

Abstract
Cilia regulate diverse functions such as motility, fluid balance, and sensory perception. The cilia of olfactory sensory neurons (OSNs) compartmentalize the signaling proteins necessary for odor detection; however, little is known regarding the mechanisms of protein sorting/entry into olfactory cilia. Nephrocystins are a family of ciliary proteins likely involved in cargo sorting during transport from the basal body to the ciliary axoneme. In humans, loss-of-function of the cilia–centrosomal protein CEP290/NPHP6 is associated with Joubert and Meckel syndromes, whereas hypomorphic mutations result in Leber congenital amaurosis (LCA), a form of early-onset retinal dystrophy. Here, we report that CEP290–LCA patients exhibit severely abnormal olfactory function. In a mouse model with hypomorphic mutations in CEP290 [retinal dystrophy-16 mice (rd16)], electro-olfactogram recordings revealed an anosmic phenotype analogous to that of CEP290–LCA patients. Despite the loss of olfactory function, cilia of OSNs remained intact in the rd16 mice. As in wild type, CEP290 localized to dendritic knobs of rd16 OSNs, where it was in complex with ciliary transport proteins and the olfactory G proteins Golf and Gγ13. Interestingly, we observed defective ciliary localization of Golf and Gγ13 but not of G protein-coupled odorant receptors or other components of the odorant signaling pathway in the rd16 OSNs. Our data implicate distinct mechanisms for ciliary transport of olfactory signaling proteins, with CEP290 being a key mediator involved in G protein trafficking. The assessment of olfactory function can, therefore, serve as a useful diagnostic tool for genetic screening of certain syndromic ciliary diseases.
Keywords: nephrocystin, olfaction
Cilia are microtubule-based structures that project from the surface of most cells in humans and are implicated in numerous biological functions, including motility, sensory perception, and signaling (1, 2). In most cell types, the cilium extends from the plasma membrane at a basal body, originating from the mother centriole of the centrosome complex (3). In multiciliated cells, such as in olfactory sensory neurons (OSNs), ciliogenesis requires the synthesis and assembly of multiple basal bodies formed en masse from centrioles (4). OSNs terminate in a dendritic knob containing multiple basal bodies from which sensory cilia project into the nasal mucosa. These cilia compartmentalize the signaling molecules necessary for odorant detection. Odorant signal transduction is initiated by odorant binding to G protein-coupled receptors, leading to activation of olfactory G proteins (5, 6). The G protein heterotrimer consists of the adenylyl cyclase-stimulating Gαolf as well as the βγ-subunit, including Gγ13. Odor transduction through this complex efficiently couples external stimuli to action potential generation (5, 6). The loss of olfactory cilia or deletion of selected components of the olfactory signaling cascade leads to anosmia (7–11). Although each component of this signaling cascade is enriched in olfactory cilia, little is known regarding the mechanisms of their trafficking and subcellular localization.
Cilia of OSNs lack the necessary machinery for protein synthesis. Therefore, nascent proteins must be transported from the cell body into the cilium. Movement along the ciliary axoneme is tightly regulated and most likely involves evolutionarily conserved intraflagellar transport (IFT) proteins, whereas multiprotein complexes at the basal body act as a barrier to diffusion and restrict access to the cilium (1, 12). Importantly, not all proteins can access the ciliary compartment because there appears to be a selective gate at the base that regulates entry. Despite ever-increasing knowledge of IFT components, the mechanisms regulating protein sorting/entry are poorly understood.
Genetic mutations in ciliary, basal body, and centrosomal proteins lead to pleiotropic human diseases, including polycystic kidney disease, Bardet–Biedl syndrome, Senior–Loken syndrome, Meckel syndrome, and retinitis pigmentosa (RP) (2, 7, 13–15). Nephrocystins are a family of ciliary proteins that are likely involved in cargo sorting during transport from the basal body to the ciliary axoneme (16–20). Loss-of-function mutations in CEP290 are associated with Joubert syndrome, which is characterized by nephronophthisis, retinal degeneration, and cerebellar vermis hypoplasia (21, 22). Interestingly, an in-frame deletion of exons 35–39 in CEP290 causes early-onset retinal degeneration in the rd16 mouse, without associated cerebellar or kidney abnormalities (23). This deletion led to the identification of hypomorphic CEP290 mutations in >20% of patients with Leber congenital amaurosis (LCA), a severe retinal dystrophy characterized by visual impairment from birth (24, 25). As in the rd16 mouse, these LCA patients did not exhibit cerebellar or renal abnormalities. CEP290 is localized to the connecting cilium of retinal photoreceptors, the basal body of an inner medullary collecting duct cell line (IMCD-3), and the centrosome of mitotic cells (21–23). Furthermore, the basal body of mammalian olfactory cilia is enriched in γ-tubulin and resembles the Caenorhabditis elegans transition zone, which is enriched in nephrocystin proteins (7, 16–18). Hence, mutations in CEP290 may alter ciliary microtubule transport.
