CaM, calmodulin; FBS, fetal bovine serum; PBS, phosphate-buffered saline; mTOR, mammalian target of rapamycin; EGF, epidermal growth factor.
H. Zhang and C. C. Yun, unpublished data.
References
- 1.An S, Dickens MA, Bleu T, Hallmark OG, Goetzl EJ. Biochem Biophys Res Commun. 1997;231:619–622. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- 2.An S, Bleu T, Hallmark OG, Goetzl EJ. J Biol Chem. 1998;273:7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 3.Aoki J, Bandoh K, Inoue K. Ann N Y Acad Sci. 2000;905:263–266. doi: 10.1111/j.1749-6632.2000.tb06556.x. [DOI] [PubMed] [Google Scholar]
- 4.van Corven EJ, Groenink A, Jalink K, Eichholtz T, Moolenaar WH. Cell. 1989;59:45–54. doi: 10.1016/0092-8674(89)90868-4. [DOI] [PubMed] [Google Scholar]
- 5.Tigyi G, Dyer DL, Miledi R. Proc Natl Acad Sci U S A. 1994;91:1908–1912. doi: 10.1073/pnas.91.5.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goetzl EJ, Dolezalova H, Kong Y, Zeng L. Cancer Res. 1999;59:4732–4737. [PubMed] [Google Scholar]
- 7.Gaits F, Salles JP, Chap H. Kidney Int. 1997;51:1022–1027. doi: 10.1038/ki.1997.143. [DOI] [PubMed] [Google Scholar]
- 8.Kim J, Keys JR, Eckhart AD. Cell Signal. 2006;18:1695–1701. doi: 10.1016/j.cellsig.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Sorensen SD, Nicole O, Peavy RD, Montoya LM, Lee CJ, Murphy TJ, Traynelis SF, Hepler JR. Mol Pharmacol. 2003;64:1199–1209. doi: 10.1124/mol.64.5.1199. [DOI] [PubMed] [Google Scholar]
- 10.Cechin SR, Dunkley PR, Rodnight R. Neurochem Res. 2005;30:603–611. doi: 10.1007/s11064-005-2747-4. [DOI] [PubMed] [Google Scholar]
- 11.Dixon RJ, Brunskill NJ. Kidney Int. 1999;56:2064–2075. doi: 10.1046/j.1523-1755.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 12.Yang M, Zhong WW, Srivastava N, Slavin A, Yang J, Hoey T, An S. Proc Natl Acad Sci U S A. 2005;102:6027–6032. doi: 10.1073/pnas.0501535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radhika V, Hee Ha J, Jayaraman M, Tsim ST, Dhanasekaran N. Oncogene. 2005;24:4597–4603. doi: 10.1038/sj.onc.1208665. [DOI] [PubMed] [Google Scholar]
- 14.Moolenaar WH. Ann N Y Acad Sci. 2000;905:1–10. doi: 10.1111/j.1749-6632.2000.tb06532.x. [DOI] [PubMed] [Google Scholar]
- 15.Smith KJ, Johnson KA, Bryan TM, Hill DE, Markowitz S, Willson JK, Paraskeva C, Petersen GM, Hamilton SR, Vogelstein B, Kinzlen KW. Proc Natl Acad Sci U S A. 1993;90:2846–2850. doi: 10.1073/pnas.90.7.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinzler KW, Vogelstein B. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 17.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 18.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 19.Schuh R, Aicher W, Gaul U, Cote S, Preiss A, Maier D, Seifert E, Nauber U, Schroder C, Kemler R, Jaäckle H. Cell. 1986;47:1025–1032. doi: 10.1016/0092-8674(86)90817-2. [DOI] [PubMed] [Google Scholar]
- 20.Turner J, Crossley M. Trends Biochem Sci. 1999;24:236–240. doi: 10.1016/s0968-0004(99)01406-1. [DOI] [PubMed] [Google Scholar]
