

Abstract
AMP-activated protein kinase (AMPK) represents a key energy-sensing molecule in many cell types. Because astrocytes are key mediators of metabolic signaling in the brain, we have initiated studies on the expression and activation of AMPK in these cells. Treatment of cultured rat cortical astrocytes with a pharmacological AMPK activator, AICA-riboside (AICAR) resulted in a time- and concentration-dependent increase in phosphorylation of AMPK and acetyl-CoA carboxylase (ACC), a direct substrate. AICAR treatment also induced a transition from epithelioid to stellate morphology in a time- and concentration-dependent manner. As stellation is indicative of actin cytoskeletal reorganization, the formation of stress fibers and focal adhesions in response to AICAR was assessed. AICAR-induced stellation correlated with F-actin disassembly and focal adhesion dispersal. Furthermore, transient transfection of an activated RhoA construct prevented AICAR-induced stellation, indicating a mechanism upstream of RhoA. Use of pharmacological inhibitor compound C prevented AICAR-induced stellation demonstrating necessity of AMPK activity for the response. Our findings suggest that AMPK mediates morphological alterations of astrocytes in response to energy depletion.
Keywords: AMPK, AICAR, astrocyte, stellation
INTRODUCTION
Astrocytes exhibit multiple branched processes in vivo. In pathological states, reactive astrocytes undergo characteristic morphological changes such as greater branching and thickening of processes and somatic hypertrophy [34]. In contrast, cultures of primary rodent cortical astrocytes exhibit a flat and polygonal shape. Knowledge about the mechanisms that govern astrocyte morphology changes comes from the study of agents that induce cultured astrocytes to exhibit a process bearing, or “stellate”, morphology. Agents that induce stellation include cyclic AMP [37,48]; neuromodulators, such as ATP and adenosine; growth factors, such as FGF2; hormones, such as vasopressin; PKC activators, such as phorbol esters; cytokines, such as TNF alpha [36]; and peptides, such as β amyloid [32]. Because these molecules are also released in brain injury or upregulated during gliosis [15], they could underlie morphological plasticity of astrocytes in vivo.
Recent studies revealed PI3K inhibition as the mechanism governing cyclic AMP-induced stellation [41]. Downstream of PI3K signaling, Akt phosphorylates and inhibits tuberous sclerosis complex 2, TSC2 [20,29]. AMP-activated protein kinase (AMPK) also phosphorylates TSC2, at sites distinct from Akt, resulting in its activation [30]. One function of TSC2 is to antagonize mTOR signaling [28]. There are two functionally distinct mTOR complexes, mTORC1, responsible for cell growth and proliferation, and mTORC2, which signals through Rho GTPases to stimulate actin stress fiber formation [31]. TSC2 can also inhibit Rho by binding to TSC1, and thus promote stress fiber disassembly and focal adhesion remodeling [23]. Therefore, AMPK modulates substrates that potentially control actin stress fiber dynamics. Indeed, stable overexpression SNARK, a member of the AMPK catalytic subunit family, caused HepG2 cells to exhibit a rounded morphology that was attributed to conversion of F-actin filaments to G-actin monomers [50]. SNARK overexpression also prevented focal adhesion kinase phosphorylation in response to glucose deprivation in these cells, implying energy levels modulate the actin cytoskeleton [50].
In vivo, severe hypoglycemia results in gliosis after neuronal necrosis [6]. Energy depletion by hypoglycemia, hypoxia, and ischemia activates AMPK [17,35] to restore ATP levels [26]. Interestingly, brain injury increases AMPK catalytic subunit expression in reactive astrocytes [53]. In the present study, we investigate the consequence of AMPK activation on morphology of cultured astrocytes using AICAR, a pharmacological AMPK activator.
RESULTS
To activate AMPK in cultured astrocytes, we used AICA-riboside (AICAR), a widely used pharmacological activator. To confirm that AMPK activation had occurred, we assessed the phosphorylation of AMPK and its direct substrate, acetyl-CoA carboxylase (ACC), indicative of AMPK pathway activation. Treatment with AICAR induced a time- and concentration-dependent increase in AMPK and ACC phosphorylation (Fig. 1). AMPK and ACC phosphorylation was evident at 30 min and persisted for at least 8 h (Fig. 1A). At 24 h, phosphorylation of AMPK returned to baseline but that of ACC remained increased (Fig. 1A). Possible explanations for the divergence of AMPK and ACC phosphorylation include differential dephosphorylation kinetics by the relevant phosphatases or activation of other pathways by AICAR resulting in ACC phosphorylation. Maximal AMPK and ACC phosphorylation was evident at a concentration of 0.5mM AICAR (Fig. 1B).
Figure 1. Treating cultured rat cortical astrocytes with a pharmacological AMPK activator, AICAR, induces phosphorylation of AMPK and its direct substrate, acetyl-CoA carboxylase (ACC).
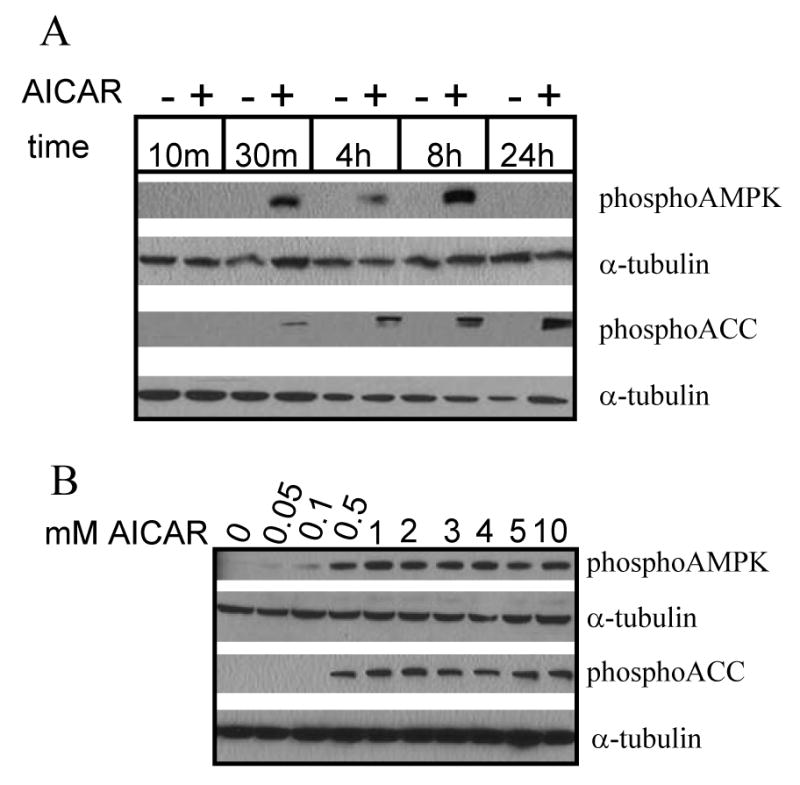
(A) Time course of AICAR-induced AMPK and ACC phosphorylation. Cells were rinsed and maintained in Hank's balanced salt solution (HBSS) containing vehicle (dH2O) or 2mM AICAR for the times indicated. Western blot reveals AICAR-induced AMPK and ACC phosphorylation after 30 min incubation, which persists for at least 8 h. (B) Concentration-dependence of AICAR-induced AMPK and ACC phosphorylation. Cells were rinsed and maintained in HBSS containing vehicle or concentrations of AICAR indicated for 2 h. Western blot reveals AICAR-induced AMPK and ACC phosphorylation at a minimum concentration of 0.5mM. Tubulin is included as a loading control. Western blots are representative of 2 independent experiments.