We therefore hypothesized that CEP290 participates in regulating the transport of specific signaling proteins involved in odorant detection and that olfactory function may be impaired in blind individuals with CEP290 mutations. Here, we show that CEP290–LCA patients and the rd16 mouse exhibit severe olfactory dysfunction, likely caused by altered ciliary localization of olfactory G proteins in sensory neurons. This work describes a mechanism by which G protein trafficking along the ciliary axoneme is selectively regulated at the dendritic knobs by CEP290.
Results
Impaired Olfactory Function in LCA Patients with CEP290 Mutations.
We investigated olfactory function in a French–Canadian LCA family, which originally revealed the CEP290 mutation, a splice-site change resulting in a premature stop codon (Cys998X) (24). Patients homozygous for the mutation and heterozygous carriers were evaluated by using the Brief Smell Identification Test. All LCA patients examined exhibited severely abnormal olfactory function (Fig. 1), whereas CEP290 Cys998X heterozygous carriers exhibited mild to severe microsmia (data not shown). Retinopathy patients with mutations in RPGR, RPGRIP, and USH2A, three other cilia and/or centrosomal proteins, demonstrated normal olfactory function (Fig. 1). These data have been stratified by age as shown in supporting information (SI) Fig. 7.
Fig. 1.

Assessment of olfactory function in LCA patients. Individuals with retinopathies (LCA or RP) were evaluated for olfactory defects with the Brief Smell Identification Test (B-SIT) (B-SIT); scores >8 were considered normal, and scores ≤8 were considered abnormal. Patients with LCA (CEP290 Cys998X/Cys998X) (n = 3) exhibited severely abnormal olfactory function, and patients with mutations in other retinopathy genes were unaffected.
Olfactory Dysfunction in rd16 Mice Carrying In-Frame Deletion of CEP290.
To investigate the mechanism of anosmia associated with hypomorphic CEP290 alleles in LCA, we assessed olfactory function in 1-month-old rd16 mice by electro-olfactograms, which measure the odorant-induced, summated generator potential of sensory neurons from the mucosal surface. At all doses of amyl acetate or other odorants tested, the rd16 mice showed significantly reduced electro-olfactogram responses compared with those of WT mice (Fig. 2 A and B), validating the results from our LCA patients.
Fig. 2.

rd16 mice are anosmic. (A) Representative electro-olfactogram traces from WT (n = 9) and rd16 (n = 5) mice after exposure of the olfactory epithelium to three concentrations of amyl acetate. Arrow indicates time of odorant administration. (B) Histogram of electro-olfactogram responses from both WT (empty bars; n = 9) and rd16 (filled bars; n = 5) mice showing four different concentrations of amyl acetate, along with five other odorants all tested at a concentration of 10−3 M of the respective odorant. All responses were normalized to a pulse of pure amyl acetate given during the same trace. AA, amyl acetate; 8-AL, octanal; 7-AL, heptaldehyde; 6-AL, hexanal; Eug, eugenol; Car, carvone. *, P < 0.05 as determined by an unpaired t test.
Structural Integrity of Olfactory Cilia in rd16 Mice.
