

Abstract
What is already known about this subject
Several clinical studies have shown that the toxicity of tipifarnib in cancer patients is acceptable.
However, previous exposure vs. toxicity analyses have been only performed for a limited range of toxic effects and were based on a single study involving a limited number of patients.
What this study adds
This study represents the first comprehensive and large-scale exposure vs. response analysis based on five clinical studies, involving more than 600 patients and evaluating more than 10 different toxicities.
The results have defined the relationship between haematological toxicities and tipifarnib exposure in patients with solid tumours. The occurrence of exposure-related nonhaematological toxicities was low regardless of tumour type.
Aims
To explore the potential relationship between systemic exposure to tipifarnib and the incidence of toxicity in cancer patients.
Methods
Data from 673 subjects (540 receiving tipifarnib and 133 receiving placebo) were included in the analysis. Tipifarnib was administered in doses ranging from 100 mg to 850 mg twice daily under fed conditions for 21 days in a 28 day cycle. Univariate and multivariate logistic regression models were used to evaluate the relationships between tipifarnib exposure and haematological (neutropenia and thrombocytopenia) and nonhaematological toxicities. Tipifarnib exposure was quantified as the area under the curve during the first cycle of chemotherapy (AUC), the maximum plasma concentration (Cmax), the time above plasma tipifarnib concentrations of 400 (T400) and 600 ng ml−1 (T600), the cumulative area under the curve (AUCT), and the area under the curve density (AUCD), defined as the ratio AUCT to the duration of treatment. The nonhaematological toxicities measured were elevation of AST, ALT, bilirubin and serum creatinine, the occurrence of a skin rash, CNS or peripheral neuropathy, nausea and vomiting, diarrhoea and inflammation of the gastrointestinal tract. Odds ratios (OR) associated with drug exposure were used to measure the effect of the drug.
Results
Tipifarnib AUC exhibited a positive and significant association with neutropenia grade ≥3 (OR 1.69, 95% CI 1.47, 1.95) and thrombocytopenia grade ≥3 (OR 1.41, 95% CI 1.21, 1.63) in patients with solid tumours, but not in refractory or relapsed AML patients. The incidence of exposure-related nonhaematological toxicity is small regardless of tumour type. No association was found between tipifarnib AUC and the elevation of AST, ALT and total bilirubin, and nausea and vomiting. There was a weak relationship between tipifarnib AUC and serum creatinine elevation (OR 1.18, 95% CI 1.11, 1.26), CNS (OR 1.05, 95% CI 1.01, 1.10) and peripheral neurotoxicity (OR 1.10, 95% CI 1.03, 1.18), diarrhoea (OR 1.14, 95% CI 1.08, 1.21), gastrointestinal tract inflammation (OR 1.13, 95% CI 1.07, 1.19), and skin rash (OR 1.10, 95% CI 1.04, 1.17).
Conclusions
In those patients who develop severe toxicity, dose reduction may improve the tolerability of tipifarnib. However, an exposure-guided approach to dosage adjustment to limit haematological and nonhaematological toxicity is not warranted.
Keywords: cancer patients, pharmacokinetics, tipifarnib, toxicity
Introduction
Tipifarnib (R115777, Zarnestra®) is a potent, selective and competitive inhibitor of the enzyme farnesyltransferase (FTase) [4, 5], which is important in the processing and activation of signalling molecules linked to cell proliferation and malignant transformation, such as Ras, Rho-B, Rac, and lamin proteins. Inhibition of FTase by tipifarnib induces antileukaemic and antitumour activity, which has been demonstrated in both in vitro and in vivo animal models. Tipifarnib is believed to exert its antitumour activity, at least in part, by preventing the farnesylation of proteins involved in mechanisms that extend beyond Ras. The variety of cellular and tumour tissue responses elicited by tipifarnib treatment in vivo is consistent with the hypothesis that its antitumour effects are being derived from disruption of multiple effectors downstream of FTase inhibition [4].
Several haematological and nonhaematological dose-limiting toxicities have been reported for tipifarnib [6, 7]. Investigating the relationship between systemic exposure to antineoplastics and their safety in cancer treatment is an important aspect of the development of anticancer agents [1]. The prospective population pharmacokinetic and pharmacodynamic (PK/PD) approach has been increasingly implemented during the clinical development of anticancer agents in order to assess systemic exposure as a prognostic factor for clinical outcome. Recently modelling techniques have been used to elucidate further the nature of exposure-response relationships in cancer chemotherapy [2, 3]. The model-based integration of the exposure-response information collected during the clinical development of tipifarnib was used in early phase I/II studies in patients with solid tumours to explore the occurrence of adverse events [6–8]. The objective of the present analysis was to explore further the relationship between systemic exposure to tipifarnib and the incidence of haematological (neutropenia and thrombocytopenia) and nonhaematological (increase in AST, ALT, bilirubin and creatinine, neurotoxicity, skin rash, nausea and vomiting, diarrhoea and gastrointestinal tract inflammations) toxicities in cancer patients from five clinical studies, including subjects with a diagnosis of relapsed and refractory acute myelogenous leukaemia (r-AML).
Methods
Patients and treatments
The current analysis was based on data from subjects who had participated in five clinical studies during the development of tipifarnib. There aim was to assess the safety and effectiveness of tipifarnib as a single agent in advanced cancer [10], advanced breast cancer [8], urothelial tract transitional cell carcinoma [11], advanced colorectal cancer [13] and r-AML [12]. Table 1 provides a brief summary of the characteristics of the studies, but a more detailed description has been published elsewhere [8, 10–13]. All these studies were conducted in accordance with principles for human experimentation as defined in the Declaration of Helsinki (1983) and were approved by the Human Investigational Review Board of each study centre.
Table 1.
