

Abstract
Hypophysiotropic thyrotropin-releasing hormone- (TRH) synthesizing neurons reside in the hypothalamic paraventricular nucleus (PVN) and are the central regulators of the hypothalamic-pituitary-thyroid (HPT) axis. TRH-synthesis and release from these neurons are primarily under negative feedback regulation by thyroid hormone. Under certain conditions such as cold exposure and fasting, however, neuronal inputs from neurons in the brainstem and hypothalamic arcuate and dorsomedial nuclei alter the setpoint for negative feedback through regulation of CREB phosphorylation. Thus, during cold exposure, adrenergic neurons stimulate the HPT axis, while fasting-induced central hypothyroidism is mediated through an arcuato-paraventricular pathway. Feedback regulation of TRH neurons may also be modified by local tissue levels of thyroid hormone regulated by the activation of type 2 iodothyronine deiodinase (D2), the primary enzyme in the brain that catalyzes T4 to T3 conversion. During infection, endotoxin or endotoxin induced cytokines increase D2 activity in the mediobasal hypothalamus, which by inducing local hyperthyroidism, may play an important role in infection-induced inhibition of hypophysiotropic TRH neurons.
Keywords: thyrotropin-releasing hormone (TRH), hypophysiotropic, negative feedback, arcuate nucleus, type 2 deiodinase (D2)
Introduction
Thyrotropin-releasing hormone (TRH) is a tripeptide amide derived from the proteolytic cleavage of an approximately 26,000 MW prohormone (proTRH) [1]. The proTRH protein contains multiple Gln-His-Pro-Gly TRH progenitor sequences [1], five in rats and mice, 6 in humans and 7 in frogs [1]. ProTRH is cleaved by two prohormone convertase enzymes, PC1/3 and PC2, into TRH progenitors and multiple cryptic peptides [2]. The TRH progenitor sequences are subject to further posttranslational modifications: the C-terminal glycine is amidated, while the N-terminal glutamyl residue is converted into pyroglutamyl [3; 4]. The maturation of TRH occurs in the trans-Golgi network and in the vesicles of the regulated secretory pathway [2].
TRH-synthesizing neurons are distributed widely in the central nervous system, including many hypothalamic and brainstem nuclei and also the hippocampus [5]. While TRH serves as a neuromodulator in most of these neuronal groups, in the so called “hypophysiotropic TRH neurons” that regulate anterior pituitary TSH secretion, TRH functions primarily as a neurohormone [1].
Hypophysiotropic TRH and its role in the regulation of the hypothalamic-pituitary axes
Hypophysiotropic TRH neurons are located in the hypothalamic paraventricular nucleus (PVN), a triangular shaped nucleus located at the dorsal limits of the third ventricle [6; 7; 8]. The PVN consists of two major parts: a magnocellular and a parvocellular division [9]. Neurons of the magnocellular division project to the posterior pituitary and contain only rare TRH neurons [5]. The parvocellular division is further divided into anterior, periventricular, medial, ventral, dorsal and lateral parvocellular subdivisions [9]. While TRH-synthesizing neurons are found in all parvocellular subdivisions [5], hypophysiotropic TRH neurons are located exclusively in the periventricular and medial parvocellular subdivisions (fig. 1) [6; 7; 8]. Axons of the hypophysiotropic TRH neurons project to the external zone of the median eminence (Fig. 2) where TRH is released into the perivascular space around the capillary loops of the pituitary portal system [1; 10]. The secreted TRH can then reach its target through the long portal veins that drain to the anterior pituitary [1].
Figure 1.

Distribution of TRH-synthesizing neurons in the PVN. Low power micrographs (A–C) illustrate the TRH neurons at three rostrocaudal levels of the PVN. Schematic drawings (D–F) illustrate the subdivisions of the PVN where hypophysiotropic TRH neurons are localized (gray). AP, anterior parvocellular subdivision; DP, dorsal parvocellular subdivision; LP, lateral parvocellular subdivision; MN, Magnocellular part of PVN; MP, medial parvocellular subdivision, PV, periventricular parvocellular subdivision, III, third ventricle
Figure 2.

Distribution of TRH-immunoreactive (IR) terminals in the median eminence. While TRH-IR axons densely innervate the external zone of the median eminence, only scattered fibers can be seen in the internal zone. III, third ventricle; ME, median eminence
The two, major, TRH sensitive cell types of the pituitary are the thyrotropes and the lactotropes [11]. These two cell types comprise approximately 90% of the total cell population in the anterior pituitary that bind TRH [11]. The first recognized function of TRH is its role in maintenance of thyroid hormone homeostasis through the regulation of TSH secretion from thyrotrops [12; 13]. TRH bound to TRH receptors stimulates the gene expression of TSH subunits through the phosphatidylinositol pathway [14]. In addition, TRH regulates the biologic activity of TSH by modulation of TSH glycosylation [15]. Without TRH, animals have severe hypothyroidism with low circulating TSH and thyroid hormone levels [16]. TRH also stimulates prolactin secretion [17]. Interestingly, however, TRH does not seem to be necessary for the maintenance of basal prolactin secretion, but plays a crucial role in the suckling-induced increase in prolactin release [18].
Negative feedback regulation of hypophysiotropic TRH neurons by thyroid hormone
A common feature of hypothalamic-pituitary-endocrine systems is a closed feedback loop in which peripheral hormones secreted by the target organ exert inhibitory effects on the synthesis of their own hypophysiotropic hormone. Negative feedback regulation is also characteristic of the hypothalamic-pituitary-thyroid-axis [19]. Thyroid hormones inhibit both the gene expression and the posttranslational processing of proTRH in hypophysiotropic neurons [19; 20], but have no effect on other proTRH-synthesizing neuronal groups of the forebrain [20; 21]. When circulating levels of thyroid hormones are low, proTRH gene expression is substantially increased in the PVN (Fig. 3) [21]. This is accompanied by increased synthesis of prohormone convertase enzymes (PC1/3 and PC2) in the PVN and a decrease in the content of TRH in the median eminence due to increased secretion of the mature hormone into the portal circulation [20]. In contrast, during hyperthyroidism, when circulating thyroid hormone levels are increased above normal values, proTRH-mRNA and the synthesis of prohormone convertase enzymes are decreased in the PVN (Fig. 3) [20; 21].
Figure 3.
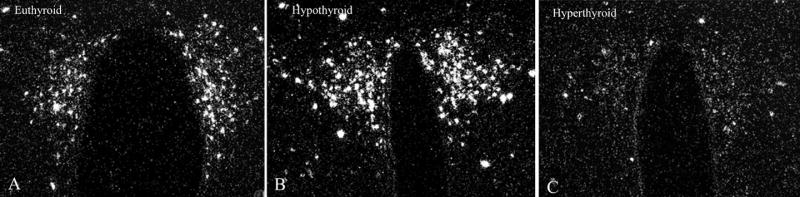
In situ hybridization autoradiograms showing the effect of hypo- and hyperthyroidism on TRH mRNA level in the medial parvocellular subdivision of the PVN. A substantial increase in silver grain accumulation is observed in the hypothyroid animal (B) compared to the fed control (A). In contrast, hyperthyroidism results in a marked reduction of TRH mRNA level in the PVN (C). (From Dyess et al [23], copyright 1988, with permission from The Endocrine Society.)
The feedback effect of thyroid hormones is exerted directly on hypophysiotropic TRH neurons. TRH neurons in the PVN express all isoforms of thyroid hormone receptors (TR) (Fig. 4) [22]. In addition, implantation of microcristals of T3 (the biologically active form of thyroid hormone) in the vicinity of the PVN results in a marked inhibition of TRH gene expression on the side of implantation, without alteration of TRH synthesis on the contralateral side [23]. While both TRα1 and TRβ2 are highly expressed in hypophysiotropic TRH neurons [22], TRβ2 would appear to be the most important thyroid hormone receptor isoform responsible for the feedback regulation of hypophysiotropic TRH neurons [24]. Targeted disruption of TRβ2 expression in mice results in increased TRH mRNA in the PVN, comparable to that observed in hypothyroid wild type (WT) mice [24]. Moreover, neither severe hypo- nor hyperthyroidism have any influence on TRH synthesis in TRβ2 knockout mice, indicating that the TRβ2 isoform is the key mediator of the thyroid hormone negative feedback effect in the PVN [24]. This is in contrast to the pituitary, where ablation of TRβ2 or the entire TRβ allele causes only partial thyroid hormone resistance of TSH secretion [25; 26; 27].
Figure 4.
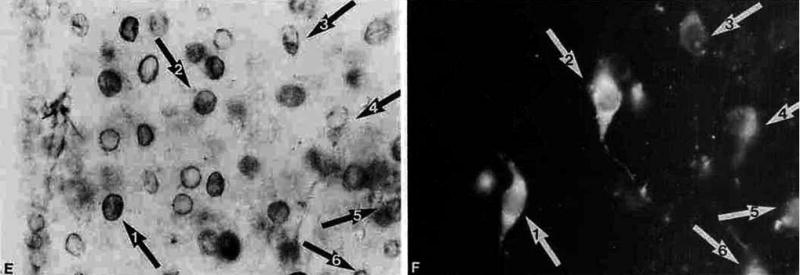
Presence of TRβ2 isoform (A) in the TRH neurons (B) in the PVN. Arrows indicate the neurons containing both TRβ2 and TRH. (From Lechan et al [22], copyright 1994, with permission from The Endocrine Society.)
Thyroid hormones also regulate the synthesis of a second peptide, cocaine- and amphetamine regulated transcript (CART), in hypophysiotropic TRH neurons [8; 28]. Similar to the regulation of TRH, hypothyroidism upregulates, while hyperthyroidism downregulates the synthesis of CART [28]. Since CART and TRH are co-localized in axon terminals of the median eminence [8], the two peptides may be co-released into the portal circulation. While CART has no influence on the stimulatory effect of TRH on the secretion of TSH from thyrotrophs, CART blocks the effect of TRH to stimulate prolactin secretion from lactrotrophs in primary pituitary cell culture [28]. Thus, increased CART secretion in hypothyroid rats seems to permit differential effects of TRH on these two anterior pituitary cell types, allowing stimulation of TSH secretion but attenuation of prolactin secretion.
Neuronal regulation of the set point for negative feedback regulation of hypophysiotropic TRH neurons
Under certain physiological and pathophysiological conditions, negative feedback regulation of TRH neurons in the PVN is altered. Thus, in a cold environment, when increased peripheral levels of thyroid hormones are necessary to stimulate thermogenesis, the TRH mRNA content of hypophysiotropic neurons is increased [29; 30; 31; 32; 33]. This increase occurs despite the accompanied increase in circulating thyroid hormone levels, suggesting that a cold stimulus can increase the set point for negative feedback regulation of thyroid hormone on hypophysiotropic TRH. In contrast, fasting, infection or chronic diseases result in a fall of TRH synthesis with low circulating thyroid hormone levels [34; 35; 36; 37; 38; 39], indicating that these conditions reduce the set point for thyroid hormone feedback.
Feedback sensitivity of TRH neurons is altered by neuronal afferents that densely innervate hypophysiotropic TRH neurons. Numerous synaptic contacts between the axon terminals and the perikarya and dendrites of TRH-synthesizing neurons have been observed, including both the symmetric type, suggesting inhibitory control and the asymmetric type, suggesting excitatory control [1]. For example, hypophysiotropic TRH neurons are densely innervated by axons containing glutamic acid decarboxylase (GAD), the synthesizing enzyme of GABA that envelop, sometimes even obscure the perikarya and dendrites of TRH neurons due to the large number of contacts [40]. Ultrastructurally, GAD-IR axon varicosities establish symmetric type synapses on the perikarya and dendrites of TRH neurons [40]. On the other hand, a large number of excitatory terminals containing the vesicular glutamate transporter-2 (VGLUT2), a marker of glutamatergic axons, also form synaptic specializations with the TRH neurons [41]. In addition, adrenalin and a wide variety of neuropeptides (agouti-related protein (AGRP), neuropeptide Y (NPY), α-melanocyte stimulating hormone (α-MSH), cocaine- and amphetamine regulated transcript (CART), galanin, TRH, corticotrophin-releasing hormone (CRH), pituitary adenylate cyclase-activating polypeptide and somatostatin) are present in the afferents of TRH neurons [8; 42; 43; 44; 45; 46; 47; 48; 49].