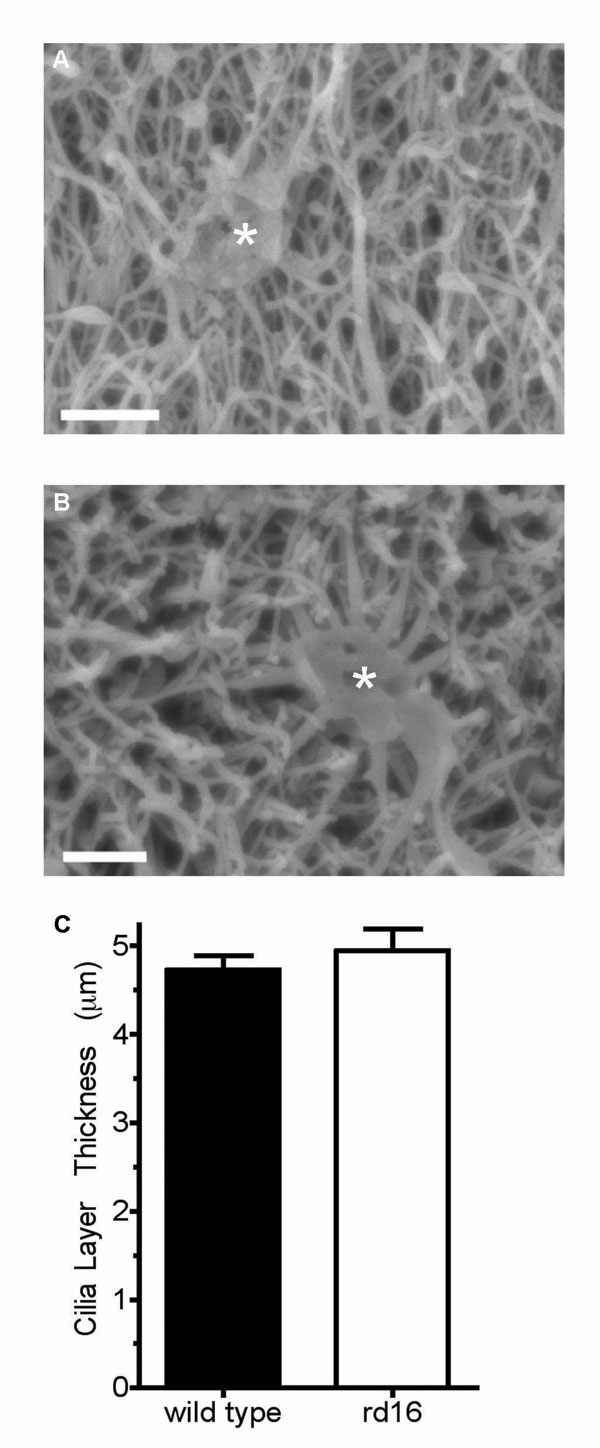
The anosmic phenotype in the Bbs4-null mice (7, 26) is caused by the disruption of the microtubule network and a deficiency in the number of olfactory cilia (7). Hence, one possible mechanism for olfactory dysfunction caused by CEP290 mutations can be the loss of cilia from OSNs. In the rd16 mice, both the cilia, as measured by acetylated α-tubulin staining (Fig. 3Center), and the dendritic knobs, marked by γ-tubulin (Fig. 3A), appeared unaltered. The localization of mutant CEP290 to the dendritic knobs also was unaffected in the rd16 OSNs (Fig. 3 B and C). Further examination by scanning electron microscopy of the olfactory epithelium surface from WT and rd16 mice demonstrated an intact cilia layer from which no discernible differences in the ultrastructure of the cilia could be detected (Fig. 3D). The dendritic endings with proximal parts of the olfactory cilia for both WT and rd16 mice are shown in SI Fig. 8 A and B. In addition, we measured the width of the ciliary layer as demarcated by acetylated tubulin staining and found no statistical difference between WT and rd16 mice (SI Fig. 8C). We, however, observed that select regions of the epithelium showed signs of dendritic microtubule disorganization. The normal parallel tracks of acetylated α-tubulin staining are observed in the dendrites of WT mice; these tracks were disordered in small clusters in the rd16 OSNs (SI Fig. 9 A and B). TUNEL staining revealed that this population of neurons may be in the early stages of neuronal apoptosis and/or epithelial degeneration (SI Fig. 9C), whereas other regions of the rd16 olfactory epithelium appear normal (SI Fig. 9D) compared with WT mice (SI Fig. 9E). Overall, our results do not provide any evidence of defects in ciliogenesis or global integrity of olfactory cilia in the rd16 mice.
Fig. 3.

Immunohistochemistry from WT and rd16 mice showing the staining pattern of CEP290 in vivo. Olfactory epithelial slices were subject to immunohistochemistry by using either CEP290 antiserum or an anti-γ-tubulin antibody. (A) γ-Tubulin localizes to dendritic knobs. (B and C) Staining patterns indicate that CEP290 localization is similar to γ-tubulin in dendritic knobs in both the WT (B) and rd16 (C) mice. (Scale bar, 10 μm.) (D) Scanning electron microscopy revealed no difference in the olfactory epithelium between WT (Left) and rd16 (Right) mice. Images are shown at ×7,400 zoom. (Scale bar, 1 μm.)
Mislocalization of Olfactory G Proteins in rd16 Mice.
