

Abstract
To clarify the pH-dependent conformational transitions of proteins, we propose an approach in which structural changes monitored by heteronuclear sequential quantum correlation (HSQC) spectroscopy were analyzed by using a principal component analysis (PCA). We use bovine β-lactoglobulin, a protein widely used in protein folding studies, as a target. First, we measured HSQC spectra at various pH values and subjected them to a PCA. The analysis revealed three apparent transitions with pKa values of 2.9, 4.9, and 6.8, consistent with previous reports using different methods. Next, Gdn-HCl-induced unfolding was examined by measuring tryptophan fluorescence at various pH values. Between pH 2 and 8, β-lactoglobulin exhibited a number of structural transitions as well as changes in stability represented by the free energy change of unfolding, ΔGU. By combining the NMR and fluorescence results, the change in ΔGU was suggested to result from the decreased pKa of some acidic residues. Notably, the native state at neutral pH is destabilized by deprotonation of Glu-89, leading to an increase in the relative population of the intermediate. Thus, the PCA of pH-dependent HSQC spectra provides a more comprehensive understanding of the stability and function of proteins.
Keywords: pH-dependent conformational change, Tanford transition
The principal component (PC) analysis (PCA) is a kind of multivariate analysis frequently used in various fields (1). In biophysical study, for example, PCA is applied for 3D structures obtained from molecular dynamics simulation to extract the structural change of the target protein (2). PCA also is applied to a series of spectra obtained during chemical reactions of a compound (3) and a protein (4) to determine how many constituents appear in the observations and to identify spectrum for each constituent. In the latter two cases, time-dependent changes in population of each constituent can also be determined. PCA is, thus, widely used to extract a small number of fundamental properties from a large set of data.
The conformation and stability of proteins depend on pH. Clarifying such dependencies is fundamental to understanding the structure and function of proteins. Tanford (5) established the basis of the pH-dependent conformational stability of proteins, in which conformational equilibrium is linked to the binding of protons. The basic mechanism is common to various conformational equilibria, which depend on the binding of ligands, such as denaturant-induced unfolding transitions. However, the analysis of conventional spectroscopic data, such as fluorescence or CD data, cannot determine which residues are responsible for the change of stability. In contrast, heteronuclear NMR spectra, such as the heteronuclear sequential quantum correlation (HSQC) spectrum, monitoring the behavior of essentially all residues, has the potential to address the contributions of individual residues (6). In practice, although NMR spectra have been used extensively to monitor pH-dependent conformational transitions, studies have focused on only a limited number of residues. Moreover, the contribution of the titration of individual residues to a conformational transition, including the intermediate state, is not precisely understood. Here, we introduced the PCA to correlate pH-dependent HSQC spectra with pH-dependent conformational transitions.
As a target protein, we used bovine β-lactoglobulin (βLG). βLG consists of 162 amino acid residues (18 kDa) and contains two tryptophan residues, Trp-19 and Trp-61 [supporting information (SI) Fig. 6A]. It is a predominantly β-sheet protein consisting of nine β-strands (A–I), of which the A–H strands form an up-and-down β-barrel, and one major α-helix at the C terminus of the molecule (7–10). The β-barrel assumes a flattened calyx (or cone) with a large cavity surrounded by hydrophobic residues, in which various kinds of hydrophobic ligands are bound. Between pH 2 and 8, βLG exhibits a number of pH-induced structural transitions as well as changes in the association state and stability. These transitions are generally summarized by a four-state mechanism between M, Q, N, and R states, with pKa,M-Q = 3, pKa,Q-N = 5, and pKa,N-R = 7, respectively (see Scheme 1) (11, 12).
Scheme. 1.
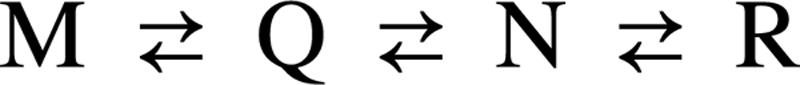
Experimental design.
