

Abstract
What is already known about this subject
According to recent literature, the pathophysiologies of allergic rhinitis and chronic idiopathic urticaria are thought to be similar in adults and children. In addition, the response to antihistamine treatment is similar in adults and children, suggesting a similar concentration-response relationship.
However, an appropriate dose selection and the pharmacokinetics of desloratadine in children of ≥6 months−≤2 years old have never been addressed in the literature.
What this study adds
This study demonstrated that desloratadine syrup offers a safe treatment option for allergic conditions in young children.
A suitable dose for children aged ≥6 months−<1 year is 1.0 mg, while the corresponding predicted dose for children aged ≥1 year−≤2 years is 1.25 mg. These paediatric doses yielded similar systemic desloratadine exposures (AUC) to those seen with a typical adult dose of 5.0 mg.
Aims
The aim of this study was to identify the dose of desloratadine in children aged ≥6 months−≤2 years that would yield a single-dose target exposure (AUC) comparable with that in adults taking 5 mg desloratadine as syrup.
Methods
In a phase 1, single-dose, open-label, pharmacokinetic study in 58 children aged ≥6 months−<1 year and ≥1 year−≤2 years were randomly assigned to desloratadine syrup 0.625 mg (1.25 ml) and 1.25 mg (2.5 ml), respectively. Because the volume of blood that could be collected from individual subjects was limited, a population pharmacokinetic approach was used to estimate the pharmacokinetics of desloratadine. Safety was assessed based on results of screening and postdose physical examinations, laboratory safety tests, vital signs, and adverse events.
Results
The apparent clearance (CL/F) of desloratadine, population estimate (%CV), in children aged ≥6 months−<1 year was 27.8 l h−1 (35) and corresponding values in children ≥1 year−≤2 years was 35.5 l h−1 (51), compared with 137 l h−1 (58) for adults. The CL/F ratios (children to adults) indicated that doses of 1 mg for ≥6 months−<1 year and 1.25 mg for ≥1 year−≤2 years would result in similar systemic exposure to that observed in adults receiving the recommended 5 mg dose. Desloratadine was well tolerated with no safety issues.
Conclusions
Doses of 1.0 and 1.25 mg in children aged ≥6 months−≤2 years should result in an exposure to desloratadine similar to that of adults receiving doses of 5 mg.
Keywords: desloratadine, dose, paediatric, pharmacokinetics, safety
Introduction
Nonsedating antihistamines are a first-line treatment for allergic rhinitis and chronic urticaria in children [1–3]. Desloratadine, a potent metabolite of loratadine, is a nonsedating, histamine H1-receptor antagonist. The pathophysiology of allergic rhinitis and chronic idiopathic urticaria is thought to be similar in adults and children [4]. In addition, the response to antihistamine treatment is similar in adults and children [5], suggesting a similar concentration-response relationship.
Following oral administration of 5 mg desloratadine once daily to healthy adult volunteers for 10 days, the mean time to maximum plasma concentrations (tmax) was approximately 3 h postdose. The mean steady-state plasma Cmax and AUC(0,24 h) after dosing were 4 ng ml−1 and 56.9 ng ml−1 h, respectively [6]. Two large placebo controlled trials were conducted to characterize the 24 h efficacy of once-daily desloratadine in adults and adolescents with seasonal allergic rhinitis and chronic idiopathic urticaria [2]. Results showed that once daily dosing with 5 mg desloratadine provides 24 h relief of signs and symptoms. Single doses of desloratadine syrup (1.25 and 2.5 mg) were safe and well tolerated in children 2–5 and 6–11 years old. Median Cmax values for the 2–5 years old and 6–11 years old groups were 2.28 and 2.05 ng ml−1, respectively, at tmax of 2 h for both age groups. Median AUC(0,last) was 38.8 and 38.2 ng ml−1 h for the 2–5 and 6–11 years old, respectively, and these values were similar to those in adults, who received 5 mg doses of desloratadine [7].