In the absence of any overt structural defects in the cilia of OSNs, we examined whether the CEP290 mutation altered its expression or ability to interact with other transport proteins. Immunoblot analysis of the olfactory epithelial protein extract revealed that, aside from the expected molecular mass shift observed in the rd16 mouse, there was no detectable change in the amount of the CEP290 protein (Fig. 4A). Immunoprecipitations from rd16 olfactory epithelial tissue showed that CEP290 remains associated with ciliary, basal body, and microtubule transport proteins, including p150Glued (a dynein motor protein), KIF3A (a kinesin motor involved in IFT), RPGRORF15 [a putative guanine–nucleotide exchange factor (GEF)], and γ-tubulin (Fig. 4B), as observed in WT retina (16). We did not detect an interaction between CEP290 and an intraflagellar transport protein, IFT88, or a Bardet–Biedl syndrome protein, BBS4 (Fig. 4B). No change was detected in the interaction of any of these proteins in the rd16 mice compared with WT. We, however, discovered that both Gγ13 and Golf (two of the components of the olfactory G protein) also immunoprecipitate with CEP290 in WT and rd16 mice (Fig. 4C), suggesting that CEP290 may regulate the transport of olfactory signal proteins.
Fig. 4.

Immunoprecipitation from WT and rd16 mouse olfactory epithelium. (A) Western blot analysis of olfactory epithelium shows CEP290 expression in WT and rd16 mice. The shift in mobility in rd16 lysate results from the in-frame 297-aa deletion in CEP290. (B) Lysates from WT and rd16 mouse olfactory epithelium were immunoprecipitated with, and blotted for the indicated antibodies. Molecular mass in kilodaltons is shown to the left of each blot. Input indicates 10% of the immunoprecipitate starting material. (C) Lysates were immunoprecipitated with the indicated antibodies and separated by SDS/PAGE. Immunoblots were probed with CEP290 antiserum, and the arrow indicates the specific immunoreactive band for CEP290. Input indicates 10% of the immunoprecipitate starting material in both WT and rd16 mice. IP, immunoprecipitation; IB, immunoblotting.
To further dissect the mechanism of impaired olfactory function, we examined the localization of olfactory signaling molecules in OSNs of the rd16 mouse. Prominent ciliary staining of Gγ13 and Golf was detected in the WT, but these G proteins were undetectable in the OSNs cilia of the rd16 mice (Fig. 5 A–D). Although the Gγ13 protein exhibited mislocalization to cell bodies and dendrites (see Fig. 5 A and B), Golf could not be detected in the extra-ciliary layers of the rd16 OSNs (Fig. 5 C and D). Interestingly, type III adenylyl cyclase (ACIII; isoform expressed in OSNs) and the α-subunit of the cyclic nucleotide-gated channel, CNGA2, remained enriched in olfactory cilia and colocalized with acetylated tubulin, consistent with the observed maintenance of ciliary integrity in rd16 mice (Fig. 5 E–H). Fig. 5I shows the averaged data in which the fluorescence signal from the immunostained images was quantitated. These results indicate that the olfactory phenotype in LCA patients and rd16 mice is likely caused by the defective transport of olfactory G proteins.
Fig. 5.

Defects in localization of olfactory G proteins in rd16 mice. Olfactory epithelial sections were immunostained with antibodies to odorant-signaling molecules enriched in cilia (green) and costained with acetylated α-tubulin (red). (A) Gγ13 is enriched in cilia of WT mouse OSNs and colocalizes with acetylated α-tubulin (merged image). (B) Gγ13 is undetectable in the ciliary layer and is mislocalized to dendrites and cell bodies in rd16 mice. (C) Similar to Gγ13 staining, Golf is enriched in olfactory cilia and colocalizes with acetylated α-tubulin. (D) In the rd16 mice, Golf is absent from olfactory cilia and, unlike Gγ13, does not appear to redistribute within OSNs. (E and F) ACIII remains enriched in both WT and rd16 mice and colocalizes with acetylated α-tubulin in OSN cilia. (G and H) CNGA2 remains enriched in both WT and rd16 mice and colocalizes with acetylated α-tubulin in OSN cilia. (Scale bar, 10 μm.) (I) Fluorescence quantitation of immunohistochemical images represented in A–H. Data are averages of at least 30 images per protein from three discontinuous regions per olfactory epithelium. Four different mice were analyzed for both WT (filled bars) and rd16 (empty bars). ***, P < 0.001 as determined by an unpaired t test.
We then tested the subcellular localization of G protein-coupled odorant receptors by immunohistochemical analysis. Unlike the G proteins, mOR28 remained localized to OSNs cilia of rd16 mice (Fig. 6 A–C), showing a similar staining pattern to WT mouse olfactory epithelium (SI Fig. 10). In addition, antibodies against two other odorant receptors, mOR256 and mOR262, marked cilia in OSNs of rd16 mice (SI Fig. 11). These data indicate that olfactory signaling components do not get transported as a preassembled complex and that the entry of G protein signaling complex into cilia is regulated by at least two distinct mechanisms.
Fig. 6.