At acidic pH, βLG assumes a monomeric form (denoted M) and exhibits remarkably high stability. It is reported that βLG dimerizes with little alteration in structure at around pH 3 (M–Q) (10, 13). Between pH 4.5 and 6, βLG is converted from the acidic Q state to the native (N) dimeric state. It has been reported that this Q–N transition involves changes in compactness (14). At pH 7.0, βLG undergoes the so-called Tanford transition (N–R) (15). The Tanford transition involves a conformational change of the EF loop (residues 85–90), which might be caused by the cleavage of hydrogen bonds between the F and G strands (16). It is noted that, although βLG undergoes a dimer-to-octamer transition between pH 4 and 5 (17, 18), the octamerization does not involve any appreciable change in its secondary structure (19).
To relate the pH-dependent stability with the conformational behavior at the residue level, pH titration and hydrogen/deuterium (H/D) exchange experiments monitored by HSQC were carried out. We applied a PCA to the chemical shift data, finding that the conformational transitions are collective motions of a number of residues. Then, we performed Gdn-HCl-induced unfolding experiments monitored using tryptophan fluorescence at various pH levels. A spectral decomposition was used to characterize the pH-dependent change of overall stability and the intermediate, which populates notably at neutral pH. Taking these results into consideration, the pH-dependent conformational stability of βLG was explained more precisely and comprehensively. Additionally, we propose that the increase in population of an intermediate at neutral pH is the underlying mechanism of the Tanford transition, relevant to the increased ligand-binding at neutral pH.
Results
pH Titration Monitored by HSQC Spectrum.
In the NMR measurements, we used a mutant βLG dimer, A34C, which provided a significant improvement in spectral quality, enabling a quantitative analysis even at neutral pH (16). To check whether A34C βLG retains the structure of wild-type βLG at pH 2.4 and pH 6.5, chemical shift differences (CSDs) were investigated at the two pH values (SI Fig. 7). There was no significant CSD over sequence at pH 6.5, whereas significant CSDs were observed at the dimer's interface at pH 2.4. At this pH, wild-type and A34C βLGs assume monomer and dimer, respectively. Thus, there can be a difference in their conformation associated with the dimerization.
We measured HSQC spectra at pH 2.4–8.1 to examine the four-state conformational transitions reported previously (Scheme 1) (11). Fig. 1A and SI Fig. 8 show the superposition of the HSQC spectra obtained at various pH values. It is evident that chemical shifts of many signals change with pH. For these residues, we observed no evident change of peak intensity, suggesting the fast exchange between conformational states. On the other hand, some residues showed a decrease in peak intensity above pH 6 without changing the chemical shift (16), suggesting a contribution of slow conformational change (see Discussion).
Fig. 1.

pH titration monitored by HSQC spectrum. (A) A close-up view of superposition of HSQC spectra acquired at pH 2.4–8.1. The red, orange, green, and blue spectra were acquired at pH 2.4, 5.0, 6.7, and 8.1, respectively. Signal displacements of residues are indicated by arrows. See SI Fig. 8 for all regions. (B) An example of signal displacements of Gln-5. The error bars for δH and δN are set to be 0.02 and 0.1 ppm, respectively, which indicate the standard linewidth of the signals. The solid lines are reconstructed traces from the results of the PCA. The letters are marked at the estimated signal positions for each state. (C and D) A Hue Plot for all of the residues (C) and the scale figure (D). The bars and circles in C indicate the positions of β-strands, the α-helix, and residues involved in the dimer interface, respectively.
To present the pH-dependent chemical shifts of each residue on one graph, we used a Hue plot, in which coloration indicates the direction and degree of signal displacement (Fig. 1C). The correspondence of the direction and depth of the color is presented in the Hue circle scale (Fig. 1D). For example, the change in the signal position of Gln-5 is shown in Fig. 1B. From pH 2 to pH 5, the signal of Gln-5 moves toward the top right of the spectrum whereas, from pH 5 to pH 8, it moves downwards. Thus, as indicated by the Hue scale, Gln-5 in the plot exhibits orange and purple below and above pH 5, respectively.
Fig. 1C indicates that individual residues show their own transitions, whose midpoints do not necessarily converge to common pKa values, indicating that the four-state transition (Scheme 1) is not a result of highly cooperative transitions throughout the molecule. Rather, the pH-dependent conformational change of βLG might be a result of collective conformational changes of many residues. To extract such collective changes from the NMR data, we performed PCA.
PCA of pH Titration Data.