Desloratadine is metabolized to 3-hydroxy (3-OH)-desloratadine which is subsequently glucuronidated. Data from clinical trials indicate that a subset of the general population has a decreased ability to form 3-OH-desloratadine, and are classed as poor metabolizers. In pharmacokinetic studies (n = 3748), approximately 6% of subjects were poor metabolizers of desloratadine (defined as a subject with an AUC ratio of 3-OH-desloratadine : desloratadine of >0.1) [8].
The aim of this study was to identify the desloratadine dose in children aged ≥6 months−≤2 years that would yield a single-dose target exposure (AUC) comparable with that in adults taking 5 mg desloratadine as syrup. Administration of 1.25 mg to subjects aged between 2 and 5 years produced comparable AUCs with adults, who received 5 mg as syrup. Based on these data it was estimated that the dose in paediatric subjects aged ≥6 months−≤2 years should be between 0.625 and 1.25 mg. The two doses tested were 0.625 mg and 1.25 mg.
Methods
Study population
The study was conducted at Arkansas Research, Little Rock, Arkansas, USA, with approval of the protocol by the Arkansas Research Human Volunteer Research Committee. The study was performed in accordance with the 1996 World Medical Association Declaration of Helsinki and subsequent amendments concerning written informed consent and the rights of human subjects. Written informed consent was obtained from each subject's parent or legal guardian at the initial screening visit.
Male and female subjects, ≥6 months−≤2 years of age, who were candidates for antihistamine therapy or had previously been treated with an antihistamine were eligible for inclusion in the study. Subjects were free from any clinically significant disease or condition that would require a physician's care and/or interfere with study evaluations or procedures and had clinical laboratory test results within normal limits or clinically acceptable to the investigator. For inclusion, physical examination and electrocardiogram findings (ECG; 12-lead recorded at 25 mm s−1 and reporting ventricular rate and PR, QRS, QT, and QTc intervals) also had to be normal or clinically acceptable. Subjects, with a clinically significant infectious disease within 4 weeks prior to study treatment, or who were positive for hepatitis B surface antigen or hepatitis C antibody, were excluded from the study. Those with a clinically significant food or drug allergy (in particular, to loratadine) were also excluded. Additional exclusion criteria included the use of any medication within 14 days prior to study treatment (except acetaminophen) or participation in a clinical trial of an investigational drug within 30 days prior to study treatment. No concomitant medications, other than acetaminophen, were permitted during the course of the study without prior approval, except in a medical emergency.
Study design and conduct
The study in children
This was a phase 1, single-dose, randomized, stratified, parallel-group, open-label study evaluating the population pharmacokinetics of desloratadine to assess the most appropriate dose of desloratadine syrup for use in children ≥6 months−≤2 years of age. Following a screening period of up to 3 weeks, subjects were stratified by age (≥6 months−<1 year and Π ≥1 year−≤2 years) and, within each age group, randomly assigned to one of two doses of desloratadine syrup (0.5 mg ml−1): 0.625 mg (1.25 ml) or 1.25 mg (2.5 ml). Doses were selected based on data from previously completed pharmacokinetic studies in older children, 2–5 years of age, which showed that a dose of 2.5 mg resulted in approximately double the exposure (AUC) seen in adults taking the recommended adult dose of 5 mg, whereas a dose of 1.25 mg produced comparable exposure [7]. Desloratadine was administered orally via a dosing syringe in the morning after a 2 h fast, followed by a further 2 h fast. Subjects remained at the study site for 24 h prior to dosing and for at least 12 h after dosing.
The study in adults
A randomized, crossover study designed to compare the bioavailability of desloratadine from syrup and the marketed-tablet formulations was used as the basis of historical adult study data (data on file at Schering-Plough, Kenilworth, New Jersey, USA). In this study, plasma concentrations for the 5 mg desloratadine syrup formulation were obtained predose and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, 36, 48, 72, 96, and 120 h postdose.
The combined number of plasma samples from the two studies was 800, obtained from 88 subjects: the 58 paediatric subjects in the current study, plus 30 adults from the earlier study.