Next, we examined the relationship between AMPK activation and astrocyte morphology. Because individual cell morphologies were difficult to assess in confluent cultures, cells were infected with GFP adenovirus to highlight a subset of cells for quantification. Stellate cells were classified as those exhibiting at least 3 discrete processes and excluded unbranched short filopodia less than one soma diameter. GFP adenovirus-infected astrocytes treated with vehicle (dH2O) exhibited rare stellate cells (Fig. 2A), whereas cells treated with AICAR exhibited widespread stellation (Fig. 2B). Stellation correlated with the time- and concentration-dependence of AMPK and ACC phosphorylation induced by AICAR. AICAR-induced stellation was evident at 1 h of incubation, with statistical significance at 2 and 4 h, and began to reverse by 6 h (Fig. 2C). These results suggest that AICAR-induced stellation was transient which was confirmed by photomicrographs of live cells taken on a marked coverslip in the same field at 4 (Fig. 2E) and 24 (Fig. 2F) h after treatment. At 4 h, cells 1 and 2 are stellate (Fig. 2E). After 24 h, cell 2 remains stellate, whereas cell 1 has returned to its original polygonal morphology (Fig. 2F). This time course of AICAR-induced stellation was similar to other stellating agents, including adenosine and phorbol myristate acetate [3,4]. Stellation was significant at 2mM AICAR with maximal effect at 10mM (Fig. 2D).
Figure 2. AICAR treatment induces stellate morphology in a time- and concentration-dependent manner in cultured rat cortical astrocytes.
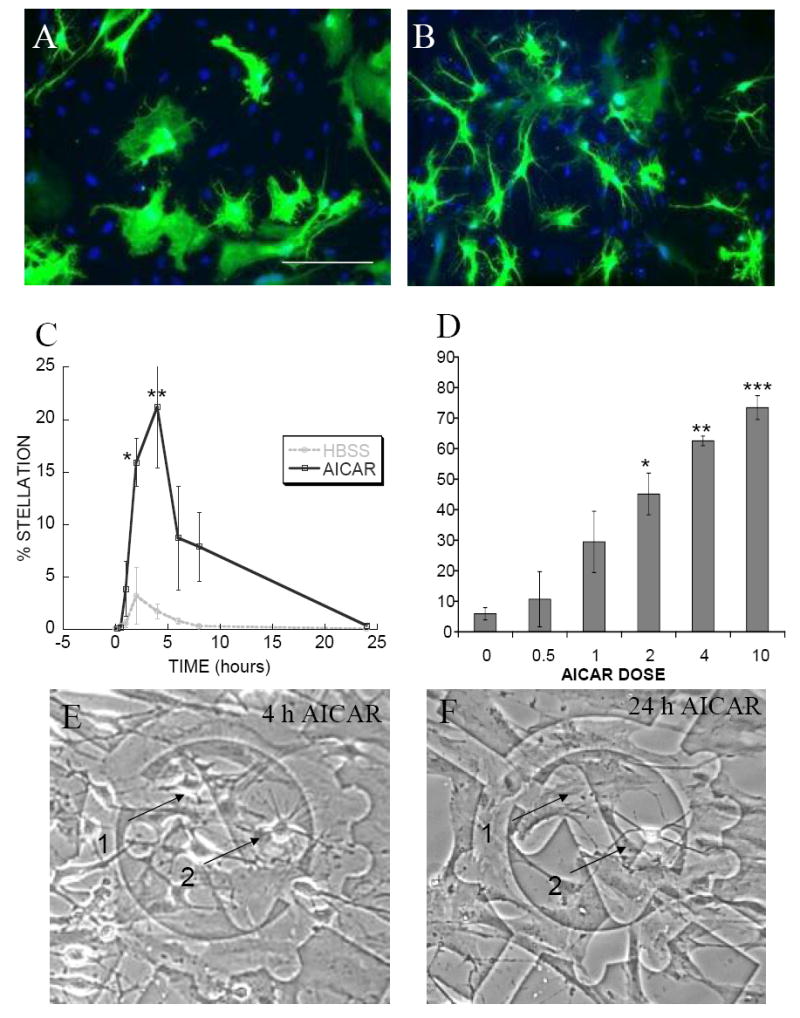
Photomicrographs show stellate morphology induced in cells treated with AICAR. Cells infected with GFP adenovirus for 48 h were rinsed and maintained in HBSS containing either (A) vehicle or (B) 4mM AICAR for 2 h. (A and B) Scale bar 50μm. (C) AICAR-induced stellation begins at 1 h of incubation and reverses by 24 h. Cells were maintained in HBSS in the presence (black solid line) or absence (gray dotted line) of 4mM AICAR for the times indicated on the abscissa. Fixed cells were stained with Oregon Green-labeled phalloidin and DAPI. Percent stellation was calculated as number of stellate cells divided by total number of DAPI nuclei. Data are mean ± SEM (bars) of 3 independent experiments. * p = 0.0017, ** p = 0.0001 (D) AICAR-induced stellation requires a minimum concentration of 2mM. Cells infected with GFP adenovirus for 48 h were rinsed and maintained for 2 h in HBSS containing the concentrations of AICAR indicated on the abscissa. Percent stellation was calculated as number of GFP-positive stellate cells divided by total number of GFP-positive cells. Data are mean ± SEM (bars) of 3 independent experiments. * p = 0.05, ** p = 0.025, *** p = 0.0178 (E and F) AICAR-induced stellation is transient. Photomicrographs show live cells that were rinsed and maintained in HBSS containing 4mM AICAR for (E) 4 h or (F) 24 h. (E) At 4 h, cells 1 and 2 are stellate. (F) After 24 h most cells, such as cell 1, have returned to their original polygonal morphology whereas occasional cells, such as cell 2, remain stellate.