Maintenance of mOR28 localization to cilia in rd16 mice. rd16 mouse olfactory epithelial slices were stained by using antibodies directed against mOR28. (A) A compressed confocal image showing multiple mOR28-expressing olfactory sensory neurons. (Scale bar, 20 μm.) (B) A ×2 zoomed image of the boxed neuron in A showing mOR28 expression in the cell body, dendrite, and dendritic knob. (Scale bar, 5 μm.) (C) A ×2.5 zoomed image of the boxed region in B showing receptor expression in OSNs cilia. Arrows indicate individual cilia. (Scale bar, 2 μm.)
Discussion
Our study shows that CEP290 participates in regulating the ciliary transport of specific signaling proteins involved in odorant detection and that olfactory function is impaired in blind individuals with CEP290 mutations. CEP290 is expressed in OSNs and is localized to dendritic knobs, which contain multiple basal bodies that form the base of extending cilia. In OSNs, hypomorphic mutations in CEP290 specifically result in the mislocalization of olfactory G proteins but not of other components of the signaling cascade tested. These results suggest that the entire complement of OSN transduction proteins does not move into cilia as a single preassembled complex. The odorant receptors traffic into cilia independently of olfactory G proteins and, therefore, may assemble into a signaling complex within the cilium.
Our studies suggest that OSNs use different mechanisms for the control of protein entry into the cilium. One interesting hypothesis is that there may be differences in the way that peripheral and transmembrane proteins localize to cilia. Although anchored, peripheral membrane proteins exist predominantly in the cytoplasm and are more likely to interact with the basal body complex found beneath the plasma membrane. As an integral component of this complex, CEP290 is positioned to regulate movement of cargo into and out of the cilium. The basal body has been proposed to act as a highly selective barrier regulating protein entry into the cilium (27). Located adjacent to the basal body is a protein-rich region of the ciliary membrane, termed the “ciliary necklace,” which has been compared functionally with the nuclear pore complex (1, 27). Selective entry through the nuclear pore requires cargo binding to a GTP-binding protein and guanine–nucleotide exchange involving a GEF to release cargo into the nucleus (27). We showed that RPGRORF15, a putative GEF (28), is a cilia/centrosomal protein that may regulate selective transport of cargo proteins from basal bodies distally along the ciliary axoneme in the photoreceptor connecting cilium (14, 29). We now show that RPGRORF15 is in complex with CEP290 in OSNs of both WT and rd16 mice, suggesting a possible role in regulating protein entry into the olfactory cilium. Furthermore, recent reports have identified Ric8B as a putative GEF, which interacts with Golf and increases functional expression of odorant receptors (30, 31). Therefore, it is tempting to hypothesize a role for GEF activity acting in complex at the basal body with CEP290 to regulate protein entry into the cilia. These results, together with our finding that cilia remain intact in rd16 OSNs, suggest that CEP290 plays multiple regulatory roles in protein entry into the cilium. It may act as a docking zone for IFT motors, such as KIF3A, while simultaneously orchestrating peripheral membrane protein trafficking into cilia.
It has been shown previously that, in rd16 mice, mutations in CEP290 result in retinal degeneration of rod, but not cone, photoreceptors (23, 32). Unlike OSNs, where G protein-coupled receptors remain localized to cilia, in rod photoreceptors both rhodopsin and arrestin are mislocalized before any degeneration in rd16 retina (23). This may reflect differences between the rod photoreceptor connecting cilium vs. the terminal cilia of OSNs. Despite these differences, it is apparent that sensory cells are much more susceptible to mutations in CEP290 than other ciliary systems. Nonsense mutations of CEP290 result in Joubert syndrome, characterized by retinal degeneration, cerebellar vermis aplasia, and nephronophthisis, and Meckel syndrome, an autosomal recessive lethal condition characterized by CNS, kidney, and liver malformations (15, 21, 22, 25). Missense mutations (such as those detected in LCA and the rd16 mice), however, appear to only affect sensory systems, resulting in retinal degeneration and olfactory dysfunction in both humans and mice, with no overt kidney or cerebellar defects.
Although mutations in different ciliary proteins can result in analogous sensory phenotypes, the underlying mechanisms appear divergent. Similar to our studies presented here, null mutations in Bbs1, Bbs2, or Bbs4 are reported to result in sensory defects, including anosmia and retinal degeneration (7, 33, 34). However, a striking difference between rd16 and Bbs-null mice is that olfactory cilia remain intact with hypomorphic mutations in CEP290. This is consistent with earlier studies in C. elegans, in which mutations in nephrocystins specifically affected chemosensation and sensory behavior without altering ciliary assembly or maintenance (18, 19, 35). Furthermore, the odorant receptors ACIII and CNGA2 remained localized to OSNs cilia, suggesting that in-frame deletion of CEP290 selectively alters protein transport into the cilia. The fact that cilia remain intact makes the rd16 mouse a powerful model to further investigate the regulatory mechanisms of protein trafficking into cilia.