Blood sampling and bioanalytical analyses
Subjects were randomly allocated, according to a computer-generated schedule, to one of two groups (A and B) that had distinct plasma sampling schedules, with five sampling times in each schedule. In group A, blood samples were obtained at 1, 3, 6, 24, and 72 h postdose. In group B, blood samples were obtained at 2, 4, 8, 12, and 48 h postdose. Deviations of 30 min for time points up to 4 h, 1 h for the 6 and 8 h samples, and 2 h for the remaining time points were allowed, with actual sample times being used in the pharmacokinetic analysis. At each time point, 2 ml blood samples were collected and stored in heparin-containing tubes.
Blood samples were centrifuged for 20 min at 4°C and approximately 1500 g within 30 min of collection. Plasma was separated and frozen to at least −20°C until and shipped frozen with dry ice to the analytical laboratory (PPD Pharmaco, Richmond, Virginia, USA). Plasma samples were then assayed for desloratadine and 3-OH-desloratadine concentrations using liquid chromatography with tandem mass spectrometric detection [9]. The lower limit of quantification (LLOQ) was 0.025 ng ml−1 for both desloratadine and 3-OH-desloratadine. The accuracy (%bias) at the lower limit of quantification (LLOQ) was −12.8 and +3.4% for desloratadine and 3-OH desloratadine, respectively. The precision (%CV) for samples at the LLOQ was 15.1 and 10.9% for desloratadine and 3-OH desloratadine, respectively.
Pharmacokinetic and statistical analyses
Individual desloratadine concentration-time data (normal or nonlog transformed) from both studies were combined and modelled simultaneously, using a one-compartment model with first-order absorption and elimination, designated model 3 in the nonlinear, mixed-effect modelling computer program WinNonMix (Version 2.0.1; Pharsight Corporation, California, USA) [10]. The model is summarized as follows:
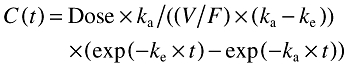 |
where ka is the absorption rate constant, V/F is the apparent volume of distribution, CL/F is the apparent clearance, and ke (terminal phase rate constant) is (CL/F)/(V/F). The intersubject variability (η) was modelled as an exponential error distribution (one η term for each structural pharmacokinetic parameter). An exponential variance function was used for the residual error model:
where g is the variance function, ýij is predicted concentration for ith subject at time j, and a is a constant (a > 0).
The covariance matrix for intersubject variability was assessed as a diagonally constrained matrix. In the initial run, the minimization method used was the first-order method (extended least squares) and the parameters obtained were included as initial estimates in a second run with first order conditional estimation.
Two conditions were tested to describe the relationship between CL/F, V/F, and ka in the previously described structural model. In the reduced model, these parameters were assumed to be the same for all age groups, thus implying that all individuals' values were derived from a single distribution. In the full model, CL/F and V/F, but not ka, were assumed to be different for each age group, thus implying that individuals' values were derived from a distribution specific to each age group. The ka could be not determined with precision in children because not all had samples at tmax due to nature of blood collection procedure, randomized sparse samples. Therefore, one ka (with η) for all three groups was used in the model. The goodness of fit criteria (identified as the objective function) for each model was used to perform a statistical comparison of the two conditions. For each run, WinNonMix computed the minimum value of the objective function; a statistically significant difference (P< 0.05 with 1 degree of freedom) was associated with a change of 3.9 in the objective function.
The dose of desloratadine (DL) required for children was calculated as:
where CL/Fchildren is the apparent clearance in children and CL/Fadults is the apparent clearance in adults. The maximum predicted plasma concentration (Cmax), time of Cmax (tmax), area under the plasma concentration–time curve (AUC; exposure), and ke values for each study subject were estimated from individual predictions for that individual's concentration-time curve. Scatter plots are presented using predicted (population) vs. observed concentration (with unity line) and weighted residuals vs. predicted concentration and weighted residuals vs. predicted concentrations.
An analysis of variance was conducted on the log-transformed predicted AUC and Cmax values of desloratadine. Point estimates and 90% confidence intervals were calculated.