We compared the stellate morphologies of astrocytes treated with AICAR with that induced by conventional stellating agents, forskolin and AMP. Forskolin-induced stellation is more complete than that induced by AMP, involving complete retraction of the cell membrane [24]. Morphology was visualized using phase contrast microscopy (Fig. 3A-D), or via immunostaining against GFAP (Fig. 3E-H) or ezrin (Fig. 3I-L), an actin-binding protein revealing the fine peripheral processes of astrocytes [14]. AICAR-induced stellation was observed to be similar to that induced by AMP, with fewer cells exhibiting complete membrane retraction. In several cases using cultured astrocytes, stellate morphology correlates with increased GFAP expression [5,39,46]. Therefore, we investigated GFAP protein levels over the time course of AICAR-induced stellation. Western blots did not reveal any increase in GFAP during AICAR incubation up to 24 h (data not shown).
Figure 3. AICAR-induced stellation is similar to AMP-induced stellation, with fewer cells exhibiting complete membrane retraction as seen in forskolin-induced stellation.
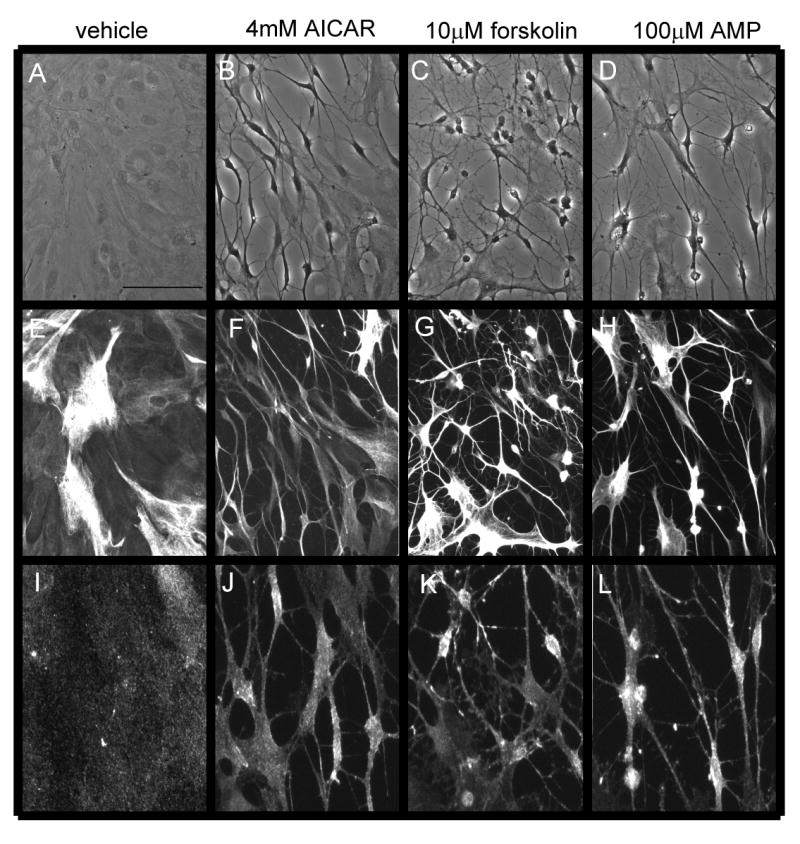
Cells were rinsed and maintained in either (A-B, E-F, I-J) HBSS or (C-D, G-H, K-L) SF DMEM containing (A, E, I) vehicle, (B, F, J) 4mM AICAR, (C, G, K) 10μM forskolin, or (D H, L) 100μM AMP for 2 h. (A-D) Photomicrographs show representative fields of cells visualized with phase contrast. (E-H) GFAP immunofluorescence. (I-L) Ezrin immunofluorescence reveals the fine processes of astrocytes. Scale bar 50μm. Photomicrographs are representative of 3 independent experiments.
AICAR enters the cell through the adenosine transporter [21]. Intracellular phosphorylation by adenosine kinase results in its AMPK activating form, “ZMP” [45]. In rat brain slices, AICAR treatment induced depression of excitatory synaptic transmission in CA1 hippocampus [21]. This effect of AICAR was due to indirect activation of adenosine receptors because AICAR competes with adenosine for access to the transporter, increasing extracellular adenosine [21]. Treating astrocytes with adenosine, and its derivatives AMP, ADP, and ATP, induces stellation by P1 purinoceptor activation [3]. To rule out this potential alternate mechanism of AICAR-induced stellation, we examined the effect of an adenosine A1 receptor inhibitor on AICAR-induced stellation. As previously reported, preincubation with 10 μM A1 adenosine receptor inhibitor, xanthine amine congener (XAC), prevented astrocyte stellation induced by 100 μM extracellular AMP (Fig. 4A) [3]. In contrast, preincubation with XAC did not diminish AICAR-induced stellation (Fig. 4B). Thus, AICAR-induced stellation does not require adenosine A1 receptor activation, supporting the hypothesis that AMPK activation induces astrocyte stellation.
Figure 4. Adenosine A1 receptor inhibitor, xanthine amine congener (XAC), blocks AMP-induced stellation with no effect on AICAR-induced stellation in cultured rat cortical astrocytes.

Cells were infected with GFP adenovirus for 48 h. After 24 h, cell media was changed to SF DMEM overnight. Then cells were rinsed and maintained in either (A) HBSS or (B) SF DMEM containing vehicle (DMSO) or 10μM XAC for 5 min. Then either (A) 100μM AMP or (B) 4mM AICAR was added for 3 h. Percent stellation was calculated as number of stellate GFP-positive cells divided by total number of GFP-positive cells. Data are mean ± SEM (bars) of 4 independent experiments. * p = 0.005, ** p = 0.0044, *** p = 0.0014
Actin stress fibers contact the extracellular matrix at focal adhesions in the plasma membrane [10]. Astrocyte stellation is usually the result of actin stress fiber disassembly [22]. To demonstrate the involvement of stress fibers and focal adhesions in AICAR-induced stellation, we stained cells with phalloidin, a probe that specifically binds to F-actin [1], and vinculin, an actin-binding protein enriched at focal adhesions [10]. AICAR-induced stellation resulted in marked loss of stress fibers as evidenced by phalloidin staining (arrows, Fig. 5A-C). Immunofluorescence with antibodies against vinculin revealed dispersal of focal adhesions after AICAR treatment (Fig. 5D-F).