Currently, three main sources of the input to TRH neurons are known, C1-3 adrenergic neurons of the brainstem, the hypothalamic arcuate nucleus and neurons of the hypothalamic dorsomedial nucleus [8; 42; 43; 50; 51]. These three neuronal groups mediate the effects of different physiological stimuli exerted on hypophysiotropic TRH neurons as described below.
Adrenergic input from the C1-3 brainstem areas
The ascending input from the brainstem adrenergic neurons are believed to mediate the stimulatory effects of cold exposure on the hypophysiotropic TRH neurons [32; 33]. Neurons of the C1-3 region send direct input to the vast majority of hypophysiotropic TRH neurons and form asymmetric type, stimulatory synapses [42]. Furthermore, the cold-induced rapid peak of TRH synthesis is associated with an increase in the hypothalamic concentration of catecholamines [31; 52], and the rise in circulating thyroid hormone levels with cold exposure does not occur within the first 10 days after birth in the rat when the hypothalamic catecholamine innervation is still immature [53]. Since an increase in thyroid hormone would ordinarily inhibit TRH gene transcription at the level of the PVN through negative feedback effects, catecholamines are believed to increase the setpoint for inhibition of TRH gene expression by T3, thereby permitting high circulating levels of thyroid hormone to contribute to increased thermogenesis. The intracellular interaction of the thyroid hormone and the adrenaline effects seem to converge at the level of the TRH promoter. Catecholamines act on TRH neurons primarily through α1 adrenergic receptors [33]. When activated, these receptors can induce the phosphorylation of CREB [54] that activates the TRH promoter by binding to a CREB response element (CRE) in the promoter [55], commonly referred to as Site 4. Site 4 also functions as a binding site for thyroid hormone receptors [55; 56]. While phosphorylated CREB (PCREB) recruits CREB-binding protein (CBP) to induce transcriptional activation through Site 4 of the promoter [55], however, liganded thyroid hormone receptors inhibit TRH transcription through the same site [56]. Therefore, it is conceivable that with cold exposure, increased levels of PCREB may compete with thyroid hormone receptors for binding to Site 4, thereby increasing the set point for feedback regulation by thyroid hormone (Fig. 5).
Figure 5.

Genomic and promoter structure of TRH. The murine, rat and human TRH genes are composed of three exons and two introns. The coding sequence for the precursor protein is present on exons 2 and 3. As depicted, the TRH promoter region precedes the transcription start site in exon 1. The proximal 250 bp sequences of the human, mouse and rat promoters are similar and share the indicated transcription factor binding sites. The location of the CREB binding site (Site 4) and sequences in human (H), mouse (M) and rat (R) are shown. (B and C) Hypothesized schematic representation of the interaction between PCREB and the thyroid hormone receptor at Site 4. (B) illustrates that in the presence of abundant PCREB, there may be less availability for binding of the thyroid hormone receptor/T3 complex, hence, an increase in TRH gene transcription. When PCREB concentrations fall as shown in (C), increased binding of the thyroid hormone receptor/T3 complex reduces TRH gene transcription. (From Lechan and Fekete [62], copyright 2006, with permission from Elsevier.)
Adrenergic fibers in contact with TRH neurons co-contain at least two neuropeptides, CART and NPY [57; 58]. CART is contained in approximately 50% and NPY in 75% of the adrenergic innervation to TRH neurons [57; 58]. The potential role of these peptides in response to cold has not been explored. However, similar to adrenaline, CART also exerts a stimulatory effect on the synthesis and release of TRH [8] and thereby may potentiate the action of adrenaline on the TRH neurons during cold exposure. In contrast, NPY exerts a potent inhibitory effect on the transcription of the TRH gene [59] through inhibition of the cAMP-CREB second messenger pathway [60]. The physiologic purpose of axons releasing both stimulatory and inhibitory regulatory signals for hypophysiotropic TRH neurons is uncertain. However, it is intriguing to hypothesize that NPY may play a role in the termination of the short term increase of TRH synthesis during cold exposure, either through direct actions on TRH neurons or by acting presynaptically on adrenergic axons. NPY can increase the number of presynaptic α2 catecholaminergic receptors and thus, may inhibit the release of adrenaline by potentiating the presynaptic effects of catecholamines [61]. Co-secretion of NPY from catecholamine terminals may also be important during infection when hypophysiotropic TRH neurons are inhibited (see below), as a mechanism to antagonize the increased release of adrenaline in the PVN.
A second site where the adrenergic fibers could influence hypophysiotropic TRH neurons is the median eminence [33]. Adrenergic axons terminate in close proximity to TRH terminals in the median eminence [33]. Moreover, superfusion of median eminence fragments with the α1-receptor agonist, phenylephrine, induces a significant rise in TRH release [33], demonstrating the direct effect of catecholamines at the level of TRH terminals.
Input from the feeding-related neurons of the arcuate nucleus
The arcuate nucleus sends a prominent peptidergic input to hypophysiotropic TRH neurons. The best understood function of the arcuato-paraventricular pathway in the regulation of HPT axis is the mediation of fasting-induced central hypothyroidism [62]. Fasting is associated with decreased synthesis of thyrotropin-releasing hormone (TRH) in the PVN and low TSH and thyroid hormone levels in the bloodstream [34; 35; 36]. Since thyroid hormones stimulate mitochondrial oxygen consumption and increase thermogenesis [63; 64], the reduction in circulating levels of thyroid hormone during fasting is presumed to be an important mechanism to conserve energy. Since peripheral administration of leptin, an adipose tissue-derived satiety factor [65], prevents fasting-induced hypothyroidism (Fig. 6), the fall of peripheral leptin levels is considered the primary peripheral signal that induces inhibition of the HPT axis [36]. The effect of leptin on TRH neurons is primarily mediated through the arcuate nucleus because chemical ablation of arcuate nucleus neurons prevents fasting-induced central hypothyroidism and also the stimulatory effects of leptin on TRH gene expression in hypophysiotric neurons [66].
Figure 6.

Darkfield illumination photomicrographs of proTRH mRNA in the hypothalamic PVN in fed (A), fasted (B), and fasted animals receiving leptin (C). Note the marked reduction in silver grains over neurons in the PVN in the fasted animals but restoration to normal in the fasted animals receiving leptin. III, third ventricle (From Legradi et al, copyright 1997 [36], with permission from The Endocrine Society.)
The arcuate nucleus contains at least two, distinct populations of neurons that are directly regulated by leptin [67; 68; 69; 70; 71], involved in the regulation of feeding behavior [71; 72], and project to TRH neurons in the PVN [8; 43; 44; 50]. The first is a medially located neuronal group that synthesizes and co-contains the two, potent, orexigenic peptides, NPY and AGRP, that are inhibited by leptin [73]. The second is a more laterally located neuronal group that synthesizes and co-contains the anorectic peptides, α-MSH and CART, that are stimulated by leptin [8; 69; 74; 75]. Both NPY and AGRP-containing fibers densely innervate all TRH neurons in the PVN (Fig. 7) [44; 45]. Approximately 80% of NPY and 100% of AGRP innervation of hypophysiotropic TRH neurons originate from the arcuate nucleus AGRP/NPY neurons [44; 50]. The ultrastructural characteristics of NPY- and AGRP-containing synapses on hypophysiotropic TRH neurons [44; 45], indicate an inhibitory nature of these contacts. Indeed, central administration of either AGRP or NPY to freely fed animals reproduces central hypothyroidism observed in fasted animals (Fig. 8) despite that AGRP- and NPY-treated animals have significantly increased food intake and elevated leptin levels [76]. The inhibitory effect of AGRP is mediated primarily through the melanocortin-4 receptor (MC4-R) [77], whereas Y1 and Y5 NPY receptors are involved in the inhibition of HPT axis by NPY [78].
Figure 7.
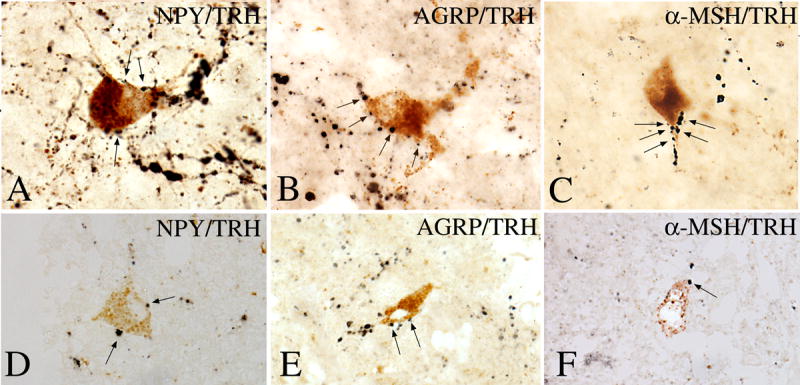
The association between axon terminals containing NPY, AGRP, and α-MSH-immunoreactivity (arrows) with TRH-positive perikarya in the PVN of human hypothalamus is shown in A–F. A multipolar TRH-IR neuron is heavily contacted by NPY-IR axon terminals (A). AGRP-IR axon varicosities establish numerous axo-somatic and axo-dendritic connections with a TRH-positive cell (B). α-MSH-IR axon varicosities densely cover the dendrite of a TRH-containing neuron (C). Similar associations between NPY-, AGRP-, and α-MSH-IR axon terminals and TRH-containing cells are shown in 1-μm thick sections in D–F. (From Mihály et al [160], copyright 2000, with permission from The Endocrine Society.)
Figure 8.
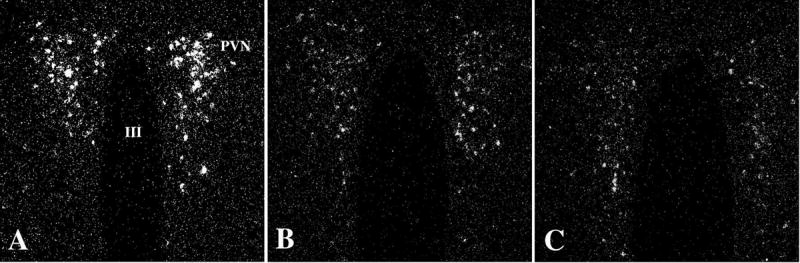
Darkfield illumination photomicrographs of proTRH mRNA in the medial parvocellular subdivision of the PVN in control (A), AGRP- (B), and NPY-treated (C) animals. Note marked reduction in silver grains over neurons in the PVN in both the AGRP- and NPY-infused groups. III, Third ventricle (From Fekete et al [76], copyright 2002, with permission from The Endocrine Society.)
Approximately 70% of TRH neurons in the periventricular parvocellular subdivision and 34% of TRH neurons in the medial parvocellular subdivision of the PVN are innervated by α-MSH-containing fibers (Fig. 7) [43]. Since all α-MSH-fibers in the PVN co-contain CART and α-MSH and CART are co-synthesized only in cells of the arcuate nucleus [8], the exclusive source of the α-MSH innervation of hypophysiotropic TRH neurons is the arcuate nucleus [8; 43]. In contrast, the arcuate nucleus is the origin of a relatively small proportion of CART fibers innervating the TRH neurons [8; 79]. Most CART fibers in contact with TRH neurons do not contain α-MSH [8] but rather originate from brainstem sources [80]. As opposed to NPY and AGRP, central administration of α-MSH or CART have stimulatory effects on proTRH gene expression [8; 43; 81] and prevent fasting-induced inhibition of proTRH mRNA in PVN (Fig. 9) [8; 43].
Figure 9.

Dark-field illumination micrographs of pro-TRH mRNA in the medial and periventricular parvocellular subdivisions of the hypothalamic PVN in fed (A, D) and fasted (B, E) animals and fasted animals receiving an intracerebroventricular infusion of α-MSH (C) or CART (F) every 6 hr for 64 hr. Note the reduction in the accumulation of silver grains over the PVN in fasted animals compared with the fed controls. Both α-MSH and CART administration prevent the fasting induced fall of TRH mRNA levels. III, Third ventricle (From Fekete et al, copyright 2000, with permission from The Endocrine Society.)