Characteristics of the studies and subjects included in the analysis
| Study | Population | Dose (mg twice daily) | Sampling schedule | Sex (M/F) | Age*(years) | Weight*(kg) | AUCb*(mg l−1 h) | Cmaxb*(mg l−1) | T > 400*(h) | T > 600*(h) | AUCD*(daily mg l−1 h) | AUCT*(g l−1 h) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A [10] | Advanced cancer | 100–850 | Full PK profile on day 1 and 21d | 20/14 | 57 (35–81) | 72 (49–124) | 8.5 (3.2–24.7) | 1.6 (0.6–5.6) | 4.3 (0–11) | 2.8 (0–8) | 3.6 (0.7–11.8) | 0.26 (0.04-1.31) |
| B [8] | Advanced breast cancer | 300 | Sparse sampling on week 2, 6 and 10: Pre-dose, 1h and another sample after 2h | 0/24 | 50 (37–82) | 64 (49–93) | 7.4 (3.2–13.3) | 1.3 (0.6–2.2) | 3.8 (0–6.8) | 0.6 (0–4.8) | 2.8 (1.4–6.7) | 0.53 (0.01–1.63) |
| C [11] | Advanced urothelial tract transitional cell carcinoma | 300 | Sparse sampling on day 8, 15 and 22: Pre-dose, 1h and another sample after 2h | 17/11 | 64 (41–82) | 75 (50–133) | 8.3 (2.5–14.5) | 1.4 (0.3–3.1) | 4.0 (0–8.3) | 1.5 (0–5.3) | 3.3 (1.1–7.0) | 0.23 (0.05–1.75) |
| D [12] | Refractory or relapsed acute myeloid leukaemia | 600 | Sparse sampling on day 1, 8, and 15:e Pre-dose, 1h and another sample after 2h | 141/101 | 62 (18–85) | 71 (41–120) | 7.4 (1.8–30.6) | 1.3 (0.3–4.8) | 7.0 (0–12) | 5.0 (0–12) | 6.2 (1.5–30.6) | 0.35 (0.01–2.54) |
| E [13] | Advanced colorectal cancer | 0a−300 | Sparse sampling on day 1, 8, and 15: Pre-dose, 1h and another sample after 2h | 223/120 | 61 (21–83) | 73 (41–119) | 9.3 (1.7–25.0)c | 1.6 (0.2–5.8)c | 4.8 (0–12)c | 2.5 (0–8)c | 3.5 (0.7–12.5) | 0.44 (0.01–3.77) |
Results expressed as median (min–max).
Subjects randomized to the placebo group.
Dose normalized (600 mg twice daily.).
Only subjects treated with tipifarnib.
Plasma samples were drawn on days 1 and 21 immediately before administration and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, and 12 h. On day 21, additional plasma samples were collected at 16, 24, 36, 48 and 72 hours after drug administration. On days 7 and 14, a plasma sample was drawn immediately prior to drug administration (isolated measurements).
In 20 subjects, plasma samples were drawn immediately before administration and at 0.5, 1, 2, 3, 5, 8, and 12 h on day 1.
Subjects were eligible if they had histological or cytological confirmation of a malignant tumour not amenable to established forms of therapy. Other eligibility criteria included a World Health Organization performance status of 0–2, anticipated life expectancy of at least 3 months, and age >18 years. Previous anticancer radiation therapy and/or chemotherapy, if given, had to be discontinued for at least 4 weeks before entry into the study, or 6 weeks in the case of pretreatment with nitrosoureas or mitomycin C. Patients had to have adequate unassisted oral or adequate enteral intake to maintain a reasonable state of nutrition, a negative pregnancy test (in female patients with reproductive potential), and normal hepatic and renal function, defined as bilirubin ≤1.5 times normal upper limit, AST and ALT ≤2.5 times normal upper limit (≤5 times normal upper limit in case of hepatic metastases), and serum creatinine ≤1.5 times normal upper limit. All patients had to have acceptable bone marrow function, defined as white blood cells >3500 µl−1, granulocyte count >1500 µl−1 and platelets >100 000 µl−1, except in patients with a diagnosis of refractory or relapsed acute myeloid leukaemia. Subjects fulfilling one or more of the following criteria were not selected: prior treatment with farnesyltransferase inhibitors, prior extensive radiation therapy (>25% of bone marrow reserve); prior bone marrow transplantation or high dose chemotherapy with marrow or stem cell rescue, concurrent radiation therapy, chemotherapy, hormonal therapy or immunotherapy, participation in a clinical trial involving an investigational drug in the past 30 days or concurrent enrolment in another investigational trial and, any coexisting medical condition that was likely to interfere with study procedures and/or results. Informed consent was obtained from each subject after being told the potential risks and benefits, as well as the investigational nature of the study.
Subjects were included in this analysis they were receiving oral tipifarnib in doses ranging from 100 mg to 850 mg twice daily under fed conditions for 3 consecutive weeks, followed by 1 week of rest, or matched placebo in the same schedule. Subjects received the treatment until disease progression or severe toxicity occurred. The 3 weeks on – 1 week off schedule was selected, as it was the recommended dosing regimen for subjects with acute myeloid leukaemia [12]. Subjects treated with tipifarnib were included in this analysis if plasma drug concentrations were available for population pharmacokinetic analysis [9]. Subjects treated with placebo were also included in the analysis, as were those who had a toxicity assessment at baseline and during the study conduct.
Pharmacokinetic analysis
A population pharmacokinetic model had been developed previously using data obtained from 15 clinical studies [9]. An open, three-compartment model with linear elimination from the central compartment was used to describe the pharmacokinetics of tipifarnib in plasma after intravenous infusion. Systemically available tipifarnib was cleared from the body in a linear fashion, which was influenced by total bilirubin concentration. The volume of the central compartment was directly correlated with body weight. Oral absorption was modelled as a sequential zero-order input into the depot compartment, followed by first-order absorption from the depot compartment to the systemic circulation, and a lag time.
As the population analysis revealed between and within subject variability on pharmacokinetic parameters and time-invariant pharmacokinetics, the individual parameters obtained at steady-state during the first course of treatment were used to derive the exposure variables used in the statistical analysis presented here. For each subject who received tipifarnib, the individual pharmacokinetic parameters were calculated from the Bayesian estimates of the random effects, by using the final parameter estimates of the population pharmacokinetic model as prior information, and the plasma tipifarnib concentration data. The NONMEM V level 1.1 software package (GloboMax, Hanover, MD, USA) was used to derive the individual pharmacokinetic parameters.
Table 1 summarizes the sampling scheme used in each of the clinical studies included in the current analysis. Tipifarnib plasma concentrations were measured using either high-performance liquid chromatography with ultraviolet detection (HPLC-UV) or liquid chromatography with tandem mass spectrometry (LC-MS/MS). A cross validation study between both techniques confirmed their consistency and interchangeability. The lower limit of quantification for the HPLC-UV and LC-MS/MS methods was 1.00 and 0.50 ng ml−1, respectively. The mean overall coefficient of variation was less than 7.1% across the validated range of concentrations, the highest of which was 5000 ng ml−1. More detailed information about the methods has been published [9].