Figure 5. AICAR-induced stellation is associated with disassembled actin stress fibers and dispersed focal adhesions.
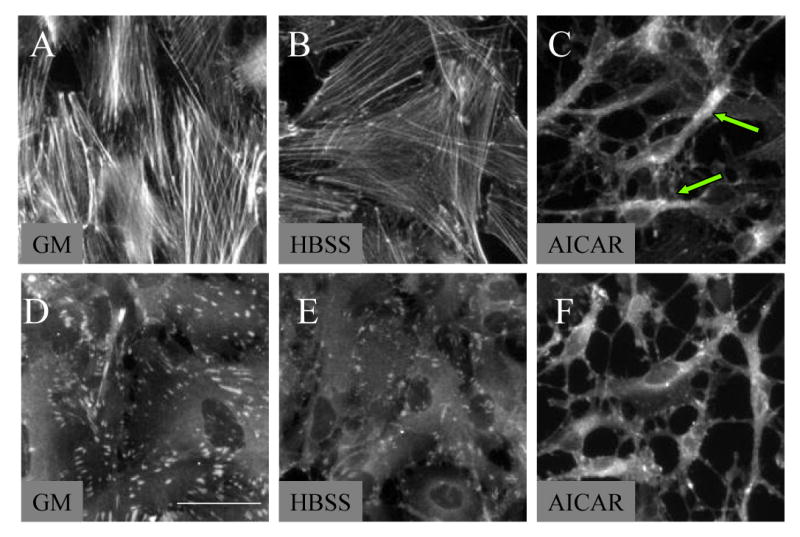
Cells were rinsed and maintained in HBSS for 2 h containing (A and D) growth media (GM), (B and E) vehicle (dH2O), or (C and F) 2mM AICAR. Cells immunostained for (A, B, C) Oregon Green labeled phalloidin toxin to visualize stress fibers or (D, E, F) monoclonal antibody against vinculin to visualize focal adhesions. Accompanying stellation, (C, arrows) stress fibers are disassembled and (F) focal adhesions are dispersed. Scale bar 25μm. Photomicrographs are representative of 3 independent experiments.
Modulation of the actin cytoskeleton and focal adhesions is directly linked to RhoA activity [25]. In addition, several modes of stellation, including adenosine and cAMP, require Rho inactivation [42,44]. Merely inhibiting Rho and/or its downstream effectors is sufficient to induce stellation in astrocytes [1,44]. To directly test whether AICAR-induced stellation was upstream of RhoA, we transiently transfected cultured rat cortical astrocytes with a plasmid expressing constitutively active RhoA (RhoV14) [40]. In cells transfected with RhoV14, AICAR-induced stellation was completely prevented, in contrast to cells transfected with GFP plasmid as a control (Fig. 6B,D). Of note, transient transfection of RhoV14 also abolished basal stellation induced by media change to HBSS (data not shown).
Figure 6. AICAR-induced stellation is prevented by constitutive RhoA activation.
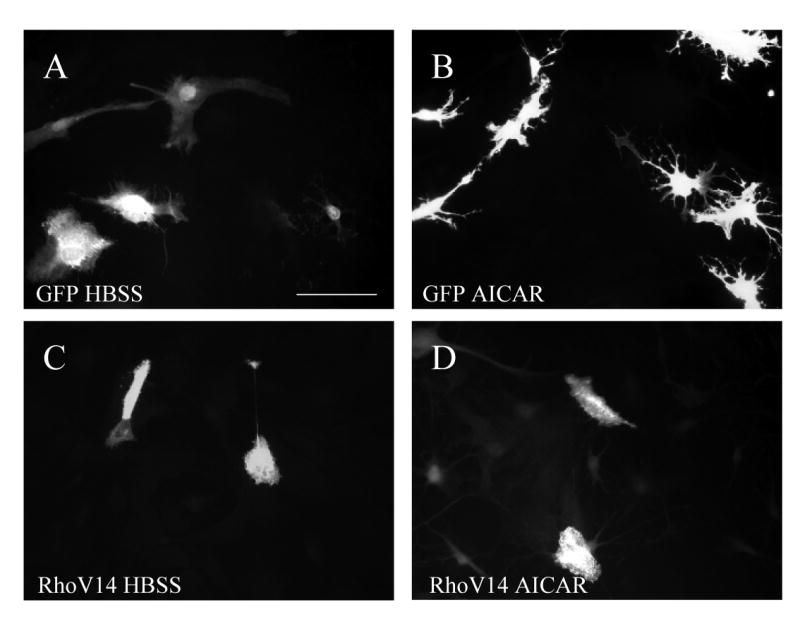
Cells were transiently transfected for 48 h with either (A and B) GFP, as control, or (C and D) myc-tagged constitutively active RhoA (RhoV14). Then cells were rinsed and maintained in HBSS containing (A and C) vehicle (dH2O) or (B and D) 2mM AICAR for 2 h. Cells were immunostained for either myc or GFP. Scale bar 50μm. Photomicrographs are representative of 3 independent experiments.
To assess whether the morphology changes induced by AICAR in cultured astrocytes required AMPK activation, we used a pharmacological AMPK inhibitor, compound C [55]. The use of compound C as an AMPK inhibitor has been previously described and characterized [55]. The mechanism of compound C action is to inhibit AMPK kinase activity, but not the activity of its upstream kinases. As expected, preincubation with compound C caused diminished AICAR-induced phosphorylation of the AMPK substrate ACC, but not of AMPK itself (Fig. 7B). In cultured rat cortical astrocytes preincubated with 40 μM compound C (CC), AICAR-induced stellation was prevented (Fig. 7A). These results suggest that AMPK activation is necessary for AICAR-induced stellation.
Figure 7. AICAR-induced stellation requires AMPK activity.