Both of the functionally antagonistic neuronal groups in the arcuate nucleus converge on the hypophysiotropic TRH neurons [43]. All hypophysiotropic TRH neurons that are innervated by fibers of the α-MSH/CART neurons are also innervated by AGRP/NPY fibers [43]. Therefore, both populations are in anatomical position, poised to fine tune the activity of hypophysiotropic TRH neurons at the level of a single TRH cell. Thus, during fasting, low circulating levels of leptin allow upregulation of NPY and AGRP and inhibition α-MSH and CART gene expression in arcuate nucleus neurons and hence, inhibition of proTRH gene expression. Conversely, after restoration of energy stores, normalized leptin levels decrease NPY and AGRP and increase α-MSH and CART synthesis in arcuate neurons and therefore, stimulate TRH gene expression in the PVN.
Two of the arcuate nucleus-derived peptides involved in hypophysiotropic TRH regulation are endogenous antagonists to each other [82]; α-MSH is an agonist of the melanocortin 3 and 4 receptors [82] whereas AGRP acts as an antagonist of these receptors [82]. Since MC3 and 4 receptors are coupled to Gs proteins [83], activation of these receptors by α-MSH can increase intracellular cAMP synthesis and result in increased CREB phosphorylation [83]. In accordance, central administration of α-MSH to fasted animals markedly increases the number of PCREB-containing TRH neurons in the PVN [84]. In contrast, AGRP decreases cAMP production through the same receptors [82]. While NPY and the melanocortins act through different receptors, the two peptidergic systems can interact at post-receptor level. Both NPY Y1 and Y5 receptors [78] are coupled to Gi proteins that lead to inhibition of cAMP production [85]. Thus, when NPY is administered centrally to fasted animals, it has little effect on the number of TRH neurons in the PVN that contain PCREB [60], but if administered to fasted animals prior to α-MSH-treatment, it markedly attenuates α-MSH-induced CREB-phosphorylation [60]. These data suggest, that the intracellular effects of α-MSH, AGRP and NPY are mediated through the cAMP-CREB second messenger system. Thus, similar to adrenaline, these peptides may also alter the set point of the thyroid hormone negative feedback machinery on hypophysiotropic TRH through modulation of PCREB.
Since the receptor of CART is still unknown, it is not clear how CART influences the activity of the TRH promoter. The fact that CART does not affect CREB phosphorylation in hypophysiotropic TRH neurons [86] suggests that CART utilizes a different second messenger system than the above peptides.
Harris et al. [87] have suggested that leptin also influences hypophysiotropic TRH neurons through direct effects. This hypothesis is supported by the findings that leptin administration induces expression of SOCS-3 mRNA, a marker of leptin receptor activation [87], in a small subset of TRH neurons in the PVN [87]. Moreover, Guo et al. [88] have reported that in vivo, leptin stimulates the binding of phosphorylated STAT3, a second messenger involved in leptin signaling [89], to the TRH promoter in PVN samples. The recent study by Perello et al. [90], showing that the leptin-induced decrease in TRH content in the median eminence and the leptin-induced increase in peripheral TSH level can be prevented by the pretreatment of animals with a melanocortin receptor antagonist, suggests that any direct action of leptin may have only a minor role in regulation of the HPT axis [90].
Neurons of the arcuate nucleus are also sensitive to insulin and glucose [91; 92; 93; 94; 95]. Similar to leptin, circulating levels of these anorexigenic substances fall during fasting [96; 97], raising the possibility that insulin and glucose may also be involved in the regulation of HPT axis during fasting through the arcuato-PVN pathway. Interestingly, however, leptin, insulin and glucose exert characteristically different effects on the hypophysiotropic TRH neurons [75]. While central administration of leptin to fasted animals completely prevents fasting-induced inhibition of TRH gene expression in the PVN, insulin has no effect and glucose has an inhibitory effect on the TRH neurons [75]. This discrepancy may be explained by the different effects each of the three substances exert on peptide synthesis in arcuate nucleus neurons [75]. In contrast to the effect of both centrally and peripherally administered leptin to inhibit NPY- and AGRP- and stimulate CART- and POMC-gene expression in the arcuate nucleus of fasted rats [75; 91], central insulin administration effects only NPY and POMC gene expression and has no effects on AGRP and CART mRNA levels [75]. Central glucose administration has an even more restricted effect, restoring only NPY gene expression in fasting rats without altering AGRP, POMC and CART-synthesis [75]. Since central AGRP administration to fed animals can mimic the effect of fasting on the HPT axis [77], and insulin has no effect on the HPT axis despite restoration of NPY and POMC gene expression, we hypothesize that inhibition of melanocortin receptors by increased AGRP synthesis and release plays a critical role in fasting-induced inhibition of hypophysiotropic TRH neurons.
Afferents from the hypothalamic dorsomedial nucleus
The hypothalamic dorsomedial nucleus (DMN) may also serve as important metabolic sensor for hypophysiotropic TRH neurons. Anterograde tract tracing studies demonstrate that the DMN projects to the vast majority of TRH-producing neurons in the medial parvocellular PVN [51]. In addition, following the systemic administration of leptin, cfos is more highly expressed in the DMN than any other region of the brain [98], overlapping with the location where α-MSH-containing axon terminals derived from arcuate nucleus neurons heavily innervate the DMN [99]. When the retrogradely transported marker substance, CTB, is injected into the PVN, numerous neurons are retrogradely labeled in the dorsal and ventral subdivisions of the DMN and a large number receive contacts by axon terminals containing α-MSH [100]. Thus, α-MSH may influence pathways that regulate TRH neurons through the DMN.
The necessity for both direct arcuate-PVN and indirect arcuate-DMN-PVN pathways to TRH neurons remains uncertain, and may simply be an alternative pathway by which leptin can regulate hypophysiotropic TRH. It is conceivable, however, that the multisynaptic pathway may have special function by influencing the circadian secretion of the HPT axis. Plasma levels of TSH and thyroid hormone are well known to have a diurnal variation in experimental animals and man [101; 102; 103] and this has also been shown for TRH mRNA expression in PVN neurons [104]. While multiple factors may contribute to the rhythmicity of thyroid hormone [105], convincing evidence would suggest the importance of the suprachiasmatic nucleus (SCN) [103]. Lesions of the SCN eliminate the diurnal peak of TSH and thyroid hormones and decrease 24h mean T4 levels [103], and recent studies have shown a major projection from neurons in the ventral subparaventricular zone (SPV), one of the main output areas of the SCN [106], to the DMN [107]. As only ~30% of TRH neurons in the medial parvocellular subdivision of the PVN are innervated by axons containing α-MSH, it is possible that α-MSH regulates hypophysiotropic TRH primarily via the DMN, and that it modulates the amplitude of the circadian rhythm generated by the SCN-SPV-DMN pathway.
Role of type 2 deiodinase (D2) in the regulation of hypophysiotropic TRH neurons
The major secretory product of the thyroid gland is thyroxine (T4), which then functions as a prohormone for T3 in most tissues [108]. While both forms of thyroid hormone are present in the circulating blood, trafficking of the less active T4 into the brain and then central thyroid hormone activation by conversion to T3 is necessary for the normal function of the thyroid hormone feedback mechanism [109]. As demonstrated by Kakucska et al. [109], restoration of normal peripheral T3 levels in hypothyroid rats by the administration of T3, alone, is not sufficient to inhibit increased TRH gene expression in hypothyroid rats. Only elevated, hyperthyroid levels of peripheral T3 can restore TRH mRNA in hypophysiotropic neurons to the euthyroid level (Fig. 10) [109]. Furthermore, Hagen and Solberg [110] have demonstrated that T4 is transported into the brain much more efficiently than T3. Indeed, more than 80% of T3 in the brain originates from local T4 to T3 conversion [111]. This process requires an enzymatic, outer ring deiodination of T4 [108], and in the central nervous system, it is primarily catalyzed by type 2 iodothyronine deiodinase (D2) [108].
Figure 10.

In situ hybridization autoradiographs of proTRH mRNA in the paraventricular nucleus (PVN) in (A) hypothyroid, (B) euthyroid and (C) hypothyroid animals receiving a constant infusion of (C) 0.5 μg or (D) 0.75 μg of T3/100 gm/bw/d. Mean plasma triiodothyronine (T3) levels (±SEM) are shown for each group at the bottom of the photomicrographs. Note that only the higher dose of T3 that raised plasma T3 levels into the supranormal range was capable of suppressing TRH mRNA to euthyroid levels. (E) Regression analysis of above experiment. Interrupted line represents the mean ln(TRH mRNA) for euthyroid animals and its intercept with the regression line estimates the plasma T3 concentration required to suppress proTRH mRNA to euthyroid levels. Ninety-five percent confidence intervals for each intercept are bracketed. Open dots denote values for hypothyroid animals and hypothyroid animals infused with graded doses of T3. Closed dots denote values for euthyroid controls. (Modified from Kakucska et al [109], copyright 1992, with permission from The Endocrine Society.)
Within the hypothalamus, D2 activity is highly concentrated in a discrete region of the mediobasal hypothalamus (MBH), specifically in the median eminence, arcuate nucleus, ventromedial nucleus (VMN) region [112]. This distribution pattern of D2 is opposed to the uniform distribution of D2 activity in hippocampal and cortical regions [112]. By in situ hybridization histochemistry, the most intense D2 hybridization signal in the rat hypothalamus is located in the mediobasal hypothalamus between the rostral and caudal poles of the median eminence and the infundibular recess [113; 114; 115]. In this region, D2 mRNA is localized primarily in the floor and infralateral walls of the third ventricle, abruptly ceasing 1/2 to 2/3 up the third ventricular wall and extending into the adjacent arcuate nucleus in long cytoplasmic processes that envelop blood vessels (Fig. 11) [113]. No hybridization is present in the roof of the infundibular recess or in ependymal cells in other regions of the third ventricle rostral to the anterior pole of the median eminence [113]. Hybridization is also present overlying a cell-clear zone above the tuberoinfundibular sulci, and in the substance of the median eminence and pituitary stalk where it extends into the external zone adjacent to the portal capillaries [113]. A similar distribution of D2 or D2 mRNA has been identified in the mouse (personal observations), chicken [116] and human hypothalamus [117]. No hybridization is seen in other regions of the hypothalamus, including the paraventricular nucleus [113].
Figure 11.

Distribution of type 2 deiodinase mRNA in tanycytes lining the wall of the third ventricle. Low power micrographs of a rostral (A) and a caudal (B) section show that silver grains denoting type 2 deiodinase mRNA are accumulated over the cells lining the wall of the third ventricle, the tuberoinfundibular sulci (arrow heads) and around blood vessels in the arcuate nucleus (arrows). Note the absence of the hybridization signal in the upper third of the ventricle (open arrow). Higher power micrographs shows the association of the silver grains with the tuberoinfundibular sulcus (arrow heads) and a blood vessel (arrows) in the arcuate nucleus (C), and the presence of type 2 deiodinase mRNA in the external zone of the median eminence (D). (Modified from Fekete et al [118], copyright 2000, with permission from Elsevier.)
Double-labeling techniques using a specific marker for tanycytes, including dopamine and cAMP-regulated phosphoprotein (DARPP-32) and vimentin, have established that tanycytes are one of the major sources for hypothalamic expression of D2 [115; 118]. D2 mRNA is expressed in both the cell bodies, processes and end feet of tanycytes [118]. The role of tanycytes in D2 synthesis has been verified using immunocytochemical techniques [119].
Tanycytes are specialized ependymal cells lining the floor and the infralateral wall of third ventricle between the rostral and caudal poles of the median eminence and the infundibular recess [120]. The long apical processes of these cells terminate on neurons and blood vessels of the median eminence-arcuate nucleus-VMN regions or on the surface of the tuberoinfundibular sulci [120]. While excellent morphological descriptions of tanycytes are present in the literature [121; 122; 123], their physiological function remains poorly understood. Originally believed to only serve as part of the blood-brain barrier, it is now believed that tanycytes have much more complicated functions and likely have an active role in endocrine regulation [120]. Based on the morphological characteristics of these cells and their ability to extract substances from the CSF and also from blood, tanycytes may provide a bi-directional, cytoplasmic conduit between the CSF and the vascular elements in the arcuate nucleus and/or the median eminence, allowing the movement of substances from one compartment to the other [124]. Moreover, tanycytes receive synaptoid-like contacts from neurons and express a long list of cytokine and hormone receptors [122; 125; 126; 127; 128; 129; 130; 131], further suggesting that tanycytes have a complex role in the regulation of hypothalamic circuits.