Data for 3-OH-desloratadine were not included in the population analysis. Individual plasma desloratadine and 3-OH-desloratadine concentration-time data were used to determine pharmacokinetic parameters using model-independent methods [11]. The AUC to the time of the last quantifiable sample (AUC(0,last)) was calculated using the linear trapezoidal method. The AUC(0,last) ratio of 3-OH-desloratadine (AUC(last)3-OH-DL) relative to desloratadine (AUC(0,last)DL) was determined as follows:
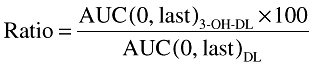 |
This was used to assess the prevalence in the study population of poor metabolizers, defined as having a ratio for exposure to 3-OH-desloratadine relative to desloratadine of <10%.
Safety assessments
Safety was assessed by monitoring adverse events, laboratory tests, physical examinations, vital signs, and ECG recordings. Details of all reported adverse events were recorded throughout the study, with severity assessed according to the common toxicity criteria (CTC; version 2) grading system, or, for events not covered by the CTC, as mild, moderate, severe, or life-threatening. Any relationship of an adverse event to treatment was determined by the investigator. Laboratory safety tests were carried out at screening, 24 h before dosing, and 48 and 72 h after dosing. A physical examination was performed at screening and 72 h after dosing. Vital signs were measured at screening, at the time of dosing, and 24 and 72 h after dosing. An ECG was performed at screening and 3 and 72 h after dosing.
Results
Subject characteristics
A total of 58 subjects aged ≥6 months−≤2 years were enrolled, and all completed the study. Twenty-nine subjects received 0.625 mg desloratadine and 29 received 1.25 mg desloratadine. Body weight ranged from 7.7 kg to 15.5 kg. Within age bands, demographic and baseline characteristics were comparable between dose groups (Table 1).
Table 1.
Demographic and baseline characteristics by desloratadine dose and age group
| Desloratadine 0.625 mg (1.25 ml) | Desloratadine 1.25 mg (2.5 ml) | |||
|---|---|---|---|---|
| ≥6 months−<1 year (n = 10) | ≥1 year−≤2 years (n = 19) | ≥6 months− <1 year (n = 10) | ≥1 year−≤2 years (n = 19) | |
| Mean age (months) (range) | 9.8 (6–11) | 20.8 (16–23) | 9.7 (7–11) | 19.9 (14–23) |
| Male/female (%) | 60.0/40.0 | 57.8/42.1 | 50.0/50.0 | 31.6/68.4 |
| Caucasian/Black (%) | 50.0/50.0 | 31.6/68.4 | 30.0/70.0 | 26.3/73.7 |
| Mean weight (kg) (range) | 9.60 (7.7–11.8) | 12.0 (9.1–15.5) | 9.68 (7.7–13.6) | 11.9 (9.1–15.0) |
| Mean height (cm) (range) | 68.2 (63.5–77.5) | 82.0 (71.1–96.5) | 70.0 (61.0–83.8) | 81.8 (71.1–94.0) |
| Mean body mass index (kg m−2) (range) | 20.7 (18.0–25.5) | 18.0 (11.3–21.8) | 19.7 (17.0–22.6) | 17.8 (15.9–21.0) |
Pharmacokinetics
The pharmacokinetic analysis was based on 800 samples obtained from 88 subjects: the 58 paediatric subjects (a total of 290 samples) in the current study, plus 30 adults (a total of 510 samples) from the earlier study. Frequency distributions for age and body weight are presented in Figure 1.
Figure 1.

Frequency distribution of age and body weight
The individual desloratadine plasma concentration-time profiles for different age groups are illustrated in Figure 2. ka could not be determined with precision in paediatric subjects owing to a lack of data during the absorption phase. Therefore, the full model was used to establish the pharmacokinetics of desloratadine, with the adult ka value (similar to children 2–11 years of age) being used for all three age groups (≥6 months−<1 year, ≥1 year−≤2 years, and adult).
Figure 2.