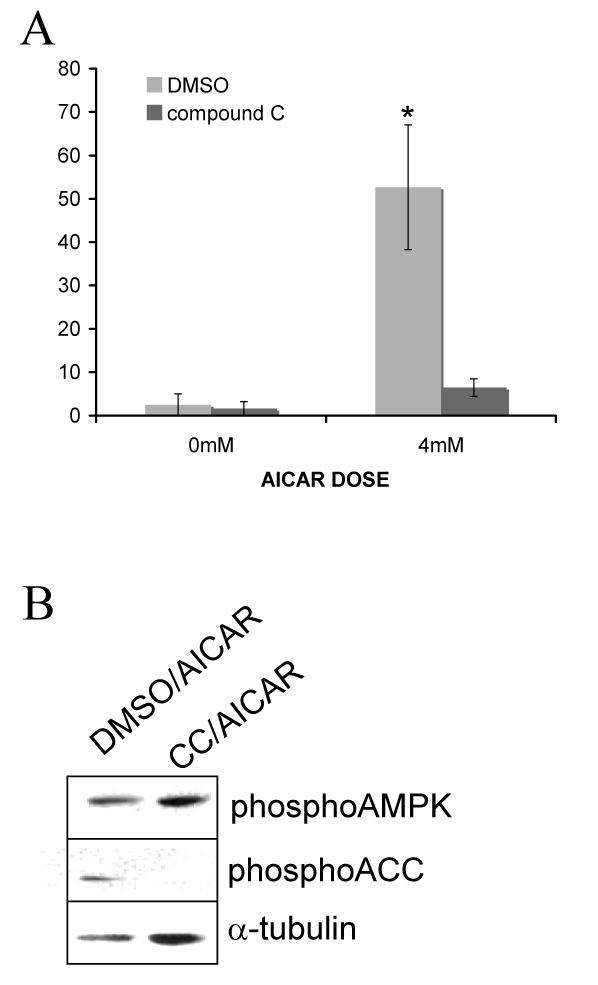
(A) Compound C prevents AICAR-induced stellation. Cells were infected with GFP adenovirus for 48 h. Then cells were rinsed and maintained in HBSS containing vehicle (DMSO) or 40μM compound C (CC) for 30 min followed by 2 h incubation with 4mM AICAR. Percent stellation was calculated as number of stellate GFP-positive cells divided by total number of GFP-positive cells. Data are mean ± SEM (bars) of 3 independent experiments. * p = 0.0216 (B) Compound C prevents AICAR-induced ACC phosphorylation. Cells were rinsed and maintained in HBSS containing DMSO or 40μM compound C for 30 min followed by 2 h incubation with 2mM AICAR. Western blot reveals diminished ACC phosphorylation in cells treated with compound C. As expected, AMPK phosphorylation is not decreased by CC since this inhibitor decreases AMPK activity but not its phosphorylation by its upstream kinase LKB. The western blot is representative of 3 independent experiments.
DISCUSSION
In this report we have shown that treatment with a pharmacological AMPK activator, AICAR, induces stellation, a characteristic morphological change in cultured rat cortical astrocytes. AICAR-induced stellation is rapid, transient, concentration-dependent, and involves disassembly of F-actin stress fibers and dispersal of focal adhesions. Our initial investigation of the mechanism of AICAR-induced stellation revealed that AICAR-induced stellation depends on RhoA inactivation. Lastly, we present evidence to suggest that AMPK activity is necessary to induce stellation in astrocytes.
Studies investigating upstream and downstream components of the AMPK pathway provide evidence that AMPK could regulate cell morphology. LKB1, recently identified as an upstream activator of AMPK [27], is a polarizing stimulus in C. elegans embryo asymmetry formations [54]. In an in vitro scratch wound assay, astrocytes polarize, with their centrosome facing the wound, before migrating to close the wound [16]. When an LKB1 expression plasmid with a disrupted ATP-binding site, rendering it kinase dead and therefore unable to activate AMPK, was injected into the leading edge of astrocytes, this wound-induced reorientation was abolished [19]. In Saccharomyces cerevisiae, pharmacological mTOR inactivation by rapamycin treatment results in depolymerization of the actin cytoskeleton [52]. In mammalian cells, mTOR signaling is antagonized by AMPK activation of TSC2. The results of this study suggest that direct activation of AMPK can alter astrocyte morphology.
In this report, we present evidence to support the fact that AICAR-induced stellation is associated with disassembly of stress fibers at focal adhesions. Stress fiber assembly and focal adhesion formation depends on RhoA activation, as initially demonstrated in Swiss 3T3 fibroblasts [33,43]. In cultured astrocytes, RhoA activation is thought to maintain polygonal morphology [2,49]. Treating pituicytes with lysophosphatidic acid, a RhoA activator, prevented stellation in response to adenosine [44]. Astrocytes expressing constitutively activated RhoA fail to undergo cAMP induced stellation [42]. Manganese-induced stellation in cultured rat cortical astrocytes was recently demonstrated to be dependent on RhoA pathway inactivation [12]. In this study, astrocytes transiently transfected with a constitutively active RhoA construct, RhoV14, failed to undergo stellation when treated with AICAR. Thus, AICAR-induced stellation, like manganese and cAMP, acts upstream of the RhoA pathway in astrocytes.
Treating astrocytes with an AMPK inhibitor, compound C, prevented AICAR-induced ACC phosphorylation and stellation, supporting the necessity of AMPK activation for morphology changes. Interestingly, lower doses of AICAR resulting in AMPK phosphorylation were not sufficient to induce significant stellation. The higher doses of AICAR required to induce stellation suggest that another pathway may cooperate with AMPK. Possible candidates would be atypical PKC isoforms. Incubation of isolated rat extensor digitorum longus muscles with two different pharmacological activators of AMPK, AICAR and dinitrophenol, resulted in PKC zeta activation [13]. In epithelial cells, polarity is regulated by activation of Par4, a LKB1 homolog [7]. When LKB1 is activated it then leads to activation of a network of Par proteins, some of which recruit Rho GTPases and others recruit atypical PKCs, to modulate actin cytoskeleton dynamics. Similarly in astrocytes, activation of the Par6/PKC zeta complex is essential for polarization of migrating cells during in vitro wound healing [16].
AICAR-induced stellation of cultured astrocytes represents a useful model to study morphological changes that may occur in vivo in response to energy deprivation. AMPK-mediated regulation of the actin cytoskeleton could play an important role in coupling metabolic signals to morphological plasticity of astrocytes. The terminal processes of astrocytes that ensheath blood vessels, termed endfeet, regulate cerebral blood flow in response to increased neuronal activity [18,38,51]. Astrocytic release of vasoactive molecules is a response to regional energy depletion because neurons need a continuous supply of glucose to produce ATP and maintain excitability, but their effectiveness is determined by proximity of astrocyte processes [18]. Astrocytes also respond to pathological energy depletion, such as hypoglycemia, by releasing lactate [9]. Interestingly, several agents that induce stellation in cultured astrocytes also stimulate glycogenolysis [11,47]. Thus, changes in astrocyte morphology upon energy deprivation may be indicative of underlying neuroprotective responses. As energy depletion activates AMPK, this suggests a novel role to modulate the actin cytoskeleton in astrocytes.