The AUC of tipifarnib at steady state during the first cycle of treatment was determined from the expression Fabs × Davg/CL, where Fabs and CL represent the Bayesian estimates of the absolute bioavailability and systemic clearance, and Davg represents the average of the twice daily dose administered to each patient during the first cycle of tipifarnib treatment, which takes into account compliance to therapy and/or missing doses during the first cycle of tipifarnib treatment. Davg was calculated from the following equation:
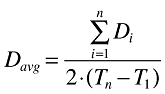 |
(1) |
where Di is the amount of tipifarnib administered at the ith dose of the first course of treatment; n is the total number of tipifarnib doses administered from day 1 (T1) to the last day of dosing during the first course of treatment (Tn), which corresponds to day 22. The predicted time that plasma tipifarnib concentrations were above threshold values of 400 ng ml−1 (T400) and 600 ng ml−1 (T600), together with the maximum plasma concentration (Cmax) at steady state during the first cycle of treatment were estimated for a dose equal to Davg based on the individual estimates of pharmacokinetic parameters. Threshold values of 400 and 600 ng ml−1 were selected because they were similar to the 33th and 66th percentiles of the steady-state trough concentrations obtained after the administration of tipifarnib 600 mg twice daily.
In order to take into account the differences in the duration of the treatment between patients, cumulative AUC and AUC density were also analyzed as exposure variables. The total or cumulative AUC (AUCT) was calculated from the expression Fabs × DT/CL, where DT is the total or cumulative dose administered until the worst grade of toxicity occurred. The AUC density (AUCD) was obtained by dividing the AUCT by the time between the start of the tipifarnib treatment and the occurrence of the worst grade toxicity, or the end of the study.
Tipifarnib toxicity
Exposure vs. toxicity analyses were conducted on selected haematological and nonhaematological toxicity outcomes. Two haematological toxicities, neutropenia and thrombocytopenia, and 10 nonhaematological toxicities, including elevation of AST, ALT, bilirubin or serum creatinine, and the presence of central nervous system (CNS) or peripheral neuropathy, skin rash, nausea and vomiting, diarrhoea and gastrointestinal tract inflammation, were selected.
The dependent variable in this analysis was the presence or absence of a given toxicity during the study. For haematological toxicities, this was defined as the presence or absence of toxicity grade ≥3, according to the definitions provided by the National Cancer Institute Common Toxicity Criteria version 2.0 (NCI CTC). For nonhaematological toxicities, the dependent variable was defined as the presence or absence of toxicity grade ≥2 at any time during the study, according to the definitions provided by the NCI CTC and using the adverse event groupings based on World Health Organization (WHO) preferred terms, when deemed appropriate. The preferred terms for CNS neuropathy were: aggressive reactions, agitation, amnesia, anxiety, apathy, aphasia, ataxia, bradykinesia, cerebellar syndrome, concentration impaired, confusion, convulsions, convulsions grand mal, co-ordination abnormal, delirium, delusion, dementia, depression, depression aggravated, dizziness, dysphonia, emotional lability, gait abnormal, hallucination, hyperkinesia, hypokinesia, insomnia, nervousness, paranoid reaction, paroniria, sleep disorder, somnolence, speech disorder, stupor, abnormal thinking, tremor, and vertigo. Those for peripheral neuropathy were: dysaesthesia, hyperaesthesia, hypertonia, hypoaesthesia, hyporeflexia, hypotonia, lower motor neurone lesion, muscle weakness, neuralgia, neuritis, neuropathy, neuropathy peripheral, paraesthesia, reflexes abnormal, sensory disturbance, and temperature changed sensation. The preferred terms for diarrhoea were: diarrhoea, ileal motility increased, diarrhoea Clostridium difficile, increased stool frequency and diarrhoea bloody. Those for gastrointestinal tract inflammation were: dyspepsia, stomatitis, mucositis nos, stomatitis ulcerative, gastritis, gastroesophageal reflux, colitis, enteritis, gingivitis, enanthema, gastroenteritis, stomatitis aphthous, cheilitis diverticulitis, glossitis, and enterocolitis.
Statistical analysis
Univariate logistic regression analysis was performed using systemic exposure to tipifarnib as the independent variable. For a given variable, such as the AUC, the univariate logistic model was described by the following equation:
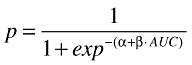 |
(1a) |
where p is the predicted probability for the presence of toxicity, and α and β are the model parameters estimated from the binary data. The results of the logistic regression analyses are presented in terms of an odds-ratio (OR), which is equivalent to eβ, and represents the factor by which the risk of toxicity is increased with an increase in one unit of exposure (for instance, 1 mg ml−1 h in AUC). The analysis for nonhaematological variables was conducted using the complete dataset. However, as haematological toxicity in r-AML subjects might be a consequence of the disease and/or drug treatment, univariate logistic regression analyses were performed separately in solid tumours and r-AML patients.
In addition, a multivariate logistic regression model, which included the effect of the treatment duration (DUR), age, tumour type (TYPE) and baseline (BSL) toxicity assessment, was fitted to the combined data. In order to assess these effects, patients were classified according to age (<65 years or ≥65 years), whether the baseline values of their laboratory parameter were or were not within normal limits, and tumour type (r-AML or solid tumour). In the case of skin rash, CNS and peripheral neurotoxicity, nausea and vomiting, diarrhoea and gastrointestinal tract inflammation, the baseline value of the assessment was not used, as these toxicities were absent at the beginning of the studies.
For a given exposure variable, such as the AUC, the multivariate logistic model is described as follows:
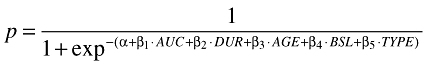 |
(2) |
Similarly, a multivariate logistic regression model was fitted separately to the solid tumour and r-AML patients for neutropenia and thrombocytopenia data.
Logistic regression analyses were performed under the assumption that there were a sufficient number of events for a meaningful analysis. P values lower than 0.05 were considered as statistically significant and no correction for multiple statistical testing was implemented. The nature of the current analysis was purely exploratory or a hypothesis generating exercise. Thus, the estimates of the parameters of interest, 95% confidence intervals and P values were determined to assist in evaluating the exposure-response relationships and therefore should be cautiously interpreted. Statistical analysis was performed using S-PLUS Professional, Version 6.0.3 for Windows (Insightful, Seattle, WA, USA).
Results
In total, data from 723 subjects were analyzed. Five hundred and ninety (81.6%) patients received oral tipifarnib twice daily, whereas the remaining 133 (18.4%) were included in the colorectal cancer study and received placebo. Fifty subjects (6.9%) receiving tipifarnib were excluded from the analysis due to a lack of pharmacokinetic data. Therefore, 673 subjects were eligible for the present analysis. A summary of the patient characteristics is presented in Table 1. The median age was 61 years (range 18–85), 263 (39%) subjects were ≥65 years old, and 58 (8.6%) were ≥75 years old. The median weight was 72 kg (range 41–133) and 40% of the subjects were female. Two hundred and forty-two (36%) subjects were diagnosed with r-AML, whereas the remaining 431 (64%) had advanced solid tumours. At baseline, the percentage of subjects with AST, ALT, total bilirubin and serum creatinine values higher than the upper limit of the normal range was 27.7%, 26.2%, 10.1% and 12.6%, respectively. These results were consistent across different tumour types (solid tumour vs. r-AML) and treatments (tipifarnib vs. placebo).