Individual observed plasma desloratadine concentration–time profiles for subjects aged ≥6 months−<1 year (A), ≥1 year−≤2 years (B) and adults (C)
A plot of individual observed concentrations of desloratadine vs. concentrations predicted by the full model demonstrated a good fit of data (Figure 3). Furthermore, when weighted residuals were plotted vs. predicted concentrations, there was no systematic bias, confirming that the model described the data appropriately (Figure 4). Scatter plots of individual observed concentrations vs. individual predicted concentrations for both age groups are shown in Figure 5, as a good fit of individual age group data. Pharmacokinetic parameter estimates and doses by age group are shown in Table 2.
Figure 3.

Individual observed concentrations of desloratadine (DL) vs. concentrations predicted by the full model
Figure 4.

Weighted residual vs. population-model predicted desloratadine concentrations
Figure 5.

Individual observed vs. predicted plots by age group
Table 2.
Population mean (% coefficient of variation) of desloratadine pharmacokinetic parameter estimates and predicted dose by age group
| Age group | V/F (l) | ka (h−1) | CL/F (l h−1) | Predicted dose (mg) (range)a |
|---|---|---|---|---|
| ≥6 months−<1 year | 470 (15) | 27.8 (35) | 1.01 (0.66–1.37) | |
| 0.922 (12) | ||||
| ≥1 year−≤2 years | 499 (13) | 35.5 (51) | 1.29 (0.63–1.96) | |
| Adult | 2249 (23) | 137 (58) | – |
Range based on ± 1 SEM.
In general desloratadine parameters were reasonably well estimated and intersubject variability, expressed as a coefficient of variation, was moderate (Table 3). The population analysis indicated that, if subjects aged ≥6 months to <1 year and ≥1 year−≤2 years received doses of 1 mg and 1.25 mg of desloratadine, respectively, their exposure (Cmax and AUC) would be similar to that observed in adults receiving 5 mg. The model-predicted mean (%CV) Cmax and AUC from time 0 to infinity (AUC(0, ∞)) for paediatric subjects were comparable with adult values. Mean Cmax and AUC (%CV) values for ≥6 months−<1 year were 1.69 (49) ng ml−1 and 38.7 (65) ng ml−1 h, respectively, for ≥1 year−≤2 years were 1.56 (46) ng ml−1 and 34.4 (101) ng ml−1 h, respectively, and corresponding values for adults were 1.94 (42) ng ml−1 and 43.2 (81) ng ml−1 h, respectively (Table 4 and Figure 6). Point estimates from the analysis of variance performed on the log-transformed predicted AUC and Cmax values indicated that the 0.625 mg dose appeared to be suboptimal for both age groups, particularly with respect to Cmax (Table 5). The 90% confidence intervals are not expected to show bioequivalence due to the small size. The doses of 1 mg and 1.25 mg selected for children ≥6 months−<1 year and ≥1 year−≤2 years, respectively, are predicted to provide comparable exposure (AUC) to that observed in adults. Individual predicted pharmacokinetic parameters by final model were compared with those calculated by noncompartmental methods (Figure 7). There was an excellent correlation between the results of calculation by noncompartmental analysis and individual estimates from the population model. Adult median noncompartment Cmax, tmax and AUC values are added as a footnote to Table 4. The noncompartmental values are comparable with the median values of model predicted parameters.
Table 3.
Intersubject and residual variability
| Parameter | Magnitude of variability (% CV) | % SEM |
|---|---|---|
| Random effect | ||
| CL/F_η | 37 | 29 |
| V/F_η | 32 | 27 |
| ka_η | 69 | 48 |
| Residual variability | 9.67 | 36 |
% CV % coefficient of variation.
Table 4.