EXPERIMENTAL PROCEDURE
Cell culture
Cortical astrocytes were prepared from the cerebral cortices of 1-day-old Sprague-Dawley rats and maintained as described previously [8]. The resultant cells were used for experiments 2-7 weeks after preparation. Cells were trypsinized, replated onto 18mm glass coverslips at an average density of 7 × 104 cells per coverslip, and grown in Dulbecco's Modified Eagle media (DMEM) containing antibiotic-antimycotic and 10% fetal bovine serum until confluent. Coverslips were not coated with poly-L-lysine because this substrate may induce morphological changes in astrocytes [24]. Where indicated, the culture medium was switched to serum-free DMEM (SF DMEM) 24 h before the start of experiments. For experiments, cells were incubated in SF DMEM or Hank's Balanced salt solution (HBSS). In typical cultures >98% of the cells were identified as astrocytes by immunostaining for glial fibrillary acidic protein (GFAP) and by their flattened, polygonal appearance. HBSS was purchased from Hyclone (Logan, UT, USA). The formulation for HBSS is as follows (in g/L): 8 NaCl, 0.05 HNa2PO4, 0.4 KCl, 0.06 KH2PO4, 0.098 MgSO4, 0.14 CaCl2, 1 D-glucose, 0 phenol red. DMEM, FBS, trypsin-EDTA, and antibiotic-antimycotic were purchased from GIBCO (Grand Island, NY, USA). Cell culture 12 well plastic plates, 60-mm plastic dishes, and glass coverslips were purchased from Fisher Scientific Company (Newark, DE, USA). CELLocate marked glass coverslips were purchased from Eppendorf (square size 175 μm; Hamburg, Germany).
Materials
AICA-Riboside (AICAR) and compound C (CC) were from Calbiochem (La Jolla, CA, USA). Xanthine amine congener (XAC) and adenosine monophosphate (AMP) were from Sigma (St Louis, MO, USA).
Transient transfections
Plasmid DNA expressing myc-tagged constitutively active RhoA (RhoV14), a gift from Dr. Alan Hall (Memorial Sloan-Kettering Cancer Center), was created as described previously [40]. The pmaxGFP control plasmid was from Amaxa (Gaithersburg, MD, USA). Plasmid expression was confirmed by immunofluorescence with a mouse monoclonal antibody against c-myc clone 9E10 (1:100, BD Biosciences, NY, USA) or a rabbit polyclonal antibody against GFP (1:2500, Abcam, Cambridge, MA, USA). Astrocytes were transfected, according to manufacturer's instructions, with Transfast reagent (Promega, Madison, WI, USA) at a DNA/liposome ratio of 1:3 and fixed 48 h after transfection.
Adenoviral infections
Control adenovirus expressing EGFP was purchased from Baylor College of Medicine Vector Core Facility. Astrocytes were incubated for 48 h with adenovirus at a concentration of 2.5 × 109 cfu/ml to infect 30–50% of cells.
Immunofluorescence
Cells were rinsed with phosphate-buffered saline (PBS) and fixed for 20 min at room temperature with 4% paraformaldehyde in PBS. Following two wash steps, each for 5 min in PBS, fixed cells were incubated for 1 h in blocking solution containing 0.1% Triton X-100, 2% goat serum, and 0.1% Na azide dissolved in PBS. Subsequently, the cells were incubated with primary antibody diluted in blocking solution for 1 h at room temp. Following three wash steps, Tetramethylrhodamine- or Oregon Green 488-labeled goat anti-rabbit or anti-mouse (1:200, Molecular Probes, Eugene, OR, USA) was applied in blocking solution for 1 h at room temp. DAPI (1μg/ml, Sigma, St. Louis, MO, USA) was included in the secondary antibody solution for nuclear staining. In the case of Oregon Green 514-labeled phalloidin (1:100, Molecular Probes, Eugene, OR, USA), the cells were blocked and then incubated in PBS containing 0.1% Triton X-100 for 30 min at room temperature. Cells stained with fluorescent-conjugated secondary antibody were washed again in PBS and mounted onto slides with GELMOUNT (Biomeda, Foster City, CA, USA). Micrographs were taken with an Olympus BX40 upright microscope equipped with a Scion color Firewire CCD camera (Scion, Frederick, MD). Immunofluorescence was performed using the following primary antibodies: rabbit polyclonal antibodies against GFP (1:2500, Abcam, Cambridge, MA, USA), GFAP (1:5000, Dako, Carpinteria, CA, USA) and mouse monoclonal antibodies against c-myc clone 9E10 (1:100, BD Biosciences, NY, USA), HA (monoclonal 1:200, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), ezrin clone 3C12 (1:2000, a gift from Dr. Kivela, Helsinki University, Finland), and vinculin (1:200, Sigma, St Louis, MO, USA).
Western blots
After rinsing with PBS, cells were scraped in 5μl (12 well plates) or 15μl (60-mm dishes) of 2X Laemmli sample buffer. Prior to polyacrylamide gel electrophoresis, lysates were boiled for 10 min then centrifuged briefly at 200 × g. Lysates were loaded onto a 10% precast gel (Cambrex, Rockland, ME, USA), run at 125 volts for 90 min, and transferred to Hybond ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA) at 50 milliamps per gel. Nitrocellulose membranes were washed three times in Tris-buffered saline with 0.1% Tween-20 (TBST) and then incubated in TBST with 5% nonfat dry milk for 1 h at room temp. After three washes in TBST, membranes were incubated with primary antibody diluted in TBST with 5% BSA and 0.1% sodium azide for 1 h. After three washes in TBST, the membranes were incubated in TBST with 5% nonfat dry milk and goat secondary antibodies coupled to horseradish peroxidase (1:1000, Sigma, St Louis, MO, USA) for 1 h. After three washes in TBST, immunoreactive bands were visualized using enhanced chemiluminescence (West Dura reagent, PIERCE, Rockford, IL, USA). As a loading control, immunoblots were probed with a monoclonal antibody against alpha-tubulin clone DM1A (1:5000, Neomarkers, Fremont, CA, USA). Immunoblots were probed with the following primary antibodies: rabbit polyclonal antibodies against phosphothreonine 172 AMP-activated protein kinase alpha (1:1000, Upstate, Charlottesville, VA, USA) and phosphoserine 79 acetyl-CoA carboxylase (1:1000, Cell Signaling, Danvers, MA, USA).
Morphological analysis
An observer blinded to the treatment conditions analyzed stellation. Stellate cells were classified as those exhibiting at least 3 discrete processes and excluded unbranched short filopodia less than one soma diameter. For each coverslip GFP-positive cells at 10X magnification over five arbitrarily chosen areas were counted and the resulting average taken. Percent stellation was calculated as the number of stellate GFP positive cells divided by the total number of GFP positive cells.