Figure 1 displays the individual Bayesian model predicted vs. the observed tipifarnib plasma concentrations. Uniform scatter around the line of identity is apparent, which indicates the absence of bias in the Bayesian estimations. A summary of descriptive statistics for the pharmacokinetic parameters for tipifarnib is shown in Table 1. Median (range) AUC, Cmax, T400, T600, AUCD and AUCT was 5.35 mg l−1 h (0.8–30.6 mg l−1 h), 1.0 mg l−1 (0.1–4.8 mg l−1), 5.5 h (0–12 h), 3.5 h (0–12 h), 8.61 mg l−1 h day−1 (1.4–61.2 mg l−1 h day−1), 0.367 g l−1 h (0.003–3.778 g l−1 h), respectively. The median (range) of the dose-normalized (600 mg twice daily) AUC and Cmax was 8.3 mg l−1 h day−1 (1.7–30.6) and 1.5 mg l−1 (0.2–5.8), respectively. As shown in Figure 2, the correlations between Cmax, T400, T600 and AUCD vs. AUC were relatively high (r > 0.92), and were similar in patients with full pharmacokinetic profile and those with sparse pharmacokinetic samples. However, the correlation between AUCT and AUC was low (r = 0.14), contrasting with that between AUCT and the duration of tipifarnib treatment (r = 0.90).
Figure 1.

Relationship between individual Bayesian predicted and observed plasma concentrations of tipifarnib
Figure 2.

Correlations between tipifarnib exposure measurements: AUC vs. Cmax, time above 400 ng ml−1 (T400), 600 ng ml−1 (T600) and AUC density. Black circles represent patients with full pharmacokinetic profile, while open circles represent the patients with sparse pharmacokinetic samples.
The data on haematological and nonhaematological toxicity are presented in Table 2. As expected, the incidence of haematological toxicities of grade ≥3 was different in subjects with r-AML compared with those with solid tumours. More than 85% of the subjects with r-AML had neutropenia and thrombocytopenia of grade ≥3, but less than 25% of subjects with solid tumours had such a level of toxicity. Overall, the incidence of CNS neuropathy of grade ≥2, and nausea and vomiting of grade ≥2 was higher than 20%. The lowest incidence corresponded to the elevation of AST or ALT of grade ≥2 and the presence of peripheral neurotoxicity of grade ≥2, which were observed in less than 10% of the subjects.
Table 2.
Incidence of tipifarnib toxicity stratified by cancer type
| Solid tumours | r-AML | Overall | ||
|---|---|---|---|---|
| Toxicity | Placebo, N (%) | Tipifarnib, N (%) | Tipifarnib, N (%) | Tipifarnib, N (%) |
| Non-haematological grade ≥ 2 | ||||
| AST | 25 (20.6) | 20 (7.6) | 15 (7.94) | 35 (7.7) |
| ALT | 18 (14.2) | 13 (4.7) | 21 (11.2) | 34 (7.3) |
| Bilirubin | 31 (24.4) | 41 (14.6) | 40 (21.3) | 81 (17.3) |
| Creatinine | 6 (4.7) | 20 (7.0) | 59 (11.3) | 79 (15.3) |
| Skin rash | 3 (2.6) | 29 (9.7) | 42 (17.4) | 71 (13.1) |
| CNS neuropathy | 30 (22.6) | 74 (24.8) | 69 (28.5) | 143 (26.5) |
| Peripheral neuropathy | 4 (3.0) | 23 (7.7) | 29 (12.0) | 52 (9.6) |
| Nausea and vomiting | 27 (20.3) | 61 (20.5) | 68 (28.1) | 129 (23.9) |
| Diarrhoea | 4 (3.0) | 33 (11.1) | 48 (19.8) | 81 (15.0) |
| GI inflammation | 4 (3.0) | 34 (11.4) | 54 (22.3) | 88 (16.3) |
| Haematological grade ≥ 3 | ||||
| Neutropenia | 1 (0.8) | 73 (24.9) | 196 (86.3) | – |
| Thrombocytopenia | 2 (1.5) | 44 (14.7) | 224 (94.9) | – |
The incidence of neutropenia and thrombocytopenia grade ≥3 in subjects with r-AML was not associated with tipifarnib AUC (Table 3). However, statistically significant associations were found in subjects with solid tumours. Overall, statistically significant associations were found between tipifarnib AUC and grade ≥2 toxicities for serum creatinine, skin rash, CNS and peripheral neurotoxicity, diarrhoea and gastrointestinal inflammation. Notably, regardless of the type of toxicity, similar degrees of association between those toxicities and AUC, Cmax, T400, T600 and AUCD were observed, in that less than a 10% difference in the OR point estimates was found. These results are consistent with the high correlation found among these exposure variables, and justify the selection of AUC to conduct the multivariate analysis. AUC was preferred over Cmax, T400, or T600 because it was estimated with greater accuracy and precision from the sparse sampling protocols in the multiple dosing regimens analyzed. In addition, AUCD was not chosen because the duration of treatment is only precisely known when the treatment ends. It was assumed that the conclusions derived from the multivariate analysis based on AUC hold true for Cmax, T400, T600 and AUCD, and those for duration of treatment can be extrapolated to AUCT. The former was selected over the latter because it is easier to interpret the change in the incidence of toxicity expressed per unit of treatment duration than per unit of AUCT. The results of the univariate analysis are displayed graphically for the relationship between tipifarnib AUC and the incidence of haematological (Figure 3) and selected nonhaematological (Figure 4) toxicities.
Table 3.