In addition to tanycytes, D2 is also expressed in astrocytes within the hypothalamus [119], characteristic of other D2-expressing regions of the brain such as the cerebral cortex [115; 119]. Using immunocytochemical techniques, Diano et al [119] have shown D2 immunostaining in astrocytes in the arcuate nucleus. Similar to observations made by in situ hybridization histochemistry, no D2 immunostaining has been identified in neurons.
Regulation of D2 activity in the hypothalamus by thyroid hormone
The major function of the D2 in the brain is to regulate local bioavailabilty of T3. In many tissues, including the cortex and hippocampus, D2 is regulated in a way to maintain constant, local T3 levels despite the changes in local T4 concentration [132]. This function is important for neuronal proliferation, organization, arborization, synapse formation, migration, myelination during development and also for the normal performance of neuronal circuits in the adult brain [133; 134]. While the regulation of D2 in the hypothalamus by circulating levels of thyroid hormone would seem to parallel that observed in the cortex [113; 132; 135], differences have been described. For example, in some studies, hypothyroidism induces only a moderate increase in D2 mRNA in the hypothalamus of approximately 1.3 to 1.6-fold over euthyroid levels [113; 114] and no increase in D2 activity [114]. Similarly, no increase in D2 activity in the hypothalamus has been observed in association with iodine deficiency, contrary to other regions in the brain [136]. Therefore, it is not surprising that the systemic infusion of high doses of T4 into hypothyroid rats increases tissue and nuclear T3 content in the hypothalamus rather than maintaining normal levels as observed in other regions of the brain [132].
Role of D2 in the regulation of HPT axis under basal conditions
The relative insensitivity of D2 expression in the hypothalamus to thyroid hormones indicates that the maintenance of a constant T3 tissue level is not the main function of D2 in this region, but rather that D2 may allow T3 to serve as a regulatory signal for specific hypothalamic functions. Thus, in the case of hypophysiotropic neurons that do not express D2 [113], T3 produced by the tanycytes may be the primary source of T3 involved in feedback regulation of these neurons. The location of tanycytes and high concentration of D2 mRNA and D2 enzymatic activity in these cells, places them in a strategic position to extract T4 from the bloodstream by tanycyte end feet processes terminating on portal capillaries or on blood vessels in the arcuate nucleus, or from the CSF via apical specializations after T4 has traversed the choroid plexus [137]. T4 could then be converted to T3 in the cytoplasm of tanycytes and released back into the CSF. T3 released into the CSF could then diffuse into the substance of the brain by volume transmission [138], moving between ependymal cells lining the third ventricle, and provide a source of T3 to TRH neurons in the hypothalamic PVN [113]. Alternatively, tanycytes may release T3 into the median eminence for uptake and retrograde transport by axon terminals of hypophysiotropic TRH neurons, or into the arcuate nucleus through tanycyte-neuronal interactions, where it could influence the activity of arcuate nucleus neurons that have known projections to TRH neurons in the PVN [8; 43; 44; 50; 139]. It is presumed, therefore, that the relatively stable level of D2 activity in the tanycytes under basal conditions and with changes in circulating thyroid hormone levels, is critical for the normal negative feedback regulation of the HPT axis to allow hypophysiotropic TRH neurons to sense any changes in T4 output by the thyroid gland.
Role of D2 in the regulation of the hypophysiotropic TRH neurons during infection
While the suppression of the HPT axis during fasting is induced primarily by the falling peripheral leptin levels via alteration of neuronal activity in the arcuato-paraventricular peptidergic pathway [140], this mechanism does not appear to explain suppression of the HPT axis during infection. Infection increases α-MSH and CART synthesis and does not change the expression of NPY in the arcuate nucleus [141; 142; 143]. Since both α-MSH and CART have stimulatory effects on TRH synthesis [8; 43], the arcuato-paraventricular pathway would be expected to stimulate the TRH neurons during infection. In addition, by activating the catecholamine-containing neurons in the medulla [144; 145], infection would also be expected to stimulate TRH-synthesis through the ascending brainstem pathways. Despite the increased activity of these known stimulatory inputs, TRH synthesis is inhibited during infection [39]. Presumably, therefore, more potent, inhibitory, regulatory controls over the hypothalamic-pituitary-thyroid axis that supercede the stimulatory action of each of these substances on hypophysiotropic TRH must come into play under these conditions. This overriding effect does not appear to be due to increased level of glucocorticoids, since adrenalectomized animals replaced with corticosterone to simulate normal basal levels, still show suppression of TRH and TSH mRNA levels following LPS [39].
While the neuronal inputs of hypophysiotropic TRH neurons have an important role in establishing the set point for negative feedback regulation of thyroid hormone, it is conceivable that the negative feedback system could also be altered by regulating local hypothalamic T3 concentrations independently from the alterations in peripheral thyroid hormone levels. Indeed, D2 activity in the rat MBH is substantially increased by immune activation [146; 147]. Following the systemic administration of bacterial endotoxin (LPS), D2 activity in the MBH shows an early maximal response at 12 h, followed by a decline at 24 h [146; 147]. Similar findings have also been observed in mice [148]. The increase in D2 activity in this brain region is due to increased D2 gene expression exclusive to tanycytes as shown by semiquantitative in situ hybridization histochemistry (Fig. 12) [146]. LPS induces a 4-fold increase in D2 mRNA content in the MBH but also in D2 activity, suggesting that the posttranslational processing of D2 remains relatively unchanged under these conditions [146]. Because the pattern of D2 activation in tanycytes after LPS differs from the more gradual, linear or stepwise increase in D2 activity in the anterior pituitary and cortex that is maximal at 24 h after LPS coincident with the maximal fall in circulating thyroid hormone levels, D2 activation in the MBH may not be induced by hypothyroidism, but rather, influenced by a unique set of regulatory controls. Support for this hypothesis is given by studies showing that LPS administration to T4 clamped animals also induces a 4-fold increase in D2 activity in the MBH 12h after the treatment, despite the lack of hypothyroidism in this animal model [147]. In contrast, the T4 clamp prevents the LPS-induced increase in D2 activity in the cortex and anterior pituitary, demonstrating in these two areas, the LPS-induced increase in D2 activity is the result of falling thyroid hormone levels [147].
Figure 12.

Low-power dark-field micrographs from two different rostrocaudal levels of the median eminence (ME) showing the effect of LPS treatment on D2 gene expression in the MBH. A–B, Controls; C–D, LPS-treated animals. Silver grains denoting D2 mRNA are accumulated over cells lining the wall of the third ventricle (III), the tuberoinfundibular sulci (arrow heads), and accumulate in the external zone of the ME. After LPS administration, the density of silver grains denoting D2 mRNA is markedly increased, particularly in the external zone of the ME (C–D). (Modified from Fekete et al [146], copyright 2004, with permission from The Endocrine Society.)
The mechanism for LPS-induced increase in D2 gene expression in tanycytes requires further study. However, the presence of TNF-1 receptor (p55) mRNA in tanycytes in normal rats and its increase by endotoxin administration [125], and the ability of TNFα in GH3 cells and LPS in mesothelioma cells to increase endogenous D2 activity in culture [149; 150], support the concept that LPS or LPS-induced proinflammatory cytokines could directly activate D2 in tanycytes.
NF-κB plays a key role in the signaling of LPS, TNFα, and other cytokine receptors [151; 152], raising the possibility that NF-κB may mediate the effects of cytokines on the promoter of D2 gene (dio2). Indeed, co-transfection of p65, an essential component of the NF-κB complex, together with a human dio2 promoter 5’flanking region-luciferase construct, results in an approximately 150-fold increase of hdio2 promoter activity [146; 149]. By truncation analyses, the NF-κB responsiveness of hdio2 promoter appears to reside in the first 900 basepairs of the 5’flanking region of dio2 promoter [149]. Within this region, computerized analyses have identified 6 potential NF-κB response elements [149], but using semiquantitative electrophoretic mobility shift assay (EMSA), only two bind NF-κB [149]. Despite similar NF-κB binding affinity of these two sites, site-directed mutagenesis and promoter assay indicates that only one site at position –198 bp 5' to the transcriptional starting site (no. 5 site) possesses transactivation potency in the presence of the p65 subunit of NF-κB [149]. Other cytokine mediators such as signal transducer and activator of transcription-3 (STAT3) or signal transducer and activator of transcription-5 (STAT5), do not induce transcription of the dio2 gene [149]. These data suggest that cytokines stimulate D2 expression predominantly via the NF-κB pathway.
On the basis of the above, it is reasonable to presume that endotoxin or cytokines may result in tissue-specific D2-mediated thyrotoxicosis in the mediobasal hypothalamus caused by increased T4 to T3 conversion in tanycytes. The increase in T3 concentration may suppress the synthesis of TRH in hypophysiotropic neurons by feedback regulation. In addition, T3 may also be released into the portal capillary system for conveyance to the anterior pituitary and exert direct effects on anterior pituitary thyrotrops to inhibit the secretion of TSH.
D2 in fasted animals
Similar to infection, fasting also results in an increase of D2-synthesis in the MBH. Diano et al. [114] have shown an approximately 2-fold increase in D2 mRNA level in the MBH of rats fasted for 3 days. This increase is accompanied by an approximately 1.6-fold increase in D2 activity in the MBH [114]. The fasting induced increase of D2 activity is independent of changes in peripheral thyroid hormone levels, but rather the result of the fasting-induced inverse shift in circulating corticosterone and leptin levels [153]. As both WT and TRβ2-KO mice show a similar reduction in TRH mRNA levels in the PVN [24], it is unlikely that changes in local thyroid hormone availability during fasting have major direct effects on the hypophysiotropic TRH neurons. The increase of T3 concentration in the hypothalamus, however, may influence the HPT axis through effects on arcuate nucleus neurons. Tanycyte end processes surround many thyroid hormone receptor-containing neurons in the arcuate nucleus that synthesize AGRP and NPY [154]. Therefore, an increase in T3 in the vicinity of AGRP/NPY neurons may inhibit hypophysiotropic TRH neurons by modulating the activity of these neurons that have direct projections to the PVN.
Role of thyroid hormone transporters in the regulation of hypophysiotropic TRH neurons
Despite their lipophylic nature, the transport of thyroid hormones into the cells requires active transport [155]. A long list of transporters have been demonstrated to facilitate the transport of thyroid hormones through the cell membrane [155]. The two most important transporter families that are involved in the thyroid hormone transport in the brain are the organic anion transporting polypeptide (OATP) and the monocarboxylate transporter (MCT) families. One member of the OATP family, OATP 14, is expressed in the PVN [156], and OATP1C1, which has widespread distribution in the brain, shows high affinity as a transporter to both T4 and T3 [157]. Regulation of OATP1C1 by thyroid hormone has been described, but currently little is known about the exact function of this transporter in the regulation of T3 availability in the brain and in hypophysiotropic TRH neurons. In contrast, several lines of evidences demonstrate the important role of MCT8, a member of MCT family, in central thyroid hormone transport. MCT8 is widely expressed in the central nervous system, primarily in neurons and in the D2 expressing tanycytes [117; 156]. Mutations of MCT8 in humans results in severe neurological abnormalities, increased circulating T3 levels, mildly decreased T4 and normal to elevated TSH [155]. MCT8-null mice have a less severe neurological phenotype, but feedback regulation of the HPT axis is also markedly altered in these animals [158; 159], manifested by increased circulating T3, normal TSH and increased TRH gene expression in hypophysiotropic neurons [158]. These data indicate that the lack of MCT8 results in local hypothyroidism in TRH neurons despite the increase in peripheral T3 concentration, suggesting that MCT8 is necessary for the normal feedback regulation of TRH neurons. Currently it is unknown whether changes of MCT8 expression under physiological conditions contribute to regulation of hypophysiotropic neurons.