- 21.Dang DT, Pevsner J, Yang VW. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feinberg MW, Lin Z, Fisch S, Jain MK. Trends Cardiovas Med. 2004;14:241–246. doi: 10.1016/j.tcm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conkright M, Wani M, Anderson K, Lingrel J. Nucleic Acids Res. 1999;27:1263–1270. doi: 10.1093/nar/27.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang DT, Bachman KE, Mahatan CS, Dang LH, Giardiello FM, Yang VW. FEBS Lett. 2000;476:203–207. doi: 10.1016/s0014-5793(00)01727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun R, Chen X, Yang VW. J Biol Chem. 2001;276:6897–6900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yun CC, Chen Y, Lang F. J Biol Chem. 2002;277:7676–7683. doi: 10.1074/jbc.M107768200. [DOI] [PubMed] [Google Scholar]
- 28.Chanchevalap S, Nandan MO, McConnell BB, Charrier L, Merlin D, Katz JP, Yang VW. Nucleic Acids Res. 2006;34:1216–1223. doi: 10.1093/nar/gkl014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laborda J. Nucleic Acids Res. 1991;19:3998. doi: 10.1093/nar/19.14.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yun CC, Sun H, Wang D, Rusovici R, Castleberry A, Hall RA, Shim H. Am J Physiol. 2005;289:C2–C11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 31.Du JX, Yun CC, Bialkowska A, Yang VW. J Biol Chem. 2007;282:4782–4793. doi: 10.1074/jbc.M603413200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziemer LT, Pennica D, Levine AJ. Mol Cell Biol. 2001;21:562–574. doi: 10.1128/MCB.21.2.562-574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon HS, Chen X, Yang VW. J Biol Chem. 2003;278:2101–2105. doi: 10.1074/jbc.M211027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shida D, Kitayama J, Yamaguchi H, Okaji Y, Tsuno NH, Watanabe T, Takuwa Y, Nagawa H. Cancer Res. 2003;63:1706–1711. [PubMed] [Google Scholar]
- 35.Ilyas M, Tomlinson IPM, Rowan A, Pignatelli M, Bodmer WF. Proc Natl Acad Sci U S A. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usui S, Sugimoto N, Takuwa N, Sakagami S, Takata S, Kaneko S, Takuwa Y. J Biol Chem. 2004;279:12300–12311. doi: 10.1074/jbc.M305025200. [DOI] [PubMed] [Google Scholar]
- 37.Chanchevalap S, Nandan MO, Merlin D, Yang VW. FEBS Lett. 2004;578:99–105. doi: 10.1016/j.febslet.2004.10.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Oncogene. 2004;23:3404–3413. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang X, Yu S, Tanyi JL, Lu Y, Woodgett JR, Mills GB. Mol Cell Biol. 2002;22:2099–2110. doi: 10.1128/MCB.22.7.2099-2110.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cummings R, Zhao Y, Jacoby D, Spannhake EW, Ohba M, Garcia JGN, Watkins T, He D, Saatian B, Natarajan V. J Biol Chem. 2004;279:41085–41094. doi: 10.1074/jbc.M404045200. [DOI] [PubMed] [Google Scholar]
- 42.Zachos NC, Tse M, Donowitz M. Ann Rev Physiol. 2005;67:411–443. doi: 10.1146/annurev.physiol.67.031103.153004. [DOI] [PubMed] [Google Scholar]
- 43.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 44.Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, Erdjument-Bromage H, Tempst P, Parmer TG, Prostko CR, Germino FJ, Hait WN. Proc Natl Acad Sci U S A. 1997;94:4884–4889. doi: 10.1073/pnas.94.10.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engelender S, De Meis L. Mol Pharmacol. 1996;50:1243–1252. [PubMed] [Google Scholar]
- 47.Nagai R, Suzuki T, Aizawa K, Shindo T, Manabe I. J Thromb Haemostasis. 2005;3:1569–1576. doi: 10.1111/j.1538-7836.2005.01366.x. [DOI] [PubMed] [Google Scholar]