Estimated odds ratio (95% confidence interval) from univariate analysis of tipifarnib exposure as a predictor of toxicity
| AUC | CMax | T > 400 ng ml−1 | Tipifarnib exposure† T > 600 ng ml−1 | AUCD | AUCT | |
|---|---|---|---|---|---|---|
| Non-haematological toxicityGrade ≥ 2 | ||||||
| AST | 0.92 (0.85, 1.00) | 0.90 (0.83, 0.99)* | 0.95 (0.87, 1.03) | 0.96 (0.86, 1.06) | 0.94 (0.89, 1.00) | 1.03 (0.99, 1.07) |
| ALT | 1.00 (0.92, 1.08) | 0.98 (0.90, 1.08) | 1.01 (0.93, 1.10) | 1.05 (0.95, 1.17) | 0.99 (0.94, 1.05) | 1.05 (1.01, 1.09)* |
| Bilirubin | 1.03 (0.97, 1.09) | 1.02 (0.96, 1.09) | 1.06 (1.00, 1.13) | 1.07 (1.00, 1.16) | 1.02 (0.98, 1.06) | 1.06 (1.02, 1.11)* |
| Creatinine | 1.16 (1.09, 1.23)* | 1.17 (1.09, 1.25)* | 1.19 (1.11, 1.28)* | 1.23 (1.13, 1.33)* | 1.06 (1.03, 1.09)* | 1.13 (1.08, 1.18)* |
| Skin rash | 1.09 (1.03, 1.16)* | 1.11 (1.04, 1.09)* | 1.12 (1.04, 1.20)* | 1.12 (1.02, 1.22)* | 1.06 (1.03, 1.10)* | 0.93 (0.88, 0.97)* |
| CNS neuropathy | 1.04 (1.00, 1.09)* | 1.03 (0.98, 1.09) | 1.08 (1.02, 1.13)* | 1.07 (1.00, 1.14) | 1.02 (0.99, 1.04) | 0.97 (0.94, 1.00) |
| Peripheral neuropathy | 1.10 (1.03, 1.17)* | 1.12 (1.03, 1.20)* | 1.13 (1.04, 1.23)* | 1.14 (1.04, 1.26)* | 1.04 (1.00, 1.07) | 1.04 (1.01, 1.07)* |
| Nausea and vomiting | 1.02 (0.97, 1.06) | 1.03 (0.97, 1.08) | 1.02 (0.97, 1.08) | 1.01 (0.95, 1.08) | 1.01 (0.99, 1.04) | 0.95 (0.92, 0.98)* |
| Diarrhoea | 1.14 (1.08, 1.21)* | 1.18 (1.10, 1.26)* | 1.19 (1.11, 1.28)* | 1.20 (1.10, 1.30)* | 1.06 (1.03, 1.10)* | 1.00 (0.97, 1.03) |
| GI Inflammation | 1.12 (1.06, 1.19)* | 1.16 (1.09, 1.24)* | 1.16 (1.08, 1.24)* | 1.16 (1.07, 1.25)* | 1.07 (1.04, 1.10)* | 0.97 (0.93, 1.00) |
| Haematological toxicityGrade ≥ 3 | ||||||
| Neutropenia (Solid tumour) | 1.60 (1.42, 1.79)* | 1.53 (1.41, 1.79)* | 1.57 (1.40, 1.75)* | 1.73 (1.52, 1.97)* | 1.21 (1.14, 1.27)* | 2.03 (1.73, 2.38)* |
| Thrombocytopenia (Solid tumour) | 1.50 (1.33, 1.68)* | 1.44 (1.28, 1.61)* | 1.52 (1.34, 1.71)* | 1.65 (1.43, 1.90)* | 1.17 (1.11, 1.24)* | 1.36 (1.23, 1.49)* |
| Neutropenia (AML) | 1.07 (0.93, 1.23) | 1.09 (0.92, 1.29) | 1.14 (0.96, 1.35) | 1.11 (0.92, 1.35) | 1.03 (0.96, 1.10) | 1.54 (1.09, 2.18)* |
| Thrombocytopenia (AML) | 1.07 (0.78, 1.45) | 0.95 (0.71, 1.26) | 1.16 (0.81, 1.68) | 1.18 (0.78, 1.80) | 1.04 (0.89, 1.22) | 1.58 (0.81, 3.09) |
P < 0.05
Odds Ratios (OR) reported correspond to an increase in tipifarnib AUC and AUCD of 1 mg ml−1 h, T > 400 ng ml−1 and T > 600 ng ml−1 of 1 h, and AUCT of 56 mg ml−1 h, which corresponds to the cumulative mean exposure for a week with 600 mg twice daily dosing regimen.
Figure 3.

Observed and predicted incidence of the worst grade of haematological toxicities as a function of plasma tipifarnib AUC and tumour type. Dotted lines represent 95% confidence intervals on the model prediction
Figure 4.

Observed and predicted incidence of the worst grade of nonhaematological toxicities as a function of plasma tipifarnib AUC. Dotted lines represent 95% confidence intervals on the model prediction
The multivariate analysis between baseline toxicity, age, tumour type, tipifarnib AUC and duration of treatment, and the incidence of haematological toxicities is shown in Table 4, and confirms the results of the univariate analysis (Table 2). Tipifarnib AUC was associated with neutropenia and thrombocytopenia of grade ≥3 in subjects with solid tumours, but not in r-AML subjects. In patients with solid tumours, an increase of 1.0 mg l−1 h in tipifarnib AUC was associated with a 1.69-fold and 1.41-fold increase in the odds of the neutropenia and thrombocytopenia grade ≥ 3, respectively. In this population, for each additional week on treatment, a 15% increase in the odds of the worst grade of neutropenia or thrombocytopenia grade was observed. The duration of the treatment was the only variable associated with an increased incidence of neutropenia of grade ≥3 in r-AML subjects. Regardless of tumour type, baseline neutrophil and platelet counts below the normal range were associated with a higher incidence of neutropenia and thrombocytopenia of grade ≥3. The exception was neutropenia in solid tumours, where this effect could not be evaluated because only two subjects had a baseline neutrophil count below the normal range. No association between age and the occurrence of the haematological toxicities was found in subjects with solid tumours and r-AML.
Table 4.
Estimated odds ratio (95% confidence interval) from the multivariate analysis of baseline, age, tipifarnib AUC and treatment duration as predictors of haematological toxicity
| Haematological toxicity | Solid tumour | AML |
|---|---|---|
| Neutropenia | ||
| Baseline PC (Grade ≥1 vs. Grade 0) | – | 4.03 (2.65, 6.96)* |
| Age (≥65 years vs <65 years) | 1.66 (0.91, 3.00) | 1.01 (0.41, 2.51) |
| Exposure (AUC, mg l−1 h) | 1.69 (1.47, 1.95)* | 1.03 (0.90, 1.18) |
| Duration (week) | 1.13 (1.05, 1.21)* | 2.22 (1.29, 3.83)* |
| Thrombocytopenia | ||
| Baseline PC (Grade ≥1 vs. Grade 0) | 2.63 (1.55, 4.48)* | 3.84 (1.85, 7.98)* |
| Age (≥65 years vs <65 years) | 1.35 (0.67, 2.72) | 1.91 (0.48, 7.68) |
| Exposure (AUC, mg l−1 h) | 1.41 (1.21, 1.63)* | 1.16 (0.91, 1.49) |
| Duration (week) | 1.16 (1.06, 1.26)* | 1.21 (0.88, 1.67) |
P < 0.05.