Conclusions
In addition to the classical components of the HPT axis, regulation of the thyroid hormone negative feedback machinery involves at least two additional components: neuronal afferents to hypophysiotropic TRH neurons from the brainstem, hypothalamic arcuate nucleus and dorsomedial nucleus, and the D2-expressing, third ventricular tanycytes. The neuronal afferents modify the sensitivity of TRH neurons to thyroid hormones via intracellular interactions between the cAMP-CREB and thyroid hormone receptor signaling pathways, and may be important to increase the set point for feedback regulation by thyroid hormone during cold exposure, to reduce the set point for feedback regulation by thyroid hormone during fasting and in the circadian variation of thyroid hormone. Tanycytes may permit hypothalamic neurons to sense changes in peripheral T4 concentrations by maintaining relatively constant D2 activity under basal conditions. However, during infection, increased D2 activity in the tanycytes may contribute to the mechanism of central hypothyroidism observed in the nonthyroidal illness syndrome by increasing the hypothalamic content of T3. A summary diagram illustrating the feedback system regulating the hypothalamic-pituitary-thyroid axis is shown in Fig. 13.
Figure 13.
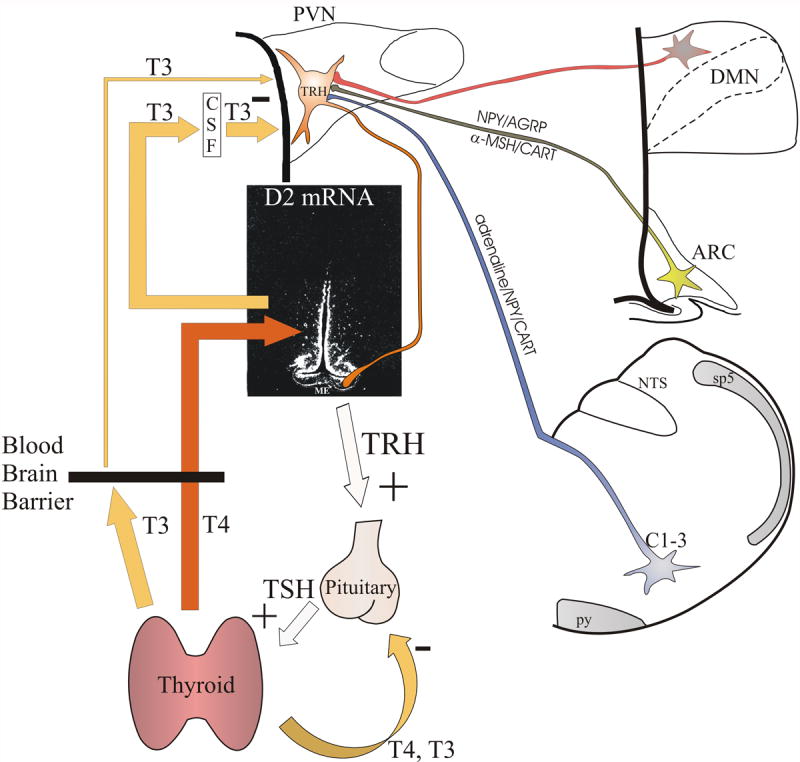
Schematic illustration of the feedback system regulating the hypothalamic-pituitary-thyroid axis. Thyroid hormones exert negative feedback effect at the level of the pituitary and hypophysiotropic TRH neurons. The central feedback effect of thyroid hormones primarily depends on the circulating T4 levels. In the hypothalamus, T4 is converted to T3 by D2 in tanycytes. By volume transmission, T3 secreted from tanycytes reaches the hypophysiotropic TRH neurons, where T3 inhibits the proTRH gene expression via TRβ2 receptors. The setpoint of the feedback regulation can be altered by two mechanisms: 1) Regulation of D2 activity in tanycytes may alter the hypothalamic T3 availability independently from the peripheral T4 concentration. 2) Neuronal afferents can alter the PCREB concentration in the hypophysiotropic TRH neurons that can change the setpoint of feedback regulation through competition of PCREB and thyroid hormone receptors for the multifunctional binding site (Site 4) of the TRH promoter. ARC, hypothalamic arcuate nucleus; C1-3, C1-3 adrenergic area of the brainstem; CSF, cerebrospinal fluid; DMN, hypothalamic dorsomedial nucleus; ME, median eminence; NTS, nucleus tractus solitarius; PVN, hypothalamic paraventricular nucleus; py, pyramidal tract; sp5, spinal trigeminal tract
Acknowledgments
This work was supported by Grants OTKA T046492 and NIH DK-37021
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lechan RM. Update on thyrotropin-releasing hormone. Thyroid Today. 1993;16:1–12. [Google Scholar]
- 2.Nillni EA, Sevarino KA. The biology of prothyrotropin-releasing hormone-derived peptides. Endocr Rev. 1999;20:599–648. doi: 10.1210/edrv.20.5.0379. [DOI] [PubMed] [Google Scholar]
- 3.Bradbury AF, Finnie MD, Smyth DG. Mechanism of C-terminal amide formation by pituitary enzymes. Nature. 1982;298:686–688. doi: 10.1038/298686a0. [DOI] [PubMed] [Google Scholar]
- 4.Fischer WH, Spiess J. Identification of a mammalian glutaminyl cyclase converting glutaminyl into pyroglutamyl peptides. Proc Natl Acad Sci U S A. 1987;84:3628–3632. doi: 10.1073/pnas.84.11.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lechan RM, Segerson TP. Pro-TRH gene expression and precursor peptides in rat brain. Observations by hybridization analysis and immunocytochemistry. Ann N Y Acad Sci. 1989;553:29–59. doi: 10.1111/j.1749-6632.1989.tb46630.x. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa K, Taniguchi Y, Inoue K, Kurosumi K, Suzuki M. Immunocytochemical delineation of thyrotrophic area: origin of thyrotropin-releasing hormone in the median eminence. Neuroendocrinology. 1988;47:384–388. doi: 10.1159/000124943. [DOI] [PubMed] [Google Scholar]
- 7.Merchenthaler I, Liposits Z. Mapping of thyrotropin-releasing hormone (TRH) neuronal systems of rat forebrain projecting to the median eminence and the OVLT. Immunocytochemistry combined with retrograde labeling at the light and electron microscopic levels. Acta Biol Hung. 1994;45:361–374. [PubMed] [Google Scholar]
- 8.Fekete C, Mihaly E, Luo LG, Kelly J, Clausen JT, Mao Q, Rand WM, Moss LG, Kuhar M, Emerson CH, Jackson IM, Lechan RM. Association of cocaine- and amphetamine-regulated transcript-immunoreactive elements with thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and its role in the regulation of the hypothalamic-pituitary-thyroid axis during fasting. J Neurosci. 2000;20:9224–9234. doi: 10.1523/JNEUROSCI.20-24-09224.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swanson LW, Kuypers HG. The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol. 1980;194:555–570. doi: 10.1002/cne.901940306. [DOI] [PubMed] [Google Scholar]
- 10.Lechan RM, Jackson IM. Immunohistochemical localization of thyrotropin-releasing hormone in the rat hypothalamus and pituitary. Endocrinology. 1982;111:55–65. doi: 10.1210/endo-111-1-55. [DOI] [PubMed] [Google Scholar]
- 11.Yu R, Ashworth R, Hinkle PM. Receptors for thyrotropin-releasing hormone on rat lactotropes and thyrotropes. Thyroid. 1998;8:887–894. doi: 10.1089/thy.1998.8.887. [DOI] [PubMed] [Google Scholar]
- 12.Schally AV, Bowers CY, Redding TW, Barrett JF. Isolation of thyrotropin releasing factor (TRF) from porcine hypothalamus. Biochem Biophys Res Commun. 1966;25:165–169. doi: 10.1016/0006-291x(66)90574-2. [DOI] [PubMed] [Google Scholar]
- 13.Sakiz E, Guillemin R. Inverse effects of purified hypothalamic TRF on the acute secretion of TSH and ACTH. Endocrinology. 1965;77:797–801. doi: 10.1210/endo-77-5-797. [DOI] [PubMed] [Google Scholar]
- 14.Gershengorn MC. Mechanism of signal transduction by TRH. Ann N Y Acad Sci. 1989;553:191–196. doi: 10.1111/j.1749-6632.1989.tb46641.x. [DOI] [PubMed] [Google Scholar]
- 15.Weintraub BD, Gesundheit N, Taylor T, Gyves PW. Effect of TRH on TSH glycosylation and biological action. Ann N Y Acad Sci. 1989;553:205–213. doi: 10.1111/j.1749-6632.1989.tb46643.x. [DOI] [PubMed] [Google Scholar]
- 16.Yamada M, Saga Y, Shibusawa N, Hirato J, Murakami M, Iwasaki T, Hashimoto K, Satoh T, Wakabayashi K, Taketo MM, Mori M. Tertiary hypothyroidism and hyperglycemia in mice with targeted disruption of the thyrotropin-releasing hormone gene. Proc Natl Acad Sci U S A. 1997;94:10862–10867. doi: 10.1073/pnas.94.20.10862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freeman ME, Kanyicska B, Lerant A, Nagy G. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80:1523–1631. doi: 10.1152/physrev.2000.80.4.1523. [DOI] [PubMed] [Google Scholar]
- 18.Yamada M, Shibusawa N, Ishii S, Horiguchi K, Umezawa R, Hashimoto K, Monden T, Satoh T, Hirato J, Mori M. Prolactin secretion in mice with thyrotropin-releasing hormone deficiency. Endocrinology. 2006;147:2591–2596. doi: 10.1210/en.2005-1326. [DOI] [PubMed] [Google Scholar]
- 19.Segerson TP, Kauer J, Wolfe HC, Mobtaker H, Wu P, Jackson IM, Lechan RM. Thyroid hormone regulates TRH biosynthesis in the paraventricular nucleus of the rat hypothalamus. Science. 1987;238:78–80. doi: 10.1126/science.3116669. [DOI] [PubMed] [Google Scholar]
- 20.Perello M, Friedman T, Paez-Espinosa V, Shen X, Stuart RC, Nillni EA. Thyroid hormones selectively regulate the posttranslational processing of prothyrotropin-releasing hormone in the paraventricular nucleus of the hypothalamus. Endocrinology. 2006;147:2705–2716. doi: 10.1210/en.2005-1609. [DOI] [PubMed] [Google Scholar]
- 21.Lechan RM, Kakucska I. Feedback regulation of thyrotropin-releasing hormone gene expression by thyroid hormone in the hypothalamic paraventricular nucleus. Ciba Found Symp. 1992;168:144–158. doi: 10.1002/9780470514283.ch10. [DOI] [PubMed] [Google Scholar]
- 22.Lechan RM, Qi Y, Jackson IM, Mahdavi V. Identification of thyroid hormone receptor isoforms in thyrotropin-releasing hormone neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1994;135:92–100. doi: 10.1210/endo.135.1.7516871. [DOI] [PubMed] [Google Scholar]
- 23.Dyess EM, Segerson TP, Liposits Z, Paull WK, Kaplan MM, Wu P, Jackson IM, Lechan RM. Triiodothyronine exerts direct cell-specific regulation of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology. 1988;123:2291–2297. doi: 10.1210/endo-123-5-2291. [DOI] [PubMed] [Google Scholar]
- 24.Abel ED, Ahima RS, Boers ME, Elmquist JK, Wondisford FE. Critical role for thyroid hormone receptor beta2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–1023. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forrest D, Hanebuth E, Smeyne RJ, Everds N, Stewart CL, Wehner JM, Curran T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor beta: evidence for tissue-specific modulation of receptor function. Embo J. 1996;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss RE, Forrest D, Pohlenz J, Cua K, Curran T, Refetoff S. Thyrotropin regulation by thyroid hormone in thyroid hormone receptor beta-deficient mice. Endocrinology. 1997;138:3624–3629. doi: 10.1210/endo.138.9.5412. [DOI] [PubMed] [Google Scholar]
- 27.Abel ED, Boers ME, Pazos-Moura C, Moura E, Kaulbach H, Zakaria M, Lowell B, Radovick S, Liberman MC, Wondisford F. Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291–300. doi: 10.1172/JCI6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raptis S, Fekete C, Sarkar S, Rand WM, Emerson CH, Nagy GM, Lechan RM. Cocaine- and amphetamine-regulated transcript co-contained in thyrotropin-releasing hormone (TRH) neurons of the hypothalamic paraventricular nucleus modulates TRH-induced prolactin secretion. Endocrinology. 2004;145:1695–1699. doi: 10.1210/en.2003-1576. [DOI] [PubMed] [Google Scholar]
- 29.Zoeller RT, Kabeer N, Albers HE. Cold exposure elevates cellular levels of messenger ribonucleic acid encoding thyrotropin-releasing hormone in paraventricular nucleus despite elevated levels of thyroid hormones. Endocrinology. 1990;127:2955–2962. doi: 10.1210/endo-127-6-2955. [DOI] [PubMed] [Google Scholar]
- 30.Uribe RM, Redondo JL, Charli JL, Joseph-Bravo P. Suckling and cold stress rapidly and transiently increase TRH mRNA in the paraventricular nucleus. Neuroendocrinology. 1993;58:140–145. doi: 10.1159/000126523. [DOI] [PubMed] [Google Scholar]