We postulate that olfactory dysfunction may represent an underappreciated clinical manifestation of ciliary disease. In humans, mutations in members of the nephrocystin family of proteins result in pleiotropic phenotypes, including nephronophthisis, Senior–Loken syndrome, retinitis pigmentosa, hepatic fibrosis, and situs inversus (36–38). However, olfactory function has not been reported in these populations. Importantly, LCA represents a disease in which patients exhibit previously uncharacterized impaired olfactory function unrelated to age or head trauma. Intriguingly, in our study, all CEP290–LCA patients queried before olfactory testing presented with a self-assumed normal olfactory function. This false supposition indicates that olfactory dysfunction may be much more prevalent in patients with ciliary diseases. We suggest that assessment of olfactory function may be a useful screening tool for early diagnosis of pleiotropic ciliopathies.
Materials and Methods
Individuals with CEP290 Mutations.
LCA patients with CEP290 mutations are described in ref. 24.
Antibodies.
Antibodies used in this study were procured as follows: monoclonal anti-acetylated α-tubulin antibody and a polyclonal anti-γ-tubulin antibody (Sigma–Aldrich, St. Louis, MO), anti-Kif3A and anti-p150Glued (BD Biosciences, San Jose, CA), anti-adenylyl cyclase III, anti-Golf polyclonal antibodies, and HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), polyclonal anti-mOR28 (R. Axel, Columbia University, New York, NY; see ref. 39), polyclonal anti-mOR256–14 and mOR262 (H. Breer, University of Hohenheim, Stuttgart, Germany; see ref. 40), polyclonal anti-Gγ13 (R. Margolskee, Mount Sinai School of Medicine of New York University, New York, NY; see ref. 41), polyclonal IFT88 antibody (B. Yoder University of Alabama, Birmingham, AL; see ref. 42), and secondary antibodies conjugated to Alexa Fluor dyes (Invitrogen, Carlsbad, CA). The polyclonal anti-CEP290 and polyclonal anti-RPGR-ORF15CP antibodies are described in ref. 23. Anti-CNGA2 antibody was purchased from Alomone Labs (Jerusalem, Israel).
Western Blots.
Olfactory epithelia were dissected from C57BL/6 WT and rd16 mutant mice and homogenized with a Dounce homogenizer in 50 mM Tris/5 mM EDTA (pH 8.0) containing Complete protease inhibitor tablets (Roche Applied Science, Indianapolis, IN). Insoluble material was removed by centrifugation at 1,000 × g for 5 min. Soluble protein was quantified, and equal amounts of protein were analyzed by SDS/PAGE and immunoblotting using appropriate antibodies. Blots were visualized with the Enhanced Chemiluminescence kit from Pierce (Rockford, IL).
Immunoprecipitation.
Olfactory epithelia were dissected from mice, and coimmunoprecipitation was performed as described (23).
Electro-Olfactograms.
Vapor-phase odor stimuli were generated by placing 10 ml of solution in a sealed 50-ml glass bottle. Odorants were delivered as a 20-ml pulse injected into a continuous stream of humidified air flowing over the tissue sample. Electrodes ranging from 4 to 8 MΩ in resistance were placed on either turbinate II or IIb for recording. We analyzed data with Clampfit (Molecular Devices, Sunnyvale, CA) and determined peak heights from prepulse baseline. Data were normalized to a pulse of pure amyl acetate given later in that same trace. For the rd16 mice, data were normalized to the average response of the WT mouse for a specific dose of the odorant. At least three mice were tested for each condition.
Sectioning and Immunohistochemistry.
After the preparation for electro-olfactograms, the other hemisphere of the mouse head was immersion-fixed in 4% paraformaldehyde for 2–12 h. For cryosections, the tissue was incubated in 30% sucrose in PBS for 24 h at 4°C, frozen in OCT compound (Sakura Finetek, Torrance, CA), and cut into sections (12–14 μm) on a cryostat.
Immunohistochemistry was performed on adult olfactory tissue sections after washing the sections with PBS three times for 5 min each and then incubating them in blocking buffer containing 10% normal serum/0.1% Triton X-100 (Fisher Scientific, Pittsburgh, PA) in PBS for 1 h at room temperature. Sections were then incubated with primary antibody for 1 h at room temperature or overnight (CEP290) at 4°C. We used primary antibodies at the following dilutions: ACIII at 1:500; Gγ13 at 1:500; CEP290 at 1:200; γ-tubulin at 1:500; Golf at 1:200; and acetylated α-tubulin at 1:1,000. Sections were washed in PBS three times for 5 min each at room temperature and visualized by secondary antibody binding with Alexa Fluor-conjugated secondary antibodies. Coverslips were mounted with ProLong Gold (Invitrogen). Images from at least three discontinuous regions from three different mice were collected on an Olympus (Melville, NY) Fluoview 500 confocal microscope with a ×100 1.35 NA oil objective.