The results of the multivariate analysis for nonhaematological toxicities are shown in Table 5. Less than 10% difference in the OR point estimates was found between univariate and multivariate analysis with respect to all the nonhaematological toxicities. Baseline values of AST, ALT, total bilirubin and serum creatinine above the normal range were clearly associated with higher incidence of toxicity of grade ≥2 associated with the elevation of these variables. However, no statistically significant effect of age on the incidence of nonhaematological toxicities was found, except for the elevation of bilirubin and the occurrence of nausea and vomiting. As expected, the incidence of the latter in older patients was lower relative to younger patients (OR 0.63, 95% CI 0.43, 0.93).
Table 5.
Estimated odds ratio (95% confidence interval) from the multivariate analysis of baseline, age, tumour type and tipifarnib AUC and treatment duration as predictor of nonhaematological toxicities
| Liver and renal toxicities | AST | ALT | Bilirubin | Creatinine |
|---|---|---|---|---|
| Baseline (Grade ≥1 vs Grade 0) | 3.50 (2.53, 4.85)* | 2.86 (2.07, 3.94)* | 2.48 (1.87, 3.31)* | 2.68 (1.96, 3.65)* |
| Age (≥65 years vs. <65 years) | 0.53 (0.27, 1.04) | 0.52 (0.26, 1.06) | 0.58 (0.36, 0.93)* | 0.98 (0.57, 1.69) |
| Cancer type (r-AML vs. solid tumour) | 1.41 (0.62, 3.23) | 1.33 (0.60, 2.95) | 1.30 (0.74, 2.29) | 4.37 (2.31, 8.27)* |
| Exposure (AUC, mg l−1 h) | 0.88 (0.78, 0.99)* | 0.93 (0.82, 1.04) | 0.98 (0.91, 1.06) | 1.07 (0.99, 1.15) |
| Treatment duration (weeks) | 1.05 (0.99, 1.11) | 1.03 (0.97, 1.10) | 1.07 (1.02, 1.11)* | 1.12 (1.07, 1.18)* |
| GI toxicities | Nausea and vomiting | Diarrhoea | GI inflammation |
|---|---|---|---|
| Age (≥65 years vs. <65 years) | 0.63 (0.43, 0.93)* | 1.02 (0.64, 1.64) | 0.74 (0.54, 1.02) |
| Cancer type (r-AML vs. solid tumour) | 1.55 (0.97, 2.49) | 1.59 (0.90, 2.83) | 1.72 (1.14, 2.59)* |
| Exposure (AUC, mg l−1 h) | 0.99 (0.93, 1.05) | 1.11 (1.03, 1.19)* | 1.02 (0.97, 1.08) |
| Treatment duration (weeks) | 0.95 (0.92, 0.98)* | 0.98 (0.94, 1.01) | 1.00 (0.98, 1.02) |
| Other toxicities | Skin rash | CNS toxicity | Peripheral neurotoxicity |
|---|---|---|---|
| Age (≥65 years vs. <65 years) | 0.99 (0.59, 1.64) | 1.04 (0.73, 1.49) | 1.63 (0.93, 2.84) |
| Cancer type (r-AML vs. solid tumour) | 1.93 (1.03, 3.63)* | 0.91 (0.58, 1.43) | 1.67 (0.84, 3.30) |
| Exposure (AUC, mg l−1 h) | 1.05 (0.98, 1.13) | 1.06 (1.00, 1.12) | 1.07 (0.98, 1.16) |
| Treatment duration (week) | 0.91 (0.86, 0.96)* | 0.96 (0.93, 0.98)* | 1.03 (1.01, 1.06)* |
p < 0.05.
Grade ≥2 elevation of serum creatinine, and presence of GI inflammation or skin rash were associated with tumour type, but not with the tipifarnib AUC. However, most of the patients receiving tipifarnib 600 mg twice daily had r-AML, and only a few with solid tumours received such a high dose. In addition, there was no evidence to support a higher incidence of these toxicities in patients with r-AML compared with those with solid tumours. Therefore, the covariate, tumour type, was excluded from the multivariate model, in order to estimate the effect of tipifarnib exposure on the incidence of these toxicities. An increase of 1 mg l−1 h in the AUC of tipifarnib was associated with an increase in the odds of the worst grade of toxicity (≥2) of 1.18 (95% CI 1.11, 1.26), 1.13 (95% CI 1.07, 1.19), and 1.10 (95% CI 1.04, 1.17) for serum creatinine, GI inflammation and skin rash, respectively. Whereas a longer duration of treatment was associated with a higher incidence of grade ≥2 elevation of serum creatinine, a shorter duration was paradoxically associated with a higher incidence of grade ≥2 gastrointestinal inflammation and skin rash. This phenomenon was also observed for CNS toxicity and nausea and vomiting, and was probably a confounding effect due to the decrease in dose implemented as a consequence of the other earlier toxicities. The mean time to the occurrence of the worst grade (≥2) of gastrointestinal inflammation (334 days), skin rash (356 days), CNS toxicity (243) and nausea and vomiting (272 days) was much longer than that for the worst grade of serum creatinine (139 days) and bilirubin (105 days) elevation or thrombocytopenia (71 days) and neutropenia (50 days).
For CNS and peripheral neurotoxicity, the relationship between tipifarnib AUC and the incidence of grade ≥2 found in the univariate analysis (Table 3), was not confirmed in the multivariate analysis (Table 5). When tumour type was excluded from the multivariate model, the associations between tipifarnib AUC and CNS (OR 1.05, 95% CI 1.01, 1.10) and peripheral neurotoxicity (OR 1.10, 95% CI 1.03, 1.18) grade ≥2 were statistically significant. Finally, the relationship between tipifarnib AUC and diarrhoea was also confirmed in the multivariate analysis presented in Table 5. An increase of 1 mg l−1 h in the AUC of tipifarnib was associated with a 1.11-fold increase in the odds of the worst grade of diarrhoea toxicity (≥2).
Discussion
An exploratory exposure vs. response analysis was conducted on data from five clinical studies in cancer patients receiving tipifarnib under fed conditions over a dose range of 100–850 mg twice daily for 3 consecutive weeks, followed by 1 week of rest for each treatment cycle, or in the colorectal study, receiving matching placebo. Even though dose is the marker of drug exposure most often used in clinical trials, plasma concentration measurements are hypothesized to be more directly related to the concentration of the drug at the target site and thus to the clinical effect. For this reason, a sparse sampling strategy combined with a population pharmacokinetic and a Bayesian analysis were used to determine exposure to tipifarnib in cancer subjects, as suggested by the FDA Guidance for Industry on the Exposure-Response Relationships [14]. Such an approach permits the estimation of individual exposure when only a small number of plasma concentrations are available in a particular subject, and also takes account between subject variability in pharmacokinetics.