We performed a PCA of the pH titration data monitored by HSQC. Details are described in Materials and Methods and SI Fig. 9. As a result of the singular value decomposition, we obtained 13 PCs and corresponding contribution ratios. The accumulative contribution ratio of the first three PCs was 0.86 (SI Fig. 10). Thus, the three dominant PCs are likely to describe most contributions of the signal changes. Indeed, although we also performed the following fitting with the first four PCs, no apparent improvement was detected, consistent with the profile that the amplitude of PC4 was small over the pH range studied (Fig. 2).
Fig. 2.

Fitting of PCs. The pH dependencies of the first, second, third, and fourth PCs are shown. The lines are theoretical curves based on Eq. 2, in which only the first, second, and third PCs were considered.
We approximated the pH dependency of PCs with a simple four-state mechanism, S1⇄S2⇄S3⇄S4. The relations between these species are described as follows.
where, Ka,i is the acid dissociation constant for species Si. In the present scheme, Ka,i with an i of 1, 2, and 3 should be considered. From these relations, fractions of species i, fSi, as a function of pH were calculated. The PCs are described with the fractions for each species:
 |
where, I([H+]) is a 3-dimensional vector containing the first, second, and third PCs. ISi is a 3-dimensional vector describing the corresponding species. The PC data were fitted by using Eq. 2, in which variable parameters were Ka,i and ISi. The result of the fitting was quite satisfactory (Fig. 2, solid lines). Furthermore, by using the first three PCs, we can reconstruct four basis spectra (SI Fig. 11) and the chemical shift data for all residues (examples shown in SI Fig. 12). It should be noted that the PCA assumed a linear combination of the basis spectra. For NMR chemical shifts, this means the fast exchange between different conformational states (SI Fig. 13). Most of the peaks with the pH-dependent chemical shift change exhibited a single sharp shape over a wide range of pH, suggesting the validity of the assumption. However, for several residues, e.g., those on the D and E strands, showing a decrease in intensity above pH 7.0 (16), we have to consider the slow conformational change (see Discussion).
Importantly, the pKa values obtained, 2.9, 4.9, and 6.8, were consistent with those obtained previously with different methods (11, 12). From the PCA, CSDs for each transition, S1–S2, S2–S3, and S3–S4, were also obtained (Fig. 3A–C), and the results were mapped on the crystal structure (Fig. 3 D–F). Thus, it was further confirmed that the pH-dependent global transition of βLG is a result of the collective behavior of individual residues.
Fig. 3.

The result of the PCA. (A–C) CSDs for the transitions at pH 2.9 (A), 4.9 (B), and 6.8 (C). Heights and colorations of the bars indicate the distances and the directions of the signal displacements, respectively. The colorations obeying the Hue scale were indicated in Fig. 1C. (D–F) Mapping of the obtained CSDs at each pH on the crystal structure.
pH and Gdn-HCl Dependencies of Tryptophan Fluorescence.
The Gdn-HCl denaturation of wild-type and A34C βLGs also was measured at various pH values by monitoring tryptophan fluorescence (Fig. 4 A and B, at pH 3.0 and 7.0, respectively). Assuming certain baselines for the native (N) and unfolded (U) states, spectral change could be approximated by a two-state transition at a pH below 6.0 (16, 20). On the other hand, above a pH of 6.0, even if such a baseline were assumed, the spectral change could not be reproduced by a two-state transition, suggesting the accumulation of an intermediate state (I).
Fig. 4.

pH- and Gdn-HCl-dependent fluorescence spectra of βLG. (A and B) Fluorescence spectra of wild-type βLG in the presence of various concentrations of Gdn-HCl at pH 3.0 (A) and 7.0 (B). The concentration of Gdn-HCl increases from 0 M (red) to 6.0 M (purple) in steps of 0.24 M as guided by arrows. (C) The result of the global fitting. The markers indicate the fluorescence intensities at 330 nm at pH 2.0 (red), 4.0 (green), 6.0 (blue), and 8.0 (purple), respectively, and the lines indicate the theoretical curves. (D) The ΔG profiles for the N (red) and I (blue) states. The solid and broken lines are of the wild-type and A34C βLG, respectively.
Characterization of the Intermediate at Neutral pH.