TUNEL Staining.
TUNEL staining was performed on at least three discontinuous sections from three different mice for each genotype tested according to the manufacturer's instructions (Millipore, Billerica, MA).
Scanning Electron Microscopy.
The samples were fixed in 2.5% glutaraldehyde in 0.1 M Sorenson's buffer at 4°C overnight. The samples were then washed twice in buffer and dehydrated in ascending concentrations of ethanol, treated with 100% ethanol, and dried in hexamethyldisilazane overnight. Dried samples were mounted on the aluminum stubs and coated with gold by using a Polaron Sputter Coater (Ringmer, U.K.). The samples were examined with an Amray (Drogheda, Ireland) 1910FE field emission scanning electron microscope at 5 kV. Images were recorded digitally with Semicaps software.
Supplementary Material
Acknowledgments
We thank Richard Axel, Gilad Barnea (Columbia University), Heinz Breer, Robert Margolskee, and Bradley Yoder for the generous gift of antibodies; Sasha Meshinchi at the University of Michigan Microscopy and Image Analysis Laboratory for help with the scanning electron microscopy images; and Bo Chang, Frans Cremers, John Heckenlively, Friedhelm Hildebrandt, and Anneke den Hollander for valuable discussions. This research was supported by National Institutes of Health Grants EY007961, EY007003, T32 DC00011, and GM07767, Research to Prevent Blindness, the Foundation Fighting Blindness of USA and Canada, and the Fonds de la Recherche en Santée du Québec.
Abbreviations
- OSN
olfactory sensory neuron
- IFT
intraflagellar transport
- RP
retinitis pigmentosa
- LCA
Leber congenital amaurosis
- GEF
guanine–nucleotide exchange factor
- ACIII
adenylyl cyclase type III.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. R.R.R. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0704140104/DC1.
References
- 1.Rosenbaum JL, Witman GB. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 2.Badano JL, Mitsuma N, Beales PL, Katsanis N. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 3.Davis EE, Brueckner M, Katsanis N. Dev Cell. 2006;11:9–19. doi: 10.1016/j.devcel.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Satir P, Christensen ST. Annu Rev Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 5.Buck L, Axel R. Cell. 1991;65:175–187. doi: 10.1016/0092-8674(91)90418-x. [DOI] [PubMed] [Google Scholar]
- 6.Reed RR. Neuron. 1992;8:205–209. doi: 10.1016/0896-6273(92)90287-n. [DOI] [PubMed] [Google Scholar]
- 7.Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, Lupski JR, Beales PL, Reed RR, Katsanis N. Nat Genet. 2004;36:994–998. doi: 10.1038/ng1418. [DOI] [PubMed] [Google Scholar]
- 8.Zufall F, Munger SD. Trends Neurosci. 2001;24:191–193. doi: 10.1016/s0166-2236(00)01765-3. [DOI] [PubMed] [Google Scholar]
- 9.Belluscio L, Gold GH, Nemes A, Axel R. Neuron. 1998;20:69–81. doi: 10.1016/s0896-6273(00)80435-3. [DOI] [PubMed] [Google Scholar]
- 10.Brunet LJ, Gold GH, Ngai J. Neuron. 1996;17:681–693. doi: 10.1016/s0896-6273(00)80200-7. [DOI] [PubMed] [Google Scholar]
- 11.Wong ST, Trinh K, Hacker B, Chan GC, Lowe G, Gaggar A, Xia Z, Gold GH, Storm DR. Neuron. 2000;27:487–497. doi: 10.1016/s0896-6273(00)00060-x. [DOI] [PubMed] [Google Scholar]
- 12.Scholey JM. Annu Rev Cell Dev Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- 13.Hildebrandt F, Otto E. Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 14.Khanna H, Hurd TW, Lillo C, Shu X, Parapuram SK, He S, Akimoto M, Wright AF, Margolis B, Williams DS, et al. J Biol Chem. 2005;280:33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, et al. Am J Hum Genet. 2007;81:170–179. doi: 10.1086/519494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fliegauf M, Horvath J, von Schnakenburg C, Olbrich H, Muller D, Thumfart J, Schermer B, Pazour GJ, Neumann HP, Zentgraf H, et al. J Am Soc Nephrol. 2006;17:2424–2433. doi: 10.1681/ASN.2005121351. [DOI] [PubMed] [Google Scholar]