Tipifarnib plasma AUC, Cmax, T400, T600, AUCD and AUCT were selected as the exposure variable for this analysis. However, as Cmax, T400, T600, AUCD were highly correlated (r > 0.90) with AUC, only this variable was used in the multivariate analysis. Consequently, conclusions derived from the AUC data were extrapolated to the other exposure variables considered in the univariate analysis.
The relationships between the AUC of tipifarnib during the first cycle and the incidence of liver, renal, gastrointestinal and haematological toxicities, skin rash and CNS and peripheral neurotoxicity were explored using static (or nondynamic) logistic regression models that accounted for the duration of the treatment until the development of toxicity.
However, the results of the effect of treatment duration on the toxicities should be interpreted with caution because the studies were not designed for this purpose, especially for those toxic effects that take longer to occur. As all the subjects were on tipifarnib treatment (or matched placebo) until disease progression or the occurrence of severe toxicity, withdrawing patient with early toxicties from the clinical study may result in selection bias in favour of those who responded and/or tolerated treatment. This is particularly important when diseases with different progression rates and different dose levels are analyzed together. Thus, in the present analysis, the subjects with r-AML received a high dose (600 mg twice daily) of tipifarnib and had a faster disease progression than subjects with colon cancer, who received only 300 mg twice daily.
The results of the multivariate analysis suggested that for toxicities with a rapid onset (for instance, haematological effects), patients who had been treated for a long period of time, had a higher risk of toxicity than patients undergoing a short period of treatment. It has been established that the development of severe haematological toxicity occurs during the first three cycles of treatment, which is faster than the progression of the disease evaluated in the current analysis or the appearance of other drug-related toxicities. In this case, the potential bias from subjects who experience disease progression or severe toxicity would be minimal.
However, for effects with a slow onset (for instance, skin rash or CNS toxicity), patients who previously experienced other severe toxicities may be withdrawn from a study, resulting in a selection bias in favour of patients who tolerate treatment. On the other hand, it is also possible that patients who previously experienced other less severe toxicity may have their dose reduced. If so, a longer duration of treatment could paradoxically be associated with a lower incidence of toxicity. In other words, after the first cycle of treatment, the dose reduction and the duration of the treatment are (as per protocol) linked to the development of toxicity, in addition to disease progression. Therefore, the dose and the exposure to the drug are temporally related to the development of toxicity. As a consequence, the inclusion of cumulative exposure (or a similar variable pertaining to the duration of the treatment) would lead to biased results, which might give rise to paradoxical results as happened for nonhaematological toxicities with slow onset. Therefore, the duration of the treatment might be a useful explanatory covariate, but with limited predictive performance. Therefore, only tipifarnib exposure during the first cycle of treatment was investigated as a predictor for drug toxicity.
In addition, no relationship was observed between tipifarnib AUC and the occurrence of neutropenia and thrombocytopenia of grade ≥3 in r-AML patients. The large number of AML patients with neutropenia and thrombocytopenia of grade ≥3, and the small number with toxicity of grade ≤2 may explain the absence of an association. Furthermore, haematological abnormalities observed in treatment refractory or relapsed AML patients may have been induced by tipifarnib, occur as a consequence of their underlying disease, or both. Therefore, an exposure-guided dose adjustment for tipifarnib to limit neutropenia and thrombocytopenia of grade ≥3 in patients with r-AML is not recommended.
The incidence of neutropenia and thrombocytopenia grade ≥3 in patients with solid tumours receiving tipifarnib was 24.9 and 14.7%, respectively. Platelet counts at baseline below the normal range were predictive of thrombocytopenia of grade ≥3 occurring at any time during the study. In addition, both tipifarnib AUC and the duration of treatment exhibited a positive and significant association with neutropenia and thrombocytopenia of grade ≥3 in patients with solid tumours. The probability of these patients having neutropenia of grade ≥3 when receiving tipifarnib 300 mg twice daily was predicted to be 14.5% for the median value of tipifarnib AUC, and 3.82 mg l−1 h, in subjects with bilirubin concentrations between 7.5 and 15 µM. As a comparison, patients with solid tumours receiving the same treatment and with bilirubin concentrations at a baseline value below 7.5 mM and above 15 mM would have a median tipifarnib AUC of 3.60 mg l−1 h and 4.15 mg l−1 h, respectively [9], which would be associated with probabilities of neutropenia of grade ≥3 of 13.1% and 16.8%, respectively. The relevance of these differences for dosage adjustment is limited. A 15% reduction in dose to 44 patients with solid tumours with bilirubin values at baseline higher than 15 µM would be needed to prevent one additional episode of neutropenia grade ≥3 relative to patients with bilirubin baseline concentrations between 7.5 and 15 mM. Therefore, in patients with solid tumours, tipifarnib dosage adjustments to limit the incidence of neutropenia of grade ≥3 on the basis of bilirubin measurements, is not warranted. This also applies to those toxicities that have an incidence of less than 25% and a similar steepness in the exposure–response curve, and to those with an incidence of 25%, but a less steep exposure–response curve.
Older patients (≥ 65years) did not show an increased incidence of haematological toxicity of grade ≥3 or nonhaematological toxicity of grade ≥2 relative to younger patients. Similar results were obtained when age was used as a quantitative covariate (data not shown). AML patients younger than 65 years old have response rates to remission induction chemotherapy of approximately 75% and approximately 35–40% of them survive following diagnosis for 5 years or more. In contrast, patients with AML who are more than 65 years old have an overall response rate to induction therapy of 45–55%, and fewer than 10% of these survive for a minimum of 5 years. Whereas treatment has improved steadily over the last 20 years in younger adults, no significant change in outcome has been noted in patients older than 65 years of age. The reasons for the poorer outcome most probably relate to the increased frequency of unfavourable cytogenetic profiles in older patients with AML, and a greater frequency of antecedent myelodysplasia and of drug resistance phenotypes. However, the different underlying biology in older patients with AML was not related to differences in the pharmacokinetics of tipifarnib [9] or in the incidence of toxicity, as has been shown in the current analysis.
The incidence of the worst grade toxicity based on AST, ALT and total bilirubin (≥2) was lower than 20% in all tumour types. No direct relationship between the tipifarnib AUC and these types of toxicity was found within the dose range of 100–850 mg. In contrast, the baseline values of each parameter were predictive of toxicity grade ≥2 throughout the study.