We characterized the I state by spectral decomposition. βLG has two tryptophan residues: Trp-19 and Trp-61 (SI Fig. 6A). Spectral changes of wild-type βLG and a mutant, W61R βLG, with only one tryptophan residue (Trp-19) were measured at 0 and 6 M Gdn-HCl (SI Fig. 6B). The spectrum of W61R βLG indicates the environment of Trp-19. On the other hand, the difference spectrum between the wild-type and W61R βLGs would show the contribution by Trp-61. The spectrum of wild-type βLG and the contributing spectra of Trp-19 and Trp-61 are compared in SI Fig. 6B. The results indicated that, in the N state (i.e., at 0 M Gdn-HCl), the fluorescence of Trp-19 contributes most to the spectrum of wild-type βLG. On the other hand, in the U state (i.e., at 6 M Gdn-HCl), the fluorescence of Trp-61 was comparable with that of Trp-19.
Here, considering the spectral change at neutral pH (Fig. 4B), we assumed that in the I state the region around Trp-61 is unfolded but the region around Trp-19 remains native. Then, the spectrum in the I state was assumed to be a sum of the spectrum of Trp-19 in the N state and that of Trp-61 in the U state (SI Fig. 6C). With the calculated spectrum of the I state and a three-state mechanism, a series of spectra obtained at neutral pH (Fig. 4B) were reproduced satisfactorily (SI Fig. 6D). Thus, we concluded that the intermediate is indeed in a partially unfolded state as we assumed.
Analysis of the pH-Dependent Fluorescence Change.
We performed a global fitting of the pH- and Gdn-HCl-dependent changes of tryptophan fluorescence. The theoretical equation for the fitting is derived by combining the pH dependence of unfolding equilibrium with its Gdn-HCl dependence (21). As an equation describing the pH dependence of unfolding equilibrium, we used that elaborated by Tanford (5), in which the pKa differences between the N and U states produce the pH dependence of the free energy change of unfolding, ΔGU. For the unfolding transition of βLG, we assumed a three-state mechanism with the N, I, and U states. It is impractical to determine the pKa values for all titratable groups by analyzing fluorescence data. Therefore, we made several assumptions. The relevant titratable groups were assumed to be only carboxyl groups. The pKa values of these residues in the U state (pKa,U) were fixed at 4.0 (the median of pKa,Glu and pKa,Asp). Similarly, the pKa,N values of the residues except for Glu-89 were set to be the same. The pKa value of Glu-89 was fixed at 7.5 in the N state according to the previous report (15), whereas, in the I state, it was fixed at 4.0, the same as in the U state. The m values were assumed to depend on pH and were optimized at each pH. With these assumptions, the equilibrium constants between N and U or I and U were represented by
 |
and
 |
where pKa,N and n are the pKa values and the number of relevant residues other than Glu-89, respectively. KNU,0 and KIU,0 are equilibrium constants when [H+] is sufficiently low and [Gdn-HCl] = 0. From these equilibrium constants, the fractions in the N, I, and U states (fN, fI, and fU, respectively) are calculated. The observed spectrum can be constructed from the following equation.
where, SN and SU are the spectra obtained at 0 M and 6 M Gdn-HCl, respectively, and SI is the estimated spectrum for the I state. The global fitting was quite satisfactory, reproducing all of the observed fluorescence spectra (Fig. 4C). Fig. 4D shows the changes in ΔGNU and ΔGIU as a function of pH. Here, the ΔGNU and ΔGIU values can be obtained by the standard relation: ΔGNU(IU) = −RTlnKNU(IU), where R and T are the gas constant and temperature in Kelvin, respectively. The fitting parameters and their errors are listed in Table 1.
Table 1.
Results of the global fitting of the fluorescence data
| βLGs | pKa | n | ΔGNU,0/kJ·mol−1 | ΔGIU,0/kJ·mol−1 |
|---|---|---|---|---|
| Wild type | 3.7 ± 0.2 | 8.2 ± 5.6 | 29.0 ± 0.4 | 10.6 ± 0.3 |
| A34C | 3.6 ± 0.1 | 13.9 ± 2.0 | 35.4 ± 0.5 | 11.8 ± 0.4 |
H/D Exchange Experiments.