- 31.Sanchez E, Uribe RM, Corkidi G, Zoeller RT, Cisneros M, Zacarias M, Morales-Chapa C, Charli JL, Joseph-Bravo P. Differential responses of thyrotropin-releasing hormone (TRH) neurons to cold exposure or suckling indicate functional heterogeneity of the TRH system in the paraventricular nucleus of the rat hypothalamus. Neuroendocrinology. 2001;74:407–422. doi: 10.1159/000054707. [DOI] [PubMed] [Google Scholar]
- 32.Arancibia S, Rage F, Astier H, Tapia-Arancibia L. Neuroendocrine and autonomous mechanisms underlying thermoregulation in cold environment. Neuroendocrinology. 1996;64:257–267. doi: 10.1159/000127126. [DOI] [PubMed] [Google Scholar]
- 33.Arancibia S, Tapia-Arancibia L, Astier H, Assenmacher I. Physiological evidence for alpha 1-adrenergic facilitatory control of the cold-induced TRH release in the rat, obtained by push-pull cannulation of the median eminence. Neurosci Lett. 1989;100:169–174. doi: 10.1016/0304-3940(89)90679-4. [DOI] [PubMed] [Google Scholar]
- 34.van Haasteren GA, Linkels E, Klootwijk W, van Toor H, Rondeel JM, Themmen AP, de Jong FH, Valentijn K, Vaudry H, Bauer K, et al. Starvation-induced changes in the hypothalamic content of prothyrotrophin-releasing hormone (proTRH) mRNA and the hypothalamic release of proTRH-derived peptides: role of the adrenal gland. J Endocrinol. 1995;145:143–153. doi: 10.1677/joe.0.1450143. [DOI] [PubMed] [Google Scholar]
- 35.Rondeel JM, Heide R, de Greef WJ, van Toor H, van Haasteren GA, Klootwijk W, Visser TJ. Effect of starvation and subsequent refeeding on thyroid function and release of hypothalamic thyrotropin-releasing hormone. Neuroendocrinology. 1992;56:348–353. doi: 10.1159/000126248. [DOI] [PubMed] [Google Scholar]
- 36.Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM. Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology. 1997;138:2569–2576. doi: 10.1210/endo.138.6.5209. [DOI] [PubMed] [Google Scholar]
- 37.De Groot LJ. Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab. 1999;84:151–164. doi: 10.1210/jcem.84.1.5364. [DOI] [PubMed] [Google Scholar]
- 38.Fliers E, Guldenaar SEF, Wiersinga WM, Swaab DF. Decreased Hypothalamic Thyrotropin-Releasing Hormone Gene Expression in Patients with Nonthyroidal Illness. J Clin Endocrinol Metab. 1997;82:4032–4036. doi: 10.1210/jcem.82.12.4404. [DOI] [PubMed] [Google Scholar]
- 39.Kondo K, Harbuz MS, Levy A, Lightman SL. Inhibition of the hypothalamic-pituitary-thyroid axis in response to lipopolysaccharide is independent of changes in circulating corticosteroids. Neuroimmunomodulation. 1997;4:188–194. doi: 10.1159/000097337. [DOI] [PubMed] [Google Scholar]
- 40.Fekete C, Wittmann G, Liposits Z, Lechan RM. GABA-ergic innervation of thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Brain Res. 2002;957:251–258. doi: 10.1016/s0006-8993(02)03576-x. [DOI] [PubMed] [Google Scholar]
- 41.Wittmann G, Lechan RM, Liposits Z, Fekete C. Glutamatergic innervation of corticotropin-releasing hormone- and thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Brain Res. 2005;1039:53–62. doi: 10.1016/j.brainres.2005.01.090. [DOI] [PubMed] [Google Scholar]
- 42.Liposits Z, Paull WK, Wu P, Jackson IM, Lechan RM. Hypophysiotrophic thyrotropin releasing hormone (TRH) synthesizing neurons. Ultrastructure, adrenergic innervation and putative transmitter action. Histochemistry. 1987;88:1–10. doi: 10.1007/BF00490159. [DOI] [PubMed] [Google Scholar]
- 43.Fekete C, Legradi G, Mihaly E, Huang QH, Tatro JB, Rand WM, Emerson CH, Lechan RM. alpha-Melanocyte-stimulating hormone is contained in nerve terminals innervating thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus and prevents fasting-induced suppression of prothyrotropin-releasing hormone gene expression. J Neurosci. 2000;20:1550–1558. doi: 10.1523/JNEUROSCI.20-04-01550.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Legradi G, Lechan RM. Agouti-related protein containing nerve terminals innervate thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1999;140:3643–3652. doi: 10.1210/endo.140.8.6935. [DOI] [PubMed] [Google Scholar]
- 45.Toni R, Jackson IM, Lechan RM. Neuropeptide-Y-immunoreactive innervation of thyrotropin-releasing hormone-synthesizing neurons in the rat hypothalamic paraventricular nucleus. Endocrinology. 1990;126:2444–2453. doi: 10.1210/endo-126-5-2444. [DOI] [PubMed] [Google Scholar]
- 46.Toni R, Jackson IM, Lechan RM. Thyrotropin-releasing-hormone-immunoreactive innervation of thyrotropin-releasing-hormone-tuberoinfundibular neurons in rat hypothalamus: anatomical basis to suggest ultrashort feedback regulation. Neuroendocrinology. 1990;52:422–428. doi: 10.1159/000125623. [DOI] [PubMed] [Google Scholar]
- 47.Wittmann G, Sarkar S, Hrabovszky E, Liposits Z, Lechan RM, Fekete C. Galanin- but not galanin-like peptide-containing axon terminals innervate hypophysiotropic TRH-synthesizing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;1002:43–50. doi: 10.1016/j.brainres.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 48.Legradi G, Hannibal J, Lechan RM. Association between pituitary adenylate cyclase-activating polypeptide and thyrotropin-releasing hormone in the rat hypothalamus. J Chem Neuroanat. 1997;13:265–279. doi: 10.1016/s0891-0618(97)10002-3. [DOI] [PubMed] [Google Scholar]
- 49.Liao N, Vaudry H, Pelletier G. Neuroanatomical connections between corticotropin-releasing factor (CRF) and somatostatin (SRIF) nerve endings and thyrotropin-releasing hormone (TRH) neurons in the paraventricular nucleus of rat hypothalamus. Peptides. 1992;13:677–680. doi: 10.1016/0196-9781(92)90172-y. [DOI] [PubMed] [Google Scholar]
- 50.Legradi G, Lechan RM. The arcuate nucleus is the major source for neuropeptide Y-innervation of thyrotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 1998;139:3262–3270. doi: 10.1210/endo.139.7.6113. [DOI] [PubMed] [Google Scholar]
- 51.Mihaly E, Fekete C, Legradi G, Lechan RM. Hypothalamic dorsomedial nucleus neurons innervate thyrotropin-releasing hormone-synthesizing neurons in the paraventricular nucleus. Brain Res. 2001;891:20–31. doi: 10.1016/s0006-8993(00)03094-8. [DOI] [PubMed] [Google Scholar]
- 52.Rondeel JM, de Greef WJ, Hop WC, Rowland DL, Visser TJ. Effect of cold exposure on the hypothalamic release of thyrotropin-releasing hormone and catecholamines. Neuroendocrinology. 1991;54:477–481. doi: 10.1159/000125940. [DOI] [PubMed] [Google Scholar]
- 53.Ignar D, Kuhn C. Relative ontogeny of opioid and catecholaminergic regulation of thyrotropin secretion in the rat. Endocrinology. 1988;123:567–571. doi: 10.1210/endo-123-1-567. [DOI] [PubMed] [Google Scholar]
- 54.Thonberg H, Fredriksson JM, Nedergaard J, Cannon B. A novel pathway for adrenergic stimulation of cAMP-response-element-binding protein (CREB) phosphorylation: mediation via alpha1-adrenoceptors and protein kinase C activation. Biochem J. 2002;364:73–79. doi: 10.1042/bj3640073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nillni EA, Vaslet C, Harris M, Hollenberg A, Bjorbak C, Flier JS. Leptin regulates prothyrotropin-releasing hormone biosynthesis. Evidence for direct and indirect pathways. J Biol Chem. 2000;275:36124–36133. doi: 10.1074/jbc.M003549200. [DOI] [PubMed] [Google Scholar]
- 56.Hollenberg AN, Monden T, Flynn TR, Boers ME, Cohen O, Wondisford FE. The human thyrotropin-releasing hormone gene is regulated by thyroid hormone through two distinct classes of negative thyroid hormone response elements. Mol Endocrinol. 1995;9:540–550. doi: 10.1210/mend.9.5.7565802. [DOI] [PubMed] [Google Scholar]
- 57.Wittmann G, Liposits Z, Lechan RM, Fekete C. Medullary adrenergic neurons contribute to the neuropeptide Y-ergic innervation of hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the rat. Neurosci Lett. 2002;324:69–73. doi: 10.1016/s0304-3940(02)00165-9. [DOI] [PubMed] [Google Scholar]
- 58.Wittmann G, Liposits Z, Lechan RM, Fekete C. Medullary adrenergic neurons contribute to the cocaine- and amphetamine-regulated transcript-immunoreactive innervation of thyrotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;1006:1–7. doi: 10.1016/j.brainres.2003.12.049. [DOI] [PubMed] [Google Scholar]
- 59.Fekete C, Kelly J, Mihaly E, Sarkar S, Rand WM, Legradi G, Emerson CH, Lechan RM. Neuropeptide Y has a central inhibitory action on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2001;142:2606–2613. doi: 10.1210/endo.142.6.8207. [DOI] [PubMed] [Google Scholar]
- 60.Sarkar S, Lechan RM. Central administration of neuropeptide Y reduces alpha-melanocyte-stimulating hormone-induced cyclic adenosine 5'-monophosphate response element binding protein (CREB) phosphorylation in pro-thyrotropin-releasing hormone neurons and increases CREB phosphorylation in corticotropin-releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2003;144:281–291. doi: 10.1210/en.2002-220675. [DOI] [PubMed] [Google Scholar]
- 61.Agnati LF, Fuxe K, Benfenati F, Battistini N, Harfstrand A, Tatemoto K, Hokfelt T, Mutt V. Neuropeptide Y in vitro selectivity increases the number of alpha 2-adrenergic binding sites in membranes of the medulla oblongata of the rat. Acta Physiol Scand. 1983;118:293–295. doi: 10.1111/j.1748-1716.1983.tb07273.x. [DOI] [PubMed] [Google Scholar]
- 62.Lechan RM, Fekete C. The TRH neuron: a hypothalamic integrator of energy metabolism. Prog Brain Res. 2006;153:209–235. doi: 10.1016/S0079-6123(06)53012-2. [DOI] [PubMed] [Google Scholar]
- 63.de Jesus LA, Carvalho SD, Ribeiro MO, Schneider M, Kim SW, Harney JW, Larsen PR, Bianco AC. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–1385. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lanni A, Moreno M, Lombardi A, de Lange P, Goglia F. Control of energy metabolism by iodothyronines. J Endocrinol Invest. 2001;24:897–913. doi: 10.1007/BF03343949. [DOI] [PubMed] [Google Scholar]
- 65.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 66.Legradi G, Emerson CH, Ahima RS, Rand WM, Flier JS, Lechan RM. Arcuate nucleus ablation prevents fasting-induced suppression of ProTRH mRNA in the hypothalamic paraventricular nucleus. Neuroendocrinology. 1998;68:89–97. doi: 10.1159/000054354. [DOI] [PubMed] [Google Scholar]
- 67.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest. 1996;98:1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 69.Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [DOI] [PubMed] [Google Scholar]
- 70.Mizuno TM, Mobbs CV. Hypothalamic agouti-related protein messenger ribonucleic acid is inhibited by leptin and stimulated by fasting. Endocrinology. 1999;140:814–817. doi: 10.1210/endo.140.2.6491. [DOI] [PubMed] [Google Scholar]