The incidence of the worst grade of renal toxicity (≥2) was lower than 15%. Serum creatinine at baseline was predictive of grade ≥2 toxicity throughout the study. In addition, there was a weak association between exposure to tipifarnib and duration of treatment when grade ≥2 occurred. Regardless of the tumour type, the incidence of worst grade of CNS and peripheral neurotoxicty (≥2) was 25% and 10%, respectively. There was a weak association between tipifarnib AUC and the incidence of the worst grade (≥2) of CNS and peripheral neurotoxicity over the dose range 100–850 mg. The incidence of the worst grade (≥2) of skin rash was 11% throughout the study. Tipifarnib AUC had no effect on the incidence of worst grade of skin rash.
In summary, the findings of this prospective large-scale pharmacokinetic and pharmacodynamic evaluation of tipifarnib suggest that at the dose range examined, there was only a significant association between haematological toxicity and exposure in patients with solid tumours. The incidence of exposure-related nonhaematological toxicities was limited regardless of tumour type. In some patients who develop significant toxicities, dose reduction may improve the tolerability of tipifarnib. Overall, an exposure-guided approach to dosage adjustment to limit haematological and nonhaematological toxicities in AML patients is not warranted. However, if future studies establish a relationship between dose and efficacy, there may be a place for such an approach in treatment with tipifarnib.
Acknowledgments
The authors would like to thank the hundreds of patients, investigators and their medical, nursing and laboratory staff who participated in the clinical studies included in the present study. In particular, we recognize the following lead principal investigators for each of the studies from which data were derived for the current study: L. Weiner, Fox Chase Cancer Center, Department of Medical Oncology, Philadelphia, PA; J. E. Rosenberg, Department of Medicine, University of California, San Francisco, CA; S. Johnston, Department of Medicine, Royal Marsden Hospital, London, Great Britain; D. Cunningham, Royal Marsden Hospital, London, Great Britain; J. Lancet, University of Rochester Medical Center, Rochester, NY, USA; J. L. Harousseau, Service Hematologie Clinique, CHU, Hotel Dieu, Nantes, France.
The study was supported by Johnson & Johnson Pharmaceutical Research & Development, L.L.C., Raritan, NJ.
References
- 1.Hon YY, Evans WE. Making TDM work to optimize cancer chemotherapy: a multidisciplinary team approach. Clin Chem. 1998;44:388–400. [PubMed] [Google Scholar]
- 2.Bruno R, Hille D, Riva A, Vivier N, ten Bokkel Huinnink WW, van Oosterom AT, Kaye SB, Verweij J, Fossella FV, Valero V, Rigas JR, Seidman AD, Chevallier B, Fumoleau P, Burris HA, Ravdin PM, Sheiner LB. Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol. 1998;16:187–96. doi: 10.1200/JCO.1998.16.1.187. [DOI] [PubMed] [Google Scholar]
- 3.Gieschke R, Burger HU, Reigner B, Blesch KS, Steimer JL. Population pharmacokinetics and concentration-effect relationships of capecitabine metabolites in colorectal cancer patients. Br J Clin Pharmacol. 2003;55:252–63. doi: 10.1046/j.1365-2125.2003.01765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caponigro F, Casale M, Bryce J. Farnesyl transferase inhibitors in clinical development. Expert Opin Invest Drugs. 2003;12:943–54. doi: 10.1517/13543784.12.6.943. [DOI] [PubMed] [Google Scholar]
- 5.End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, Venet M, Sanz G, Poignet H, Skrzat S, Devine A, Wouters W, Bowden C. Characterization of the antitumour effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res. 2001;61:131–7. [PubMed] [Google Scholar]
- 6.Zujewski J, Horak ID, Bol CJ, Woestenborghs R, Bowden C, End DW, Piotrovsky VK, Chiao J, Belly RT, Todd A, Kopp WC, Kohler DR, Chow C, Noone M, Hakim FT, Larkin G, Gress RE, Nussenblatt RB, Kremer AB, Cowan KH. Phase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 18:927–41. doi: 10.1200/JCO.2000.18.4.927. [DOI] [PubMed] [Google Scholar]
- 7.Crul M, de Klerk GJ, Swart M, van't Veer LJ, de Jong D, Boerrigter L, Palmer PA, Bol CJ, Tan H, de Gast GC, Beijnen JH, Schellens JH. Phase I clinical and pharmacologic study of chronic oral administration of the farnesyl protein transferase inhibitor R115777 in advanced cancer. J Clin Oncol. 2002;20:2726–35. doi: 10.1200/JCO.2002.09.116. [DOI] [PubMed] [Google Scholar]
- 8.Johnston SR, Hickish T, Ellis P, Houston S, Kelland L, Dowsett M, Salter J, Michiels B, Perez-Ruixo JJ, Palmer P, Howes A. Phase II study of the efficacy and tolerability of two dosing regimens of the farnesyl transferase inhibitor, R115777, in advanced breast cancer. J Clin Oncol. 2003;21:2492–9. doi: 10.1200/JCO.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 9.Perez-Ruixo JJ, Piotrovskij V, Zhang S, Hayes S, Thibault A, Zannikos P. Population pharmacokinetics of tipifarnib in healthy and cancer subjects. Br J Clin Pharmacol. 2006;62:81–96. doi: 10.1111/j.1365-2125.2006.02615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards H, Thibault A, Jia X. Clinical Research Report, Data on file. Johnson & Johnson Pharmaceutical Research and Development; 2001. A Phase I trial to determine the safety and pharmacokinetics of 21-day dosing of a farnesyltransferase inhibitor, tipifarnib (Zarnestra™) [Google Scholar]
- 11.Rosenberg JE, von der Maase H, Seigne JD, Mardiak J, Vaughn DJ, Moore M, Sahasrabudhe D, Palmer PA, Perez-Ruixo JJ, Small EJ. A phase II trial of R11577, an oral farnesyltransferase inhibitor (FTI), in patients with advanced urothelial tract transitional cell carcinoma. Cancer. 2005;103:2035–41. doi: 10.1002/cncr.21023. [DOI] [PubMed] [Google Scholar]
- 12.Harousseau JL, Stone R, Thomas X. Interim results from a phase II study of R115777 (Zarnestra™) in patients with relapsed and refractory acute myelogenous leukaemia. Proc Am Soc Clin Oncol. 2002;21:abstract. 265. [Google Scholar]
- 13.Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, Kourteva G, Iveson T, Andre T, Dostalova J, Illes A, Belly R, Perez-Ruixo JJ, Park YC, Palmer PA. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol. 2004;22:3950–7. doi: 10.1200/JCO.2004.10.037. 2002. [DOI] [PubMed] [Google Scholar]
- 14.US Department of Health and Human Services Food and Drug Administration. Exposure-response relationships – study design, data analysis, and regulatory applications.