Statistical analysis
Data points from individual assays represent mean values ± SEM. Statistically significant differences among groups were assessed with ANOVA followed by t-test using Bonferroni's correction for multiple comparisons, with values of p < 0.05 sufficient to reject the null hypothesis for all analyses. All experiments were designed with matched control conditions within each experiment to enable statistical comparison.
Abbreviations used in text
- AMPK
AMP-activated protein kinase
- AICAR
AICA-riboside
- ACC
acetyl-CoA carboxylase
- DMEM
Dulbecco's Modified Eagle media
- HBSS
Hank's balanced salt solution
- SF
serum-free
- DMSO
dimethyl sulfoxide
- GFAP
glial fibrillary acidic protein
- GFP
green fluorescent protein
- XAC
xanthine amine congener
- PBS
phosphate buffered saline
- TBST
Tris buffered saline with 0.1% Tween-20
- PKC
protein kinase C
- FGF2
fibroblast growth factor 2
- TNF alpha
tumor necrosis factor alpha
- TSC2
tuberous sclerosis complex 2
- PI3K
phosphatidylinositol 3-kinase
- Akt
cellular homolog of the viral oncogene v-akt
- Rheb
ras homolog enriched in brain
- PMA
phorbol myristate acetate
- CC
compound C
- LKB1
serine/threonine protein kinase 11
- MAPK
mitogen activated protein kinase
- ERK
extracellular signal regulated kinase
- SNARK
sucrose-non-fermenting protein kinase/AMPK-related protein kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Carlita B Favero, Neuroscience Graduate Program, University of Virginia, Charlottesville, VA.
James W Mandell, Department of Pathology (Neuropathology), University of Virginia, Charlottesville, VA.
References
- 1.Abe K, Misawa M. Astrocyte stellation induced by Rho kinase inhibitors in culture. Brain Res Dev Brain Res. 2003;143:99–104. doi: 10.1016/s0165-3806(03)00096-8. [DOI] [PubMed] [Google Scholar]
- 2.Abe K, Saito H. Astrocyte stellation induced by tyrosine kinase inhibitors in culture. Brain Res. 1999;837:306–308. doi: 10.1016/s0006-8993(99)01658-3. [DOI] [PubMed] [Google Scholar]
- 3.Abe K, Saito H. Effect of ATP on astrocyte stellation is switched from suppressive to stimulatory during development. Brain Res. 1999;850:150–157. doi: 10.1016/s0006-8993(99)02121-6. [DOI] [PubMed] [Google Scholar]
- 4.Abe K, Saito H. The p44/42 mitogen-activated protein kinase cascade is involved in the induction and maintenance of astrocyte stellation mediated by protein kinase C. Neurosci Res. 2000;36:251–257. doi: 10.1016/s0168-0102(99)00134-0. [DOI] [PubMed] [Google Scholar]
- 5.Ahlemeyer B, Kolker S, Zhu Y, Hoffmann GF, Krieglstein J. Cytosine arabinofuranoside-induced activation of astrocytes increases the susceptibility of neurons to glutamate due to the release of soluble factors. Neurochem Int. 2003;42:567–581. doi: 10.1016/s0197-0186(02)00164-x. [DOI] [PubMed] [Google Scholar]
- 6.Auer RN, Kalimo H, Olsson Y, Siesjo BK. The temporal evolution of hypoglycemic brain damage. I. Light- and electron-microscopic findings in the rat cerebral cortex. Acta Neuropathol (Berl) 1985;67:13–24. doi: 10.1007/BF00688120. [DOI] [PubMed] [Google Scholar]
- 7.Baas AF, Smit L, Clevers H. LKB1 tumor suppressor protein: PARtaker in cell polarity. Trends Cell Biol. 2004;14:312–319. doi: 10.1016/j.tcb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 8.Banker G. Culturing Nerve Cells. 2. MIT Press; 1998. p. 666. [Google Scholar]
- 9.Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, Ransom BR. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005;79:74–80. doi: 10.1002/jnr.20335. [DOI] [PubMed] [Google Scholar]
- 10.Burridge K, Fath K, Kelly T, Nuckolls G, Turner C. Focal adhesions: transmembrane junctions between the extracellular matrix and the cytoskeleton. Annu Rev Cell Biol. 1988;4:487–525. doi: 10.1146/annurev.cb.04.110188.002415. [DOI] [PubMed] [Google Scholar]
- 11.Cambray-Deakin M, Pearce B, Morrow C, Murphy S. Effects of neurotransmitters on astrocyte glycogen stores in vitro. J Neurochem. 1988;51:1852–1857. doi: 10.1111/j.1471-4159.1988.tb01168.x. [DOI] [PubMed] [Google Scholar]
- 12.Chen CJ, Liao SL, Huang YS, Chiang AN. RhoA inactivation is crucial to manganese-induced astrocyte stellation. Biochem Biophys Res Commun. 2005;326:873–879. doi: 10.1016/j.bbrc.2004.11.121. [DOI] [PubMed] [Google Scholar]
- 13.Chen HC, Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert M, Farese RV, Jr, Farese RV. Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-beta-D-riboside (AICAR)-stimulated glucose transport. J Biol Chem. 2002;277:23554–23562. doi: 10.1074/jbc.M201152200. [DOI] [PubMed] [Google Scholar]
- 14.Derouiche A, Frotscher M. Peripheral astrocyte processes: monitoring by selective immunostaining for the actin-binding ERM proteins. Glia. 2001;36:330–341. doi: 10.1002/glia.1120. [DOI] [PubMed] [Google Scholar]
- 15.Eddleston M, Mucke L. Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 17.Evans ML, McCrimmon RJ, Flanagan DE, Keshavarz T, Fan X, McNay EC, Jacob RJ, Sherwin RS. Hypothalamic ATP-sensitive K + channels play a key role in sensing hypoglycemia and triggering counterregulatory epinephrine and glucagon responses. Diabetes. 2004;53:2542–2551. doi: 10.2337/diabetes.53.10.2542. [DOI] [PubMed] [Google Scholar]
- 18.Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006 doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- 19.Forcet C, Etienne-Manneville S, Gaude H, Fournier L, Debilly S, Salmi M, Baas A, Olschwang S, Clevers H, Billaud M. Functional analysis of Peutz-Jeghers mutations reveals that the LKB1 C-terminal region exerts a crucial role in regulating both the AMPK pathway and the cell polarity. Hum Mol Genet. 2005;14:1283–1292. doi: 10.1093/hmg/ddi139. [DOI] [PubMed] [Google Scholar]
