

Abstract
Deletion of the lissencephaly disease gene LIS-1 in humans causes an extreme disorganization of the brain associated with significant reduction in cortical neurons. Here we show that deletion or RNA interference (RNAi) of Caenorhabditis elegans lis-1 results in a reduction in germline nuclei and causes a variety of cellular, developmental, and neurological defects throughout development. Our analysis of the germline defects suggests that the reduction in nuclei number stems from dysfunctional mitotic spindles resulting in cell cycle arrest and eventually programmed cell death (apoptosis). Deletion of the spindle checkpoint gene mdf-1 blocks lis-1(lf)-induced cell cycle arrest and germline apoptosis, placing the spindle checkpoint pathway upstream of the programmed cell death pathway. These results suggest that apoptosis may contribute to the cell-sparse pathology of lissencephaly.
Keywords: C. elegans, lis-1, mdf-1, programmed cell death, spindle checkpoint
Introduction
Lissencephaly is a pediatric neurological disorder that causes developmental delay, mental retardation, and epilepsy. Lissencephalic brains have a smooth cortical surface, because the cortex is cell-sparse, a defect thought to arise from abnormal neuronal migration (Shu et al., 2004). The most common cause of lissencephaly is haploinsufficiency of the LIS-1 gene; human homozygotes are presumed to miscarry. The same dosage-sensitive phenotypic pattern is seen throughout phylogeny with complete loss-of-function producing lethality in the worm, fly, and mouse, while partial loss-of-function produces neuronal defects in these organisms (Hirotsune et al., 1998; Liu et al., 2000; Cockell et al., 2004). LIS-1 encodes a microtubule-associated protein (MAP) required for intracellular motility events driven by the microtubule motor dynein (Faulkner et al., 2000; Smith et al., 2000). This function of LIS-1 likely explains its pleiotropy, dynein-mediated trafficking being required for diverse cell biological processes in a wide range of tissues.
Invertebrate homologs of LIS-1 produce pleiotropy ranging from early development through the adult nervous system. In the fly nervous system DLis1 has been shown to play a role in centrosome separation, spindle assembly, and transport of checkpoint proteins in proliferating neuroblasts (Liu et al., 2000; Siller et al., 2005) and to play a role in axonal transport and dendritic growth, branching, and maturation (Liu et al., 2000). Similarly, in the C. elegans nervous system lis-1 is required for vesicle trafficking (Williams et al., 2004). In the C. elegans one-cell embryo lis-1 phenocopies the dynein heavy chain gene (dhc-1), producing defects in centrosome separation, pronuclear migration, and spindle assembly (Cockell et al., 2004). Similarly, Drosophila Lis1 (DLis1) is required for germ cell division and nuclear positioning in oocytes (Liu et al., 1999; Swan et al., 1999).
We have isolated deletion alleles of lis-1 and have used RNAi to study the role of C. elegans lis-1 in germline development. Loss of lis-1 function causes a reduction in germ cell number in both the zone of proliferation (mitotic region) and the zone of early differentiation (pachytene region). Knocking out or silencing lis-1 disrupts the bipolar spindle of mitotic nuclei inducing cell cycle arrest. Many cells then undergo ced-3-dependent death producing an increase in germline corpses and delayed corpse engulfment, a more rigorous demonstration of the link between lis-1 and apoptosis than has been reported in other systems. Finally, epistasis studies place the spindle checkpoint pathway upstream of the programmed cell death pathway and indicate that the spindle checkpoint is required for progression from mitotic catastrophe to apoptosis in cells that have suffered damage to the bipolar spindle. Our results also suggest that apoptosis may contribute to the cell-sparse pathology of lissencephaly.
Materials and methods
Nematode strains and culture conditions
We used the C. elegans Bristol strain N2 as the wild-type parent of our mutant strains and grew nematodes under standard culture conditions at 15°C – 25°C. The mutant strains used in this study are listed in Supplementary Information, Table S1.