- 71.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 72.Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139:4428–4431. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- 73.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 74.Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 75.Fekete C, Singru PS, Sanchez E, Sarkar S, Christoffolete MA, Riberio RS, Rand WM, Emerson CH, Bianco AC, Lechan RM. Differential effects of central leptin, insulin, or glucose administration during fasting on the hypothalamic-pituitary-thyroid axis and feeding-related neurons in the arcuate nucleus. Endocrinology. 2006;147:520–529. doi: 10.1210/en.2005-0956. [DOI] [PubMed] [Google Scholar]
- 76.Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, Lechan RM. Agouti-related protein (AGRP) has a central inhibitory action on the hypothalamic-pituitary-thyroid (HPT) axis; comparisons between the effect of AGRP and neuropeptide Y on energy homeostasis and the HPT axis. Endocrinology. 2002;143:3846–3853. doi: 10.1210/en.2002-220338. [DOI] [PubMed] [Google Scholar]
- 77.Fekete C, Marks DL, Sarkar S, Emerson CH, Rand WM, Cone RD, Lechan RM. Effect of Agouti-related protein in regulation of the hypothalamic-pituitary-thyroid axis in the melanocortin 4 receptor knockout mouse. Endocrinology. 2004;145:4816–4821. doi: 10.1210/en.2004-0476. [DOI] [PubMed] [Google Scholar]
- 78.Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, Beck-Sickinger A, Lechan RM. Neuropeptide Y1 and Y5 receptors mediate the effects of neuropeptide Y on the hypothalamic-pituitary-thyroid axis. Endocrinology. 2002;143:4513–4519. doi: 10.1210/en.2002-220574. [DOI] [PubMed] [Google Scholar]
- 79.Wittmann G, Liposits Z, Lechan RM, Fekete C. Origin of cocaine- and amphetamine-regulated transcript-containing axons innervating hypophysiotropic corticotropin-releasing hormone-synthesizing neurons in the rat. Endocrinology. 2005;146:2985–2991. doi: 10.1210/en.2005-0178. [DOI] [PubMed] [Google Scholar]
- 80.Fekete C, Sarkar S, Lechan RM. Relative contribution of brainstem afferents to the cocaine- and amphetamine-regulated transcript (CART) innervation of thyrotropin-releasing hormone synthesizing neurons in the hypothalamic paraventricular nucleus (PVN) Brain Res. 2005;1032:171–175. doi: 10.1016/j.brainres.2004.10.069. [DOI] [PubMed] [Google Scholar]
- 81.Kim MS, Small CJ, Stanley SA, Morgan DG, Seal LJ, Kong WM, Edwards CM, Abusnana S, Sunter D, Ghatei MA, Bloom SR. The central melanocortin system affects the hypothalamo-pituitary thyroid axis and may mediate the effect of leptin. J Clin Invest. 2000;105:1005–1011. doi: 10.1172/JCI8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 83.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 84.Sarkar S, Legradi G, Lechan RM. Intracerebroventricular administration of alpha-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res. 2002;945:50–59. doi: 10.1016/s0006-8993(02)02619-7. [DOI] [PubMed] [Google Scholar]
- 85.Cabrele C, Beck-Sickinger AG. Molecular characterization of the ligand-receptor interaction of the neuropeptide Y family. J Pept Sci. 2000;6:97–122. doi: 10.1002/(SICI)1099-1387(200003)6:3<97::AID-PSC236>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 86.Sarkar S, Wittmann G, Fekete C, Lechan RM. Central administration of cocaine- and amphetamine-regulated transcript increases phosphorylation of cAMP response element binding protein in corticotropin-releasing hormone-producing neurons but not in prothyrotropin-releasing hormone-producing neurons in the hypothalamic paraventricular nucleus. Brain Res. 2004;999:181–192. doi: 10.1016/j.brainres.2003.11.062. [DOI] [PubMed] [Google Scholar]
- 87.Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN. Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest. 2001;107:111–120. doi: 10.1172/JCI10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guo F, Bakal K, Minokoshi Y, Hollenberg AN. Leptin signaling targets the thyrotropin-releasing hormone gene promoter in vivo. Endocrinology. 2004;145:2221–2227. doi: 10.1210/en.2003-1312. [DOI] [PubMed] [Google Scholar]
- 89.Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr, Stoffel M, Friedman JM. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet. 1996;14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 90.Perello M, Stuart RC, Nillni EA. The role of intracerebroventricular administration of leptin in the stimulation of prothyrotropin releasing hormone neurons in the hypothalamic paraventricular nucleus. Endocrinology. 2006;147:3296–3306. doi: 10.1210/en.2005-1533. [DOI] [PubMed] [Google Scholar]
- 91.Schwartz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 92.Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- 93.Schwartz MW, Sipols AJ, Marks JL, Sanacora G, White JD, Scheurink A, Kahn SE, Baskin DG, Woods SC, Figlewicz DP, et al. Inhibition of hypothalamic neuropeptide Y gene expression by insulin. Endocrinology. 1992;130:3608–3616. doi: 10.1210/endo.130.6.1597158. [DOI] [PubMed] [Google Scholar]
- 94.Muroya S, Yada T, Shioda S, Takigawa M. Glucose-sensitive neurons in the rat arcuate nucleus contain neuropeptide Y. Neurosci Lett. 1999;264:113–116. doi: 10.1016/s0304-3940(99)00185-8. [DOI] [PubMed] [Google Scholar]
- 95.Wang J, Leibowitz KL. Central insulin inhibits hypothalamic galanin and neuropeptide Y gene expression and peptide release in intact rats. Brain Res. 1997;777:231–236. doi: 10.1016/s0006-8993(97)00963-3. [DOI] [PubMed] [Google Scholar]
- 96.Diengott D, Mirsky IA, Perisutti G. Effect of fasting on insulinase activity and on hypoglycemic response to insulin. Endocrinology. 1957;60:303–307. doi: 10.1210/endo-60-2-303. [DOI] [PubMed] [Google Scholar]
- 97.Fukumoto H, Seino Y, Koh G, Takeda J, Tsuji K, Kurose T, Kitano N, Tsuda K, Taminato T, Imura H. Effects of oral glucose administration on preproinsulin mRNA in rats in vivo. Biochem Biophys Res Commun. 1986;141:1201–1206. doi: 10.1016/s0006-291x(86)80172-3. [DOI] [PubMed] [Google Scholar]
- 98.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci U S A. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jacobowitz DM, O'Donohue TL. alpha-Melanocyte stimulating hormone: immunohistochemical identification and mapping in neurons of rat brain. Proc Natl Acad Sci U S A. 1978;75:6300–6304. doi: 10.1073/pnas.75.12.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Singru PS, Fekete C, Lechan RM. Neuroanatomical evidence for participation of the hypothalamic dorsomedial nucleus (DMN) in regulation of the hypothalamic paraventricular nucleus (PVN) by alpha-melanocyte stimulating hormone. Brain Res. 2005;1064:42–51. doi: 10.1016/j.brainres.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 101.Brabant G, Prank K, Ranft U, Bergmann P, Schuermeyer T, Hesch RD, von zur Muhlen A. Circadian and pulsatile TSH secretion under physiological and pathophysiological conditions. Horm Metab Res Suppl. 1990;23:12–17. [PubMed] [Google Scholar]
- 102.Wong CC, Dohler KD, Atkinson MJ, Geerlings H, Hesch RD, von zur Muhlen A. Influence of age, strain and season on diurnal periodicity of thyroid stimulating hormone, thyroxine, triiodothyronine and parathyroid hormone in the serum of male laboratory rats. Acta Endocrinol (Copenh) 1983;102:377–385. doi: 10.1530/acta.0.1020377. [DOI] [PubMed] [Google Scholar]
- 103.Kalsbeek A, Fliers E, Franke AN, Wortel J, Buijs RM. Functional connections between the suprachiasmatic nucleus and the thyroid gland as revealed by lesioning and viral tracing techniques in the rat. Endocrinology. 2000;141:3832–3841. doi: 10.1210/endo.141.10.7709. [DOI] [PubMed] [Google Scholar]
- 104.Covarrubias L, Uribe RM, Mendez M, Charli JL, Joseph-Bravo P. Neuronal TRH synthesis: developmental and circadian TRH mRNA levels. Biochem Biophys Res Commun. 1988;151:615–622. doi: 10.1016/0006-291x(88)90638-9. [DOI] [PubMed] [Google Scholar]
- 105.Behrends J, Prank K, Dogu E, Brabant G. Central nervous system control of thyrotropin secretion during sleep and wakefulness. Horm Res. 1998;49:173–177. doi: 10.1159/000023167. [DOI] [PubMed] [Google Scholar]
- 106.Watts AG, Swanson LW, Sanchez-Watts G. Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol. 1987;258:204–229. doi: 10.1002/cne.902580204. [DOI] [PubMed] [Google Scholar]
- 107.Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;23:10691–10702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. doi: 10.1210/edrv.23.1.0455. [DOI] [PubMed] [Google Scholar]
- 109.Kakucska I, Rand W, Lechan RM. Thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus is dependent upon feedback regulation by both triiodothyronine and thyroxine. Endocrinology. 1992;130:2845–2850. doi: 10.1210/endo.130.5.1572297. [DOI] [PubMed] [Google Scholar]
- 110.Hagen GA, Solberg LA., Jr Brain and cerebrospinal fluid permeability to intravenous thyroid hormones. Endocrinology. 1974;95:1398–1410. doi: 10.1210/endo-95-5-1398. [DOI] [PubMed] [Google Scholar]
- 111.Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3'-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology. 1982;110:367–375. doi: 10.1210/endo-110-2-367. [DOI] [PubMed] [Google Scholar]
- 112.Riskind PN, Kolodny JM, Larsen PR. The regional hypothalamic distribution of type II 5'-monodeiodinase in euthyroid and hypothyroid rats. Brain Res. 1987;420:194–198. doi: 10.1016/0006-8993(87)90260-5. [DOI] [PubMed] [Google Scholar]
- 113.Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, Lechan RM. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138:3359–3368. doi: 10.1210/endo.138.8.5318. [DOI] [PubMed] [Google Scholar]
- 114.Diano S, Naftolin F, Goglia F, Horvath TL. Fasting-induced increase in type II iodothyronine deiodinase activity and messenger ribonucleic acid levels is not reversed by thyroxine in the rat hypothalamus. Endocrinology. 1998;139:2879–2884. doi: 10.1210/endo.139.6.6062. [DOI] [PubMed] [Google Scholar]
- 115.Guadano-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A. 1997;94:10391–10396. doi: 10.1073/pnas.94.19.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gereben B, Pachucki J, Kollar A, Liposits Z, Fekete C. Ontogenic redistribution of type 2 deiodinase messenger ribonucleic acid in the brain of chicken. Endocrinology. 2004;145:3619–3625. doi: 10.1210/en.2004-0229. [DOI] [PubMed] [Google Scholar]
- 117.Alkemade A, Friesema EC, Unmehopa UA, Fabriek BO, Kuiper GG, Leonard JL, Wiersinga WM, Swaab DF, Visser TJ, Fliers E. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab. 2005;90:4322–4334. doi: 10.1210/jc.2004-2567. [DOI] [PubMed] [Google Scholar]