- 20.Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of RAC/protein kinase B and their influence on kinase activity. J Biol Chem. 1997;272:8474–8481. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- 21.Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- 22.Goldman JE, Abramson B. Cyclic AMP-induced shape changes of astrocytes are accompanied by rapid depolymerization of actin. Brain Res. 1990;528:189–196. doi: 10.1016/0006-8993(90)91657-3. [DOI] [PubMed] [Google Scholar]
- 23.Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gottfried C, Cechin SR, Gonzalez MA, Vaccaro TS, Rodnight R. The influence of the extracellular matrix on the morphology and intracellular pH of cultured astrocytes exposed to media lacking bicarbonate. Neuroscience. 2003;121:553–562. doi: 10.1016/s0306-4522(03)00557-8. [DOI] [PubMed] [Google Scholar]
- 25.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 26.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 27.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 30.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 31.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 32.Jalonen TO, Charniga CJ, Wielt DB. beta-Amyloid peptide-induced morphological changes coincide with increased K+ and Cl-channel activity in rat cortical astrocytes. Brain Res. 1997;746:85–97. doi: 10.1016/s0006-8993(96)01189-4. [DOI] [PubMed] [Google Scholar]
- 33.Koyama Y, Fukuda T, Baba A. Inhibition of vanadate-induced astrocytic stress fiber formation by C3 ADP-ribosyltransferase. Biochem Biophys Res Commun. 1996;218:331–336. doi: 10.1006/bbrc.1996.0058. [DOI] [PubMed] [Google Scholar]
- 34.Landis DM. The early reactions of non-neuronal cells to brain injury. Annu Rev Neurosci. 1994;17:133–151. doi: 10.1146/annurev.ne.17.030194.001025. [DOI] [PubMed] [Google Scholar]
- 35.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 36.Merrill JE. Effects of interleukin-1 and tumor necrosis factor-alpha on astrocytes, microglia, oligodendrocytes, and glial precursors in vitro. Dev Neurosci. 1991;13:130–137. doi: 10.1159/000112150. [DOI] [PubMed] [Google Scholar]
- 37.Moonen G, Heinen E, Goessens G. Comparative ultrastructural study of the effects of serum-free medium and dibutyryl-cyclic AMP on newborn rat astroblasts. Cell Tissue Res. 1976;167:221–227. doi: 10.1007/BF00224329. [DOI] [PubMed] [Google Scholar]
- 38.Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- 39.Neary JT, Baker L, Jorgensen SL, Norenberg MD. Extracellular ATP induces stellation and increases glial fibrillary acidic protein content and DNA synthesis in primary astrocyte cultures. Acta Neuropathol (Berl) 1994;87:8–13. doi: 10.1007/BF00386249. [DOI] [PubMed] [Google Scholar]
- 40.Paterson HF, Self AJ, Garrett MD, Just I, Aktories K, Hall A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J Cell Biol. 1990;111:1001–1007. doi: 10.1083/jcb.111.3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez V, Bouschet T, Fernandez C, Bockaert J, Journot L. Dynamic reorganization of the astrocyte actin cytoskeleton elicited by cAMP and PACAP: a role for phosphatidylInositol 3-kinase inhibition. Eur J Neurosci. 2005;21:26–32. doi: 10.1111/j.1460-9568.2004.03845.x. [DOI] [PubMed] [Google Scholar]
- 42.Ramakers GJ, Moolenaar WH. Regulation of astrocyte morphology by RhoA and lysophosphatidic acid. Exp Cell Res. 1998;245:252–262. doi: 10.1006/excr.1998.4224. [DOI] [PubMed] [Google Scholar]
- 43.Ridley AJ, Hall A. Signal transduction pathways regulating Rho-mediated stress fibre formation: requirement for a tyrosine kinase. Embo J. 1994;13:2600–2610. doi: 10.1002/j.1460-2075.1994.tb06550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosso L, Peteri-Brunback B, Vouret-Craviari V, Deroanne C, Troadec JD, Thirion S, Van Obberghen-Schilling E, Mienville JM. RhoA inhibition is a key step in pituicyte stellation induced by A(1)-type adenosine receptor activation. Glia. 2002;38:351–362. doi: 10.1002/glia.10072. [DOI] [PubMed] [Google Scholar]
- 45.Sabina RL, Patterson D, Holmes EW. 5-Amino-4-imidazolecarboxamide riboside (Z-riboside) metabolism in eukaryotic cells. J Biol Chem. 1985;260:6107–6114. [PubMed] [Google Scholar]
- 46.Sasaki T, Endo T. Both cell-surface carbohydrates and protein tyrosine phosphatase are involved in the differentiation of astrocytes in vitro. Glia. 2000;32:60–70. doi: 10.1002/1098-1136(200010)32:1<60::aid-glia60>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Shao Y, Enkvist MO, McCarthy KD. Glutamate blocks astroglial stellation: effect of glutamate uptake and volume changes. Glia. 1994;11:1–10. doi: 10.1002/glia.440110103. [DOI] [PubMed] [Google Scholar]
- 48.Shapiro DL. Morphological and biochemical alterations in foetal rat brain cells cultured in the presence of monobutyryl cyclic AMP. Nature. 1973;241:203–204. doi: 10.1038/241203a0. [DOI] [PubMed] [Google Scholar]
- 49.Suidan HS, Nobes CD, Hall A, Monard D. Astrocyte spreading in response to thrombin and lysophosphatidic acid is dependent on the Rho GTPase. Glia. 1997;21:244–252. doi: 10.1002/(sici)1098-1136(199710)21:2<244::aid-glia7>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki A, Kusakai G, Kishimoto A, Minegichi Y, Ogura T, Esumi H. Induction of cell-cell detachment during glucose starvation through F-actin conversion by SNARK, the fourth member of the AMP-activated protein kinase catalytic subunit family. Biochem Biophys Res Commun. 2003;311:156–161. doi: 10.1016/j.bbrc.2003.09.184. [DOI] [PubMed] [Google Scholar]
- 51.Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 52.Torres J, Di Como CJ, Herrero E, De La Torre-Ruiz MA. Regulation of the cell integrity pathway by rapamycin-sensitive TOR function in budding yeast. J Biol Chem. 2002;277:43495–43504. doi: 10.1074/jbc.M205408200. [DOI] [PubMed] [Google Scholar]
- 53.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 54.Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development. 2000;127:1467–1475. doi: 10.1242/dev.127.7.1467. [DOI] [PubMed] [Google Scholar]
- 55.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]