- 48.Daub H, Weiss FU, Wallasch C, Ullrich A. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 49.Wu J, Cunnick JM. Biochim Biophys Acta. 2002;1582:100–106. doi: 10.1016/s1388-1981(02)00143-9. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y, Goldstein BG, Nakagawa H, Katz JP. FASEB J. 2006;21:543–550. doi: 10.1096/fj.06-6694com. [DOI] [PubMed] [Google Scholar]
- 51.Goetzl EJ, An S. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- 52.Tigyi G. Prostaglandins. 2001;64:47–62. doi: 10.1016/s0090-6980(01)00107-1. [DOI] [PubMed] [Google Scholar]
- 53.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Mol Cell Biol. 1998;18:790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao Y, He D, Saatian B, Watkins T, Spannhake EW, Pyne NJ, Natarajan V. J Biol Chem. 2006;281:19501–19511. doi: 10.1074/jbc.M511224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dewael Y, Mallefet J. Comp Biochem Physiol Part C Toxicol Pharmacol. 2002;131:153–160. doi: 10.1016/s1532-0456(01)00288-5. [DOI] [PubMed] [Google Scholar]
- 56.Sengupta P, Ruano MJ, Tebar F, Golebiewska U, Zaitseva I, Enrich C, McLaughlin S, Villalobo A. J Biol Chem. 2007;282:8474–8486. doi: 10.1074/jbc.M607211200. [DOI] [PubMed] [Google Scholar]
- 57.Hughes SJ, Smith H, Ashcroft SJ. Biochem J. 1993;289:795–800. doi: 10.1042/bj2890795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diggle TA, Subkhankulova T, Lilley KS, Shikotra N, Willis AE, Redpath NT. Biochem J. 2001;353:621–626. doi: 10.1042/0264-6021:3530621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Redpath NT, Price NT, Proud CG. J Biol Chem. 1996;271:17547–17554. [PubMed] [Google Scholar]
- 60.Ryazanov AG, Shestakova EA, Natapov PG. Nature. 1988;334:170–173. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 61.Knebel A, Morrice N, Cohen P. EMBO J. 2001;20:4360–4369. doi: 10.1093/emboj/20.16.4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horman S, Beauloye C, Vertommen D, Vanoverschelde J-L, Hue L, Rider MH. J Biol Chem. 2003;278:41970–41976. doi: 10.1074/jbc.M302403200. [DOI] [PubMed] [Google Scholar]
- 63.Ryazanov AG. FEBS Lett. 2002;514:26–29. doi: 10.1016/s0014-5793(02)02299-8. [DOI] [PubMed] [Google Scholar]
- 64.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Nat Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 65.Goetzl EJ, Lee H, Azuma T, Stossel TP, Turck CW, Karliner JS. J Biol Chem. 2000;275:14573–14578. doi: 10.1074/jbc.275.19.14573. [DOI] [PubMed] [Google Scholar]
- 66.Hilal-Dandan R, Means CK, Gustafsson AB, Morissette MR, Adams JW, Brunton LL, Heller Brown J. J Mol Cell Cardiol. 2004;36:481–493. doi: 10.1016/j.yjmcc.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 67.Sturm A, Dignass AU. Biochim Biophys Acta. 2002;1582:282–288. doi: 10.1016/s1388-1981(02)00182-8. [DOI] [PubMed] [Google Scholar]
- 68.Hong KH, Bonventre JC, O’Leary E, Bonventre JV, Lander ES. Proc Natl Acad Sci U S A. 2001;98:3935–3939. doi: 10.1073/pnas.051635898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Subramanian N, Qadri A. Nat Immunol. 2006;7:583–58. doi: 10.1038/ni1336. [DOI] [PubMed] [Google Scholar]