We performed the H/D exchange experiments at pDreading (pDr) 3.0, 4.7, and 6.9. The calculated protection factors (PFs) are shown in Fig. 5. Although the absolute values depended on pH, the patterns of the PFs were similar. The residues with the highest PF values constitute the core region (22). According to the protection patterns, the core regions were assigned to the A, C, F, G, H, and I strands and were independent of pH (Fig. 5, indicated by the gray background). The PF of the core was ≈1 × 105 at pDr 3.0 and 6.9, whereas it was 2 × 106 at pDr 4.7. Assuming the EX2 mechanism (22), the ΔGglobal values are calculated to be 30.0, 37.8, and 30.0 kJ·mol−1 at pDr 3.0, 4.7, and 6.9, respectively. These values are comparable to ΔGNU of 30.8 at pH 3.4, ΔGNI of 37.1 and 25.9 kJ·mol−1 at pH 5.1 and 7.3, from the fluorescence data, respectively (Fig. 4D). The agreement between ΔGglobal and ΔGNI values at pH 4.7 and 6.9 indicates that the I state is not protected from the exchange. Probably, although some residual structures were retained, the amide protons were largely exposed to solvents. Indeed, there are some reports that the molten globule state shows quite small PFs even when the state is dominantly populated (23). Thus, it is probable that the I state is a molten globule-like state.
Fig. 5.

PFs at pDr 3.0 (red), 4.7 (green), and 6.9 (blue). The broken lines indicate the maximum values of PF obtained at each pDr. The residues with the maximum PF values at any pH are indicated by the gray background. The lower triangles indicate the residues that did not show observable H/D exchange at pDr 4.9 during the measurement (6 months).
It is thought that the PF that is not the highest is determined by local fluctuation with the equilibrium constant Klocal (22). The PF values at the D and E strands and the GH loop decreased significantly at pDr 6.9, indicating that some fluctuation around these regions occurs at neutral pH even in the N state.
Discussion
The PCA of the NMR data showed that pH-dependent four-state transition of βLG (Scheme 1), reported previously (11, 12), is the result of the collective conformational changes of individual residues. Furthermore, the PCA revealed more detailed structural images at the level of amino acid residues (Fig. 3). We address aspects of the pH-dependent conformational transitions of βLG clarified by the PCA combined with a conventional analysis by using tryptophan fluorescence and H/D exchange.
M⇄Q Transition at pH 3.
At pH 3.0, both a decrease in ΔGNU and a collective conformational change were evident from the PCA and fluorescence data, respectively. The results of global fitting suggested that βLG is in equilibrium between the N and U states at this pH. Obtained fitting parameters, pKa and n, are listed in Table 1. Although these values may involve large errors because individual pKa values for the related residues are different, the results for A34C indicate that the pKa of 14 acidic residues is lowered to 3.6 by the formation of intermolecular hydrogen bonds or for other reasons in the N state. The candidates for such residues are Asp-11, -33, -53, and -129, and Glu-51, -127, -131, and -134 because they show both large CSD (Fig. 3A) and the formation of intra- and intermolecular hydrogen bonds in the crystal structure (referring to the structure with PDB ID 1BEB; data not shown).
There is a significant difference in the n values between the wild-type and A34C, i.e., 8 and 14, respectively. Considering this difference and the significant CSD (SI Fig. 7B), it is probable that some additional intermolecular ionic bonds between monomers are formed in the N state of A34C. Consequently, it is also likely that there are some differences in stabilities and unfolding processes of the two proteins. Even with these mutational effects, we believe that A34C βLG is an excellent model to analyze the pH-dependent conformational properties of βLG.
Q⇄N Transition at pH 5.
From our analysis of the fluorescence data, we found no evidence of a substantial conformational change at pH 5. Although PCA indicated an apparent transition at this pH, we could not detect a unique CSD pattern for the Q–N transition. Rather, it slightly resembles that for the M–Q transition. At this pH, changes in compactness and hydration of the βLG molecule are reported (14). These changes are probably caused by the octamer's formation. It is understandable that βLG molecules tend to aggregate at this pH because it is near to the pI. In our NMR experiments, white precipitates were often seen in the sample tube at pH 3.9–5.0 (data not shown). Slight changes in conditions probably determine whether a specific octamer or a nonspecific aggregate is formed. Anyway, these changes in the association state do not alter the conformation and stability of the monomers.
N⇄R Transition at pH 7.