- 17.Schermer B, Hopker K, Omran H, Ghenoiu C, Fliegauf M, Fekete A, Horvath J, Kottgen M, Hackl M, Zschiedrich S, et al. EMBO J. 2005;24:4415–4424. doi: 10.1038/sj.emboj.7600885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winkelbauer ME, Schafer JC, Haycraft CJ, Swoboda P, Yoder BK. J Cell Sci. 2005;118:5575–5587. doi: 10.1242/jcs.02665. [DOI] [PubMed] [Google Scholar]
- 19.Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S. Hum Mol Genet. 2005;14:645–566. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- 20.Wolf MT, Lee J, Panther F, Otto EA, Guan KL, Hildebrandt F. J Am Soc Nephrol. 2005;16:676–687. doi: 10.1681/ASN.2003121025. [DOI] [PubMed] [Google Scholar]
- 21.Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, et al. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- 22.Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, et al. Nat Genet. 2006;38:623–665. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- 23.Chang B, Khanna H, Hawes N, Jimeno D, He S, Lillo C, Parapuram SK, Cheng H, Scott A, Hurd RE, et al. Hum Mol Genet. 2006;15:1847–1857. doi: 10.1093/hmg/ddl107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, et al. Am J Hum Genet. 2006;79:556–651. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, Defoort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, et al. Hum Mutat. 2007;28:416. doi: 10.1002/humu.9485. [DOI] [PubMed] [Google Scholar]
- 26.Iannaccone A, Mykytyn K, Persico AM, Searby CC, Baldi A, Jablonski MM, Sheffield VC. Am J Med Genet A. 2005;132:343–346. doi: 10.1002/ajmg.a.30512. [DOI] [PubMed] [Google Scholar]
- 27.Christensen ST, Pedersen LB, Schneider L, Satir P. Traffic. 2007;8:97–109. doi: 10.1111/j.1600-0854.2006.00516.x. [DOI] [PubMed] [Google Scholar]
- 28.Roepman R, van Duijnhoven G, Rosenberg T, Pinckers AJ, Bleeker-Wagemakers LM, Bergen AA, Post J, Beck A, Reinhardt R, Ropers HH, et al. Hum Mol Genet. 1996;5:1035–1041. doi: 10.1093/hmg/5.7.1035. [DOI] [PubMed] [Google Scholar]
- 29.Shu X, Fry AM, Tulloch B, Manson FD, Crabb JW, Khanna H, Faragher AJ, Lennon A, He S, Trojan P, et al. Hum Mol Genet. 2005;14:1183–1197. doi: 10.1093/hmg/ddi129. [DOI] [PubMed] [Google Scholar]
- 30.Von Dannecker LE, Mercadante AF, Malnic B. Proc Natl Acad Sci USA. 2006;103:9310–9314. doi: 10.1073/pnas.0600697103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Von Dannecker LE, Mercadante AF, Malnic B. J Neurosci. 2005;25:3793–3800. doi: 10.1523/JNEUROSCI.4595-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cideciyan AV, Aleman TS, Jacobson SG, Khanna H, Sumaroka A, Aguirre GK, Schwartz SB, Windsor EA, He S, Chang B, et al. Hum Mutat. 2007 doi: 10.1002/humu.20565. [DOI] [PubMed] [Google Scholar]
- 33.Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC. Proc Natl Acad Sci USA. 2004;101:8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, et al. Proc Natl Acad Sci USA. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jauregui AR, Barr MM. Exp Cell Res. 2005;305:333–342. doi: 10.1016/j.yexcr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 36.von Schnakenburg C, Fliegauf M, Omran H. Pediatr Nephrol. 2007;22:765–769. doi: 10.1007/s00467-007-0434-1. [DOI] [PubMed] [Google Scholar]
- 37.Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O'Toole JF, Helou J, Attanasio M, et al. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 39.Barnea G, O'Donnell S, Mancia F, Sun X, Nemes A, Mendelsohn M, Axel R. Science. 2004;304:1468. doi: 10.1126/science.1096146. [DOI] [PubMed] [Google Scholar]
- 40.Strotmann J, Levai O, Fleischer J, Schwarzenbacher K, Breer H. J Neurosci. 2004;24:7754–7761. doi: 10.1523/JNEUROSCI.2588-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang L, Shanker YG, Dubauskaite J, Zheng JZ, Yan W, Rosenzweig S, Spielman AI, Max M, Margolskee RF. Nat Neurosci. 1999;2:1055–1062. doi: 10.1038/15981. [DOI] [PubMed] [Google Scholar]
- 42.Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}