Mean (% coefficient of variation, estimated using standard deviation) and median predicted desloratadine parameters by age
| Mean (% coefficient of variation) | Median | |||||
|---|---|---|---|---|---|---|
| Parameter | ≥6 months–<1 yeara | ≥1 year–≤2 yearsa | Adultsb | ≥6 months–<1 yeara | ≥1 year–≤2 yearsa | Adultsb |
| Cmax (ng ml−1) | 1.69 (49) | 1.56 (46) | 1.94 (42) | 1.49 | 1.41 | 1.69c |
| tmax (h) | 3.16 (22) | 3.10 (28) | 3.22 (23) | 3.12 | 2.94 | 3.11c |
| AUC(0,∞) (ng ml−1 h) | 38.7 (65) | 34.4 (101) | 43.2 (81) | 29.3 | 19.0 | 34.4c |
| ka (h−1) | 0.959 (18) | 0.929 (17) | 0.956 (29) | 0.950 | 0.944 | 0.912 |
| CL/F (l h−1) | 34.7 (66) | 43.0 (55) | 155 (47) | 30.1 | 40.3 | 146 |
| t1/2 (h) | 14.6 (77) | 12.4 (88) | 12.2 (44) | 10.5 | 8.40 | 10.9 |
| V/F (l) | 494 (32) | 518 (29) | 2373 (33) | 473 | 479 | 2314 |
Parameter from combined dose (0.625 mg and 1.25 mg)
Parameter associated with 5 mg dose
Median values of estimated noncompartmental parameters: AUC(0, ∞) = 39.6 ng ml −1 h, C max = 1.84 ng ml −1, tmax = 4.5 h.
Figure 6.

Comparable AUC values in ≥6 months−<1 year and ≥1 year−≤2 years age groups, and adults
Table 5.
Point estimates and 90% confidence intervals (CI) for predicted desloratadine AUC and Cmax values by age group and dose
| Parameter | ≥6 months−≥1 yeara | <1 year−≤2 yearsb | ≥6 months−<1 yearc | ≥1 year−≤2 yearsd |
|---|---|---|---|---|
| AUC(0,∞) | ||||
| Point estimate | 0.8204 | 0.9257 | 0.5187 | 0.8989 |
| Lower 90% CI | 0.5552 | 0.6265 | 0.3791 | 0.6570 |
| Upper 90% CI | 1.212 | 1.368 | 0.7096 | 1.230 |
| Cmax | ||||
| Point estimate | 0.6337 | 1.129 | 0.5394 | 1.117 |
| Lower 90% CI | 0.5195 | 0.9256 | 0.4599 | 0.9523 |
| Upper 90% CI | 0.7729 | 1.377 | 0.6327 | 1.310 |
≥6 months−<1 year (0.625 mg, n = 10) vs. adults (5 mg, n = 30)
≥6 months−<1 year (1.25 mg, n = 10) vs. adults (5 mg, n = 30)
≥1 year– ≤2 years (0.625 mg, n = 19) vs. adults (5 mg, n = 30
≥1 year – ≤2 years (1.25 mg, n = 19) vs. adults (5 mg, n = 30).
Figure 7.

Comparison of individual parameters of desloratadine, obtained by noncompartmental methods vs. those predicted by the final model
Mean and median AUC(0,last) values and ratios for 3-OH-desloratadine are summarized in Table 6. AUC(0,last) values for 3-OH-desloratadine increased in a dose-related manner in both paediatric age groups. The AUC(0,last) ratio of 3-OH-desloratadine relative to desloratadine indicated that four subjects ≥1 year−≤2 years of age were poor metabolizers of desloratadine (ratio < 10%). All four subjects were African American (three males and one female) and their parameters are listed in Table 7.
Table 6.
Mean (% coefficient of variation) and median AUC(0,last) values for 3-OH-desloratadine by age group and dose
| ≥6 months−<1 year | ≥1 year−≤2 years | |||
|---|---|---|---|---|
| Parameter | 0.625 mg | 1.25 mg | 0.625 mg | 1.25 mg |
| Mean AUC(0,last) (ng ml−1 h) | 9.29 (47) | 18.1 (71) | 6.30 (44) | 16.5 (65) |
| Median AUC(0,last) (ng ml−1 h) | 7.30 | 16.4 | 5.85 | 15.6 |
| Mean AUC(0,last) ratio (%) | 38.0 (55) | 51.8 (61) | 42.4 (58) | 64.1 (81) |
| Median AUC(0,last) ratio (%) | 38.4 | 50.2 | 43.2 | 58.7 |
AUC(0,last) ratio = (AUC(3-OH-desloratadine) : AUC(desloratadine)) ×100.