In this pH range, the Gdn-HCl denaturation is described by the three-state mechanism. We conclude that I is a partially unfolded state around the D and E strands and the GH loop. This conclusion was based on the fact that the fluorescence data are satisfactorily described with two assumptions; that in the I state, Trp-61 is unfolded whereas Trp-19 remains native, and that the pKa value of Glu-89 is 7.5 in the N state whereas it is 4.0 in the I state, the same as the pKa value in the U state.
The decrease in ΔGNU (Fig. 4D) is explained by the anomalous pKa value of Glu-89, which is located on the EF loop. On the other hand, the pKa of Glu-89 in the I state is 4.0, the same as that in the U state. Therefore, ΔGIU does not change at this pH. As a result, as the pH increases, the relative population in the I state increases with respect to the N state, which might be the underlying mechanism of the Tanford transition.
In addition, the PF values at the D and E strands and the GH loop at pDr 6.9 were lower than those at pDr 3.0 and 4.7. This result suggests that, even in the N state, the D and E strands and the GH loop are fluctuating above pH 6.0. The PCA gave additional information: There are no significant changes at the D and E strands and the EF loop (Fig. 3C), which are characteristic of the Tanford transition (9, 16), which may be because the conformational change is too slow to be detected in the chemical shift (i.e., slower than the microsecond time scale; see SI Fig. 13). On the other hand, a cluster of residues with large CSDs is found at the G strand (Fig. 3C). Because the residues are located near the carboxyl group of Glu-89 in the structure, the chemical shift changes are due to the deprotonation of the carboxyl group. It must be noted that the residues at the G strand assume warm colors on the Hue plot. As can be seen from the Hue circle scale, the warm colors mean that the chemical shift change is mainly the decrease in δH as pH increases. It is generally regarded that the decrease of δH indicates the cleavage or attenuation of the hydrogen bond network (24). Thus, it is likely that the deprotonation of Glu-89 side chain causes the attenuation of the hydrogen bonds at the G strand, which leads to the local unfolding of the D and E strands of the intermediate. Taulier and Chalikian (12) reported that the density and compressibility of the βLG molecule are decreased upon the Tanford transition. They suggested that the βLG molecule might be loosened upon the transition. Their discussion also supports weakened hydrogen bonds at the G strand and subsequent fluctuation at the D and E strands and the GH loop.
The partially unfolded intermediate is likely relevant to the increased ligand-binding activity. Considering the KNU and KIU values from the fluorescence data, the population in the I state is quite small in the absence of denaturant. However, even in the N state, the PF values show profiles characteristic of the I state as described above. These characteristics are very similar to those in the “excited state” in the catalytic cycle of enzymes probed by relaxation dispersion experiments (25). The partially unfolded state might be the “excited” state, which could capture the hydrophobic ligand in the βLG ligand-binding reaction.
Conclusions
The PCA of chemical shift data on βLG gave a detailed and comprehensive understanding of the pH-dependent four-state conformational transition, indicating that it is a result of collective conformational changes of many residues. This approach applies to conformational transitions of various proteins monitored by heteronuclear NMR spectroscopy and will be useful to understand the molecular mechanism of the conformational transitions at the residue level.
However, when there are conformational changes on a slower time scale, like the residues on the D and E strands, PCA cannot detect such changes. To clarify the conformational change with a slower exchange, a precise model including other parameters, such as signal intensities or linewidths, must be considered. This will enable PCA to detect local conformational changes on a wider time scale.
Materials and Methods
Materials.
15N ammonia water and 13C glycerol were purchased from Shoko Tsusho (Tokyo, Japan). 13C methanol was obtained from Nippon Sanso (Tokyo, Japan). Other reagents were purchased from Nacalai Tesque (Kyoto, Japan). The methods of gene manipulation, protein expression, stable isotope-labeling, and purification were as described before (16, 26).
Gdn-HCl-Induced Unfolding Monitored by Tryptophan Fluorescence.
The tryptophan fluorescence spectra of A34C, W61R, and wild-type βLGs were measured at a protein concentration of 0.10 mg·ml−1 in 50 mM buffer. The excitation wavelength was 280 nm. The buffers used were Gly-HCl (pH 2.0–3.5), sodium acetate (pH 4.0–5.5), Mes-NaOH (pH 6.0–7.0), and Tris·HCl (pH 7.5, 8.0). The spectral changes of A34C and the wild-type βLGs induced by Gdn-HCl were measured at each pH. Extinction coefficients of mutants were calculated from amino acid sequences by the method of Gill and von Hippel (27).