- 118.Fekete C, Mihaly E, Herscovici S, Salas J, Tu H, Larsen PR, Lechan RM. DARPP-32 and CREB are present in type 2 iodothyronine deiodinase-producing tanycytes: implications for the regulation of type 2 deiodinase activity. Brain Res. 2000;862:154–161. doi: 10.1016/s0006-8993(00)02105-3. [DOI] [PubMed] [Google Scholar]
- 119.Diano S, Leonard JL, Meli R, Esposito E, Schiavo L. Hypothalamic type II iodothyronine deiodinase: a light and electron microscopic study. Brain Res. 2003;976:130–134. doi: 10.1016/s0006-8993(03)02692-1. [DOI] [PubMed] [Google Scholar]
- 120.Rodriguez EM, Blazquez JL, Pastor FE, Pelaez B, Pena P, Peruzzo B, Amat P. Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol. 2005;247:89–164. doi: 10.1016/S0074-7696(05)47003-5. [DOI] [PubMed] [Google Scholar]
- 121.Bruni JE. Scanning and transmission electron microscopy of the ependymal lining of the third ventricle. Can J Neurol Sci. 1974;1:59–73. [PubMed] [Google Scholar]
- 122.Rodriguez EM, Gonzalez CB, Delannoy L. Cellular organization of the lateral and postinfundibular regions of the median eminence in the rat. Cell Tissue Res. 1979;201:377–408. doi: 10.1007/BF00236998. [DOI] [PubMed] [Google Scholar]
- 123.Akmayev IG, Popov AP. Morphological aspects of the hypothalamic-hypophyseal system. VII. The tanycytes: Their relation to the hypophyseal adrenocorticotrophic function. An ultrastructural study. Cell Tissue Res. 1977;180:263–282. doi: 10.1007/BF00231958. [DOI] [PubMed] [Google Scholar]
- 124.Lechan RM, Fekete C. Role of thyroid hormone deiodination in the hypothalamus. Thyroid. 2005;15:883–897. doi: 10.1089/thy.2005.15.883. [DOI] [PubMed] [Google Scholar]
- 125.Nadeau S, Rivest S. Effects of circulating tumor necrosis factor on the neuronal activity and expression of the genes encoding the tumor necrosis factor receptors (p55 and p75) in the rat brain: a view from the blood-brain barrier. Neuroscience. 1999;93:1449–1464. doi: 10.1016/s0306-4522(99)00225-0. [DOI] [PubMed] [Google Scholar]
- 126.Nishibori M, Nakaya N, Mori S, Saeki K. Immunohistochemical localization of macrophage migration inhibitory factor (MIF) in tanycytes, subcommissural organ and choroid plexus in the rat brain. Brain Res. 1997;758:259–262. doi: 10.1016/s0006-8993(97)00342-9. [DOI] [PubMed] [Google Scholar]
- 127.Givalois L, Arancibia S, Alonso G, Tapia-Arancibia L. Expression of brain-derived neurotrophic factor and its receptors in the median eminence cells with sensitivity to stress. Endocrinology. 2004;145:4737–4747. doi: 10.1210/en.2004-0616. [DOI] [PubMed] [Google Scholar]
- 128.Hashemi SH, Li JY, Schindler M, Dahlstrom A. Presence of sst2(a) receptor immunoreactivity in rat ependyma and tanycytes. Neuroreport. 2001;12:1793–1797. doi: 10.1097/00001756-200107030-00007. [DOI] [PubMed] [Google Scholar]
- 129.Lerant A, Freeman ME. Ovarian steroids differentially regulate the expression of PRL-R in neuroendocrine dopaminergic neuron populations: a double label confocal microscopic study. Brain Res. 1998;802:141–154. doi: 10.1016/s0006-8993(98)00583-6. [DOI] [PubMed] [Google Scholar]
- 130.Garcia-Segura LM, Rodriguez JR, Torres-Aleman I. Localization of the insulin-like growth factor I receptor in the cerebellum and hypothalamus of adult rats: an electron microscopic study. J Neurocytol. 1997;26:479–490. doi: 10.1023/a:1018581407804. [DOI] [PubMed] [Google Scholar]
- 131.Langub MC, Jr, Watson RE., Jr Estrogen receptor-immunoreactive glia, endothelia, and ependyma in guinea pig preoptic area and median eminence: electron microscopy. Endocrinology. 1992;130:364–372. doi: 10.1210/endo.130.1.1727710. [DOI] [PubMed] [Google Scholar]
- 132.Broedel O, Eravci M, Fuxius S, Smolarz T, Jeitner A, Grau H, Stoltenburg-Didinger G, Plueckhan H, Meinhold H, Baumgartner A. Effects of hyper- and hypothyroidism on thyroid hormone concentrations in regions of the rat brain. Am J Physiol Endocrinol Metab. 2003;285:E470–480. doi: 10.1152/ajpendo.00043.2003. [DOI] [PubMed] [Google Scholar]
- 133.Dussault JH, Ruel J. Thyroid hormones and brain development. Annu Rev Physiol. 1987;49:321–334. doi: 10.1146/annurev.ph.49.030187.001541. [DOI] [PubMed] [Google Scholar]
- 134.Bernal J. Action of thyroid hormone in brain. J Endocrinol Invest. 2002;25:268–288. doi: 10.1007/BF03344003. [DOI] [PubMed] [Google Scholar]
- 135.Anguiano B, Quintanar A, Luna M, Navarro L, Ramirez del Angel A, Pacheco P, Valverde C. Neuroendocrine regulation of adrenal gland and hypothalamus 5'deiodinase activity. II. Effects of splanchnicotomy and hypophysectomy. Endocrinology. 1995;136:3346–3352. doi: 10.1210/endo.136.8.7628370. [DOI] [PubMed] [Google Scholar]
- 136.Serrano-Lozano A, Montiel M, Morell M, Morata P. 5' Deiodinase activity in brain regions of adult rats: modifications in different situations of experimental hypothyroidism. Brain Res Bull. 1993;30:611–616. doi: 10.1016/0361-9230(93)90090-x. [DOI] [PubMed] [Google Scholar]
- 137.Southwell BR, Duan W, Alcorn D, Brack C, Richardson SJ, Kohrle J, Schreiber G. Thyroxine transport to the brain: role of protein synthesis by the choroid plexus. Endocrinology. 1993;133:2116–2126. doi: 10.1210/endo.133.5.8404661. [DOI] [PubMed] [Google Scholar]
- 138.Agnati LF, Zoli M, Stromberg I, Fuxe K. Intercellular communication in the brain: wiring versus volume transmission. Neuroscience. 1995;69:711–726. doi: 10.1016/0306-4522(95)00308-6. [DOI] [PubMed] [Google Scholar]
- 139.Diano S, Naftolin F, Goglia F, Csernus V, Horvath TL. Monosynaptic pathway between the arcuate nucleus expressing glial type II iodothyronine 5'-deiodinase mRNA and the median eminence-projective TRH cells of the rat paraventricular nucleus. J Neuroendocrinol. 1998;10:731–742. doi: 10.1046/j.1365-2826.1998.00204.x. [DOI] [PubMed] [Google Scholar]
- 140.Lechan RM, Fekete C. Feedback regulation of thyrotropin-releasing hormone (TRH): mechanisms for the non-thyroidal illness syndrome. J Endocrinol Invest. 2004;27:105–119. [PubMed] [Google Scholar]
- 141.Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Res. 2001;61:1432–1438. [PubMed] [Google Scholar]
- 142.Huang QH, Hruby VJ, Tatro JB. Role of central melanocortins in endotoxin-induced anorexia. Am J Physiol. 1999;276:R864–871. doi: 10.1152/ajpregu.1999.276.3.R864. [DOI] [PubMed] [Google Scholar]
- 143.Sergeyev V, Broberger C, Hokfelt T. Effect of LPS administration on the expression of POMC, NPY, galanin CART and MCH mRNAs in the rat hypothalamus. Brain Res Mol Brain Res. 2001;90:93–100. doi: 10.1016/s0169-328x(01)00088-2. [DOI] [PubMed] [Google Scholar]
- 144.Marvel FA, Chen CC, Badr N, Gaykema RP, Goehler LE. Reversible inactivation of the dorsal vagal complex blocks lipopolysaccharide-induced social withdrawal and c-Fos expression in central autonomic nuclei. Brain Behav Immun. 2004;18:123–134. doi: 10.1016/j.bbi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 145.Sagar SM, Price KJ, Kasting NW, Sharp FR. Anatomic patterns of Fos immunostaining in rat brain following systemic endotoxin administration. Brain Res Bull. 1995;36:381–392. doi: 10.1016/0361-9230(94)00217-o. [DOI] [PubMed] [Google Scholar]
- 146.Fekete C, Gereben B, Doleschall M, Harney JW, Dora JM, Bianco AC, Sarkar S, Liposits Z, Rand W, Emerson C, Kacskovics I, Larsen PR, Lechan RM. Lipopolysaccharide induces type 2 iodothyronine deiodinase in the mediobasal hypothalamus: implications for the nonthyroidal illness syndrome. Endocrinology. 2004;145:1649–1655. doi: 10.1210/en.2003-1439. [DOI] [PubMed] [Google Scholar]
- 147.Fekete C, Singru PS, Sarkar S, Rand WM, Lechan RM. Ascending brainstem pathways are not involved in lipopolysaccharide-induced suppression of thyrotropin-releasing hormone gene expression in the hypothalamic paraventricular nucleus. Endocrinology. 2005;146:1357–1363. doi: 10.1210/en.2004-1429. [DOI] [PubMed] [Google Scholar]
- 148.Boelen A, Kwakkel J, Thijssen-Timmer DC, Alkemade A, Fliers E, Wiersinga WM. Simultaneous changes in central and peripheral components of the hypothalamus-pituitary-thyroid axis in lipopolysaccharide-induced acute illness in mice. J Endocrinol. 2004;182:315–323. doi: 10.1677/joe.0.1820315. [DOI] [PubMed] [Google Scholar]
- 149.Zeold A, Doleschall M, Haffner MC, Capelo LP, Menyhert J, Liposits Z, da Silva WS, Bianco AC, Kacskovics I, Fekete C, Gereben B. Characterization of the nuclear factor-kappa B responsiveness of the human dio2 gene. Endocrinology. 2006;147:4419–4429. doi: 10.1210/en.2005-1608. [DOI] [PubMed] [Google Scholar]
- 150.Baur A, Bauer K, Jarry H, Kohrle J. Effects of proinflammatory cytokines on anterior pituitary 5'-deiodinase type I and type II. J Endocrinol. 2000;167:505–515. doi: 10.1677/joe.0.1670505. [DOI] [PubMed] [Google Scholar]
- 151.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 152.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 153.Coppola A, Meli R, Diano S. Inverse shift in circulating corticosterone and leptin levels elevates hypothalamic deiodinase type 2 in fasted rats. Endocrinology. 2005;146:2827–2833. doi: 10.1210/en.2004-1361. [DOI] [PubMed] [Google Scholar]
- 154.Coppola A, Liu ZW, Andrews ZB, Paradis E, Roy MC, Friedman JM, Ricquier D, Richard D, Horvath TL, Gao XB, Diano S. A central thermogenic-like mechanism in feeding regulation: an interplay between arcuate nucleus T3 and UCP2. Cell metabolism. 2007;5:21–33. doi: 10.1016/j.cmet.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Jansen J, Friesema EC, Milici C, Visser TJ. Thyroid hormone transporters in health and disease. Thyroid. 2005;15:757–768. doi: 10.1089/thy.2005.15.757. [DOI] [PubMed] [Google Scholar]
- 156.Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, Bauer K. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–1706. doi: 10.1210/en.2004-1179. [DOI] [PubMed] [Google Scholar]
- 157.Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Molecular endocrinology (Baltimore, Md. 2002;16:2283–2296. doi: 10.1210/me.2001-0309. [DOI] [PubMed] [Google Scholar]
- 158.Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, Raivich G, Bauer K, Heuer H. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter. 2007;8 doi: 10.1172/JCI28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006;147:4036–4043. doi: 10.1210/en.2006-0390. [DOI] [PubMed] [Google Scholar]
- 160.Mihaly E, Fekete C, Tatro JB, Liposits Z, Stopa EG, Lechan RM. Hypophysiotropic thyrotropin-releasing hormone-synthesizing neurons in the human hypothalamus are innervated by neuropeptide Y, agouti-related protein, and alpha-melanocyte-stimulating hormone. J Clin Endocrinol Metab. 2000;85:2596–2603. doi: 10.1210/jcem.85.7.6662. [DOI] [PubMed] [Google Scholar]