Table 7.
Mean (% coefficient of variation) and median AUC(0,last) values for 3-OH-desloratadine in four poor metabolizers of desloratadine aged ≥6 months−<2 years
| Parameter | Both dose groups |
|---|---|
| Mean AUC(0,last) (ng ml−1 h) | 2.28 (123) |
| Median AUC(0,last) (ng ml−1 h) | 1.56 |
| Mean AUC(0,last) ratio (%) | 4.61 (111) |
| Median AUC(0,last) ratio (%) | 4.59 |
AUC(0,last) ratio = (AUC(3-OH-desloratadine) : AUC(desloratadine)) × 100.
Safety
Adverse events were reported in two male subjects, both receiving desloratadine 0.625 mg. Both events were mild in severity and required no additional therapy. A 23 month old subject experienced loose stools, which were considered possibly related to the study treatment, and a 10 month old subject experienced teething pain, considered unlikely to be related to treatment. No serious or unexpected adverse events were reported. There were no clinically relevant changes in laboratory parameters, vital signs, ECG recordings, or physical examination findings at the end of the study.
Discussion
The population pharmacokinetic model in paediatric and adult subjects was based on a one-compartment model with first order absorption. We combined data from this paediatric study with adult data for population pharmacokinetic analysis to determine doses of desloratadine suitable for administration to children aged ≥6 months−≤2 years in clinical and efficacy studies. Single doses of 0.625 mg (1.25 ml) and 1.25 mg (2.5 ml) desloratadine syrup were well tolerated in this age group. Because the study was designed to collect only a minimal number of blood samples, a population pharmacokinetic modelling approach was used. Results of the pharmacokinetic analysis indicate that desloratadine apparent clearance rates were slower in the paediatric group studied than in adults, and that the 0.625 mg dose appeared to be suboptimal for both children aged ≥6 months−<1 year and for those aged ≥1 year−≤2 years, particularly with respect to Cmax. Variability between subjects in the model parameters was moderate, but was consistent with estimates of pharmacokinetic variables obtained with noncompartmental analysis. No systematic bias was observed in parameter estimation. On the basis of the results of this analysis, to ensure similar desloratadine exposure to that seen in adults, the age-appropriate doses for children aged ≥6 months−<1 year and ≥1 year−≤2 years were established as 1.0 mg and 1.25 mg, respectively.
Desloratadine is extensively metabolized in the liver to its active metabolite, 3-OH-desloratadine. The enzyme responsible for this is unknown. However, it is known that some individuals, termed poor metabolizers, have a reduced ability to form the active metabolite. Exposure to desloratadine has previously been shown to be approximately six-fold greater in poor metabolizers than in the rest of the population, with a similar magnitude of reduction in the formation of 3-OH-desloratadine in adults and children given age-appropriate doses [12]. This is the first study to assess the exposure (AUC) of 3-OH-desloratadine in young children. Based on the observed AUC(0,last) ratio of 3-OH-desloratadine relative to desloratadine, the proportion of poor metabolizers in this study population was approximately 7%. The poor metabolizers are African American. This is consistent with the findings of a recent large study in older children and adults, which found an age-independent prevalence of this phenotype of 6% [8].
Desloratadine syrup has been shown to be well tolerated in children 2–5 years and 6–12 years of age who have allergic rhinitis or chronic idiopathic urticaria [7, 13]. Desloratadine syrup also appeared to be well tolerated in the younger age group enrolled in this study, with no unexpected safety issues following a single dose. Despite the difference in exposure to desloratadine in poor metabolizers, data from studies in patients as young as 2 years have shown it to be well tolerated [14]. This was also the case in this study, with no differences in the safety profile of desloratadine between poor metabolizers and other subjects.