Knockouts and RNAi
E.B. recovered n3334 and n3346 using a PCR-based deletion screening library in the laboratory of H. R. Horvitz; primers used for deletion screening are listed in Supplementary Information, Table S2. n3334 contains a deletion encompassing bases 4325–6342 of cosmid T03F6, and n3346 contains a deletion encompassing bases 4411–5874 of T03F6. For RNAi injection experiments EST yk438f10 (Y. Kohara) was used to amplify ~1 kb of lis-1 cDNA with flanking T3 and T7 promoters. The PCR mixture was used to generate sense and antisense RNA with T3 and T7 polymerase (Stratagene). Equal amounts of sense and antisense RNA were mixed, annealed, and injected into the distal gonad arm. Injected worms were transferred to new plates every 12 hours for the first 60 hours. We performed our RNAi soaking experiments as described (Maeda et al., 2001). We amplified DNA fragments from embryonic cDNA by targeting regions where exact nucleotide matches were no longer than 20 bp to reduce cross-interference; primers used for soaking RNAi are listed in Supplementary Information, Table S2. PCR products were transcribed to produce single-stranded RNAs that were then annealed to form dsRNAs for soaking.
Strain constructions and genetics
We used MT10195 to verify autosomal recessive inheritance; 16 of 60 progeny displayed the Mel and Egl phenotypes. We backcrossed n3334 to wild type 10 times to produce MT10422 and backcrossed n3346 to wild type 5 times to produce MT10281. We found that juIs73 suppresses recombination across from lis-1 on the right arm of LG III. Therefore, after backcrossing we created balanced strains using either eT1 or juIs73. Crossing N2 males with MT10422 hermaphrodites failed to rescue embryonic lethality, indicating that lis-1(n3334) produces a strict Mel phenotype. The brood size of homozygous lis-1(t1550) and (t1698) animals, as reported by Cockell et al. (2004) is ~50 (wild-type ~200). We performed a complementation test by crossing unc-69 lis-1(n3334) III/eT1 III; +/eT1 V males into unc-69 lis-1(n3346) III/eT1 III; +/eT1 V hermaphrodites and found that unc-69 n3334/unc-69 n3346 progeny were Mel, demonstrating that the two deletion alleles fail to complement. We constructed HC356 by first crossing cep-1(gk138) I males into juIs73 hermaphrodites and later crossing lis-1(n3334)III/eT1 III; +/eT1 V males into cep-1(gk138) I; juIs73 III hermaphrodites. We verified genotypes using PCR for gk138, fluorescence for juIs73, and the Mel phenotype for n3334.
Staining protocols and microscopy
We used Hoechst 33258 or DAPI to visualize DNA. For nuclear counting experiments we scored nuclei above and below the rachus in the gonad arm closest to the observer. For SYTO 12 experiments we used the staining protocol of Gumienny et al. and scored the gonad closest to the observer 24 hours after the L4 stage (Gumienny et al., 1999). We used an Axiophot microscope (Carl Zeiss) with OpenLab 3.0 software (Improvision) for both the nuclear counting and the SYTO 12 experiments. For triple-stained nuclei in the mitotic region we tested two fixation protocols that yielded similar results: 1) Worms were cut in M9 buffer to extrude the gonads; gonads were fixed on poly-L-lysine-coated slides for one hour at 4°C in 3% formaldehyde (0.1 M K2HPO4 pH 7.2) with 1 nM Taxol, fixed for 10 minutes (min) in methanol at −20°C, then washed in PBST. Incubations were then performed overnight with α-tubulin antibody (T9026; Sigma-Aldrich) diluted 1:100, for 4 hours with rhodamine-conjugated goat α-mouse secondary antibody diluted 1:100, for 2 hours with α-phospho-H3 antibody (07-145; Upstate) diluted 1:100, for one hour with FITC-conjugated donkey α-rabbit secondary antibody diluted