NMR Measurements.
NMR measurements were performed by using a AV400-M, DRX 500, or DRX 600 (Bruker BioSpin, Rheinstetten, Germany). The conditions were the same as those in our previous report (16). By using 15N, 13C double-labeled A34C, sequential assignments of main chain atoms were performed at pH 2.4 by using the 600 MHz apparatus with CBCA(CO)NH, HNCACB, and HNCACO pulse sequences. The assignments and other NMR data analyses were performed with the program SPARKY 3, a program created by T. D. Goddard and D. G. Kneller (University of California, San Francisco, CA). The assigned chemical shifts are summarized in SI Table 2.
The Hue Plot.
The δH and δN data points for each residue obtained at each pH were connected by a smooth curve that is described by a triple-sigmoidal function by the curve fitting program. This equation is not used merely to smoothly connect the data points. Based on the curve obtained by the fitting, we numerically calculated d(δH)/d(pH) and d(δN)/d(pH), that is, the chemical shift changes of H and N signals per pH unit change. At each pH point, one residue has two data, d(δH)/d(pH) and d(δN)/d(pH), and these data specify a position on the Hue circle scale, so that the coloration and the depth of color for each point on the Hue plot are determined.
PCA for pH Titration Data.
The chemical shift data from each spectrum were represented as a one-dimensional vector that contains the δH and δN values. As a pretreatment, δN values were divided by 8, and all δH and δN were subtracted by its average over the measured pH range. The vectors were used to build a two-dimensional matrix X, in which the rows are the chemical shift data from each spectrum and the columns were the variables of pH. Then, the matrix size is 304 [152(number of traceable residues) × 2(δH and δN)] × 13(measured pH points).
A singular value decomposition analysis was performed on the matrix X, which gives three matrices, M, W, and V, where M·W·V = X. V includes some uncorrelated new variables (the PCs) and W gives contribution ratios for individual PCs. For the subsequent reconstruction of the basis spectra, see Results and SI Fig. 9.
H/D Exchange Experiments.
The H/D exchange experiments were performed at 40°C and pDr 3.0, 4.7, and 6.9 by dissolving 10 mg of lyophilized protein into 1 ml of 2H2O. In the case of pH 4.7, the sample solution was filtered before the measurements because aggregates were formed in the solution. The reaction was monitored by recording a series of 15N–1H HSQC spectra for at least a month. For each spectrum, peak intensities were calculated by using SPARKY and analyzed assuming single exponential decay with time. Protection factors were calculated as a ratio between the intrinsic exchange rate constant predicted from the amino acid sequence and the observed rate constant (28, 29).
Supplementary Material
Acknowledgments
We thank Carl A. Batt (Cornell University, Ithica, NY) for valuable comments; Rossen Apostolov (Osaka University, Osaka, Japan) for instruction regarding PCA; and Hideo Akutsu, Takahisa Ikegami (Osaka University), and Masaru Hoshino (Kyoto University, Kyoto, Japan) for instructions regarding NMR measurements. This work was supported in part by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Science, Culture and Sports.
Abbreviations
- βLG
bovine β-lactoglobulin
- HSQC
heteronuclear single quantum coherence
- NMR
nuclear magnetic resonance
- CSD
chemical shift difference
- PC
principal component
- PCA
PC analysis
- PF
protection factor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702112104/DC1.
References
- 1.Jaumot J, Vives M, Gargallo R. Anal Biochem. 2004;327:1–13. doi: 10.1016/j.ab.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Z, Madrid M, Evanseck JD, Madura JD. J Am Chem Soc. 2005;127:17253–17260. doi: 10.1021/ja053973d. [DOI] [PubMed] [Google Scholar]
- 3.Jaumot J, Marchán V, Gargallo R, Grandas A, Tauler R. Anal Chem. 2004;76:7094–7101. doi: 10.1021/ac049509t. [DOI] [PubMed] [Google Scholar]
- 4.Szundi I, Liao GL, Einarsdóttir Ó. Biochemistry. 2001;40:2332–2339. doi: 10.1021/bi002220v. [DOI] [PubMed] [Google Scholar]
- 5.Tanford C. Adv Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 6.Tollinger M, Crowhurst KA, Kay LE, Forman-Kay JD. Proc Natl Acad Sci USA. 2003;100:4545–4550. doi: 10.1073/pnas.0736600100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brownlow S, Morais Cabral JH, Cooper R, Flower DR, Yewdall SJ, Polikarpov I, North AC, Sawyer L. Structure (London) 1997;5:481–495. doi: 10.1016/s0969-2126(97)00205-0. [DOI] [PubMed] [Google Scholar]
- 8.Kuwata K, Hoshino M, Forge V, Era S, Batt CA, Goto Y. Protein Sci. 1999;8:2541–2545. doi: 10.1110/ps.8.11.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin BY, Bewley MC, Creamer LK, Baker HM, Baker EN, Jameson GB. Biochemistry. 1998;37:14014–14023. doi: 10.1021/bi981016t. [DOI] [PubMed] [Google Scholar]
- 10.Uhrínová S, Smith MH, Jameson GB, Uhrín D, Sawyer L, Barlow PN. Biochemistry. 2000;39:3565–3574. doi: 10.1021/bi992629o. [DOI] [PubMed] [Google Scholar]
- 11.Sawyer L, Kontopidis G. Biochim Biophys Acta. 2000;1482:136–148. doi: 10.1016/s0167-4838(00)00160-6. [DOI] [PubMed] [Google Scholar]
- 12.Taulier N, Chalikian TV. J Mol Biol. 2001;314:873–889. doi: 10.1006/jmbi.2001.5188. [DOI] [PubMed] [Google Scholar]
- 13.Fogolari F, Ragona L, Zetta L, Romagnoli S, De Kruif KG, Molinari H. FEBS Lett. 1998;436:149–154. doi: 10.1016/s0014-5793(98)00936-3. [DOI] [PubMed] [Google Scholar]
- 14.Timasheff SN, Mescanti L, Basch JJ, Townend R. J Biol Chem. 1966;241:2496–2501. [PubMed] [Google Scholar]
- 15.Tanford C, Bunville LG, Nozaki Y. J Am Chem Soc. 1959;81:4032–4036. [Google Scholar]
- 16.Sakurai K, Goto Y. J Mol Biol. 2006;356:483–496. doi: 10.1016/j.jmb.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 17.Gottschalk M, Nilsson H, Roos H, Halle B. Protein Sci. 2003;12:2404–2411. doi: 10.1110/ps.0305903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pessen H, Purcell JM, Farrel HM., Jr Biochim Biophys Acta. 1985;828:1–12. [Google Scholar]
- 19.Casal HL, Köhler U, Mantsch HH. Biochim Biophys Acta. 1988;957:11–20. doi: 10.1016/0167-4838(88)90152-5. [DOI] [PubMed] [Google Scholar]
- 20.Sakai K, Sakurai K, Sakai M, Hoshino M, Goto Y. Protein Sci. 2000;9:1719–1729. [PMC free article] [PubMed] [Google Scholar]
- 21.Barrick D, Baldwin RL. Biochemistry. 1993;32:3790–3796. doi: 10.1021/bi00065a035. [DOI] [PubMed] [Google Scholar]
- 22.Bai Y, Sosnick TR, Mayne L, Englander SW. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forge V, Wijesinha RT, Balbach J, Brew K, Robinson CV, Redfield C, Dobson CM. J Mol Biol. 1999;288:673–688. doi: 10.1006/jmbi.1999.2687. [DOI] [PubMed] [Google Scholar]
- 24.Parker LL, Houk AR, Jensen JH. J Am Chem Soc. 2006;128:9863–9872. doi: 10.1021/ja0617901. [DOI] [PubMed] [Google Scholar]
- 25.Boehr DD, McElheny D, Dyson HJ, Wright PE. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 26.Kim TR, Goto Y, Hirota N, Kuwata K, Denton H, Wu SY, Sawyer L, Batt CA. Protein Eng. 1997;10:1339–1345. doi: 10.1093/protein/10.11.1339. [DOI] [PubMed] [Google Scholar]
- 27.Gill SC, von Hippel PH. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 28.Bai Y, Milne JS, Mayne L, Englander SW. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connelly GP, Bai Y, Jeng MF, Englander SW. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.