In conclusion, desloratadine syrup offers a safe treatment option for allergic conditions in young children, with suitable doses in those aged ≥6 months−<1 year and ≥1 year−≤2 years of 1.0 mg (2.0 ml) and 1.25 mg (2.5 ml), respectively. These paediatric dose concentrations yielded systemic desloratadine exposures similar to those seen in adults.
Acknowledgments
Competing interests: None declared.
References
- 1.American Academy of Allergy Asthma & Immunology. The Allergy Report. Accessed at http://www.theallergyreport.org/reportindex.html.
- 2.Greaves MW. Chronic urticaria in childhood. Allergy. 2000;55:309–20. doi: 10.1034/j.1398-9995.2000.00116.x. [DOI] [PubMed] [Google Scholar]
- 3.Berger WE. Allergic rhinitis in children: diagnosis and management strategies. Paediatr Drugs. 2004;6:233–50. doi: 10.2165/00148581-200406040-00003. [DOI] [PubMed] [Google Scholar]
- 4.Vuurman EF, van Veggel LM, Uiterwijk MM, Leutner D, O'Hanlon JF. Seasonal allergic rhinitis and antihistamine effects on children's learning. Ann Allergy. 1993;71:121–6. [PubMed] [Google Scholar]
- 5.Fireman P. Therapeutic approaches to allergic rhinitis: treating the child. J Allergy Clin Immunol. 2000;105:S616–S621. doi: 10.1067/mai.2000.106152. [DOI] [PubMed] [Google Scholar]
- 6.Clarinex®, (desloratadine) tablets, syrup, Reditabs®, tablets [package insert]. Kenilworth, NJ 07033, USA: Schering-Plough Corporation; [Google Scholar]
- 7.Bloom M, Staudinger H, Herron J. Safety of desloratadine syrup in children. Curr Med Res Opin. 2004;20:1959–65. doi: 10.1185/030079904x14148. [DOI] [PubMed] [Google Scholar]
- 8.Prenner B, Kim K, Gupta S, Khalilleh S, Kantasaria B, Manitpistkul P, Lorber R, Wang Z, Lusky B. Adults and paediatric poor metabolizers of desloratadine. an assessment of pharmacokinetics and safety. Experts Opin Drug Saf. 2006;5:211–23. doi: 10.1517/14740338.5.2.211. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Clement RP, Kantesaria B, Reyderman L, Beaudry F, Grandmaison C, Lorella Di Donato Masse M, Rudewicz PJ. Validation of a sensitive and automated 96-well solid-phase extraction liquid chromatography-tandem mass spectrometry method for the determination of desloratadine and 3-hydroxydesloratadine in human plasma. J Chromatograph B, Anal Technolog Biomed Life Sciences. 2003;792:229–40. doi: 10.1016/s1570-0232(03)00267-8. [DOI] [PubMed] [Google Scholar]
- 10.WinNonMix Version 2.0.1. California, USA: Pharsight Corporation; [Google Scholar]
- 11.Gibaldi M, Perrier D. Pharmacokinetics. 2. New York: Marcel Dekker, Inc; 1998. pp. 409–17. [Google Scholar]
- 12.Gupta S, Kantesaria B, Wang Z, Khalilieh S. Pharmacokinetics of desloratadine in poor metabolizers. Presented at the Annual Meeting of American College of Allergy. November 12–17, 2004, Asthma and Immunology, Boston, MA Abstract P249.
- 13.Rossi GA, Tosca MA, Passalacqua G, Bianchi B, Le Grazie C, Canonica GW. Evidence of desloratadine syrup efficacy and tolerability in children with pollen-induced allergic rhinitis. Allergy. 2005;60:416–7. doi: 10.1111/j.1398-9995.2005.00714.x. [DOI] [PubMed] [Google Scholar]
- 14.Khalilieh S, Lutsky B, Lorber R. Safety of desloratadine in poor metabolizers. Presented at the Annual Meeting of the American College of Allergy. November 12–17, 2004, Asthma and Immunology, Boston, MA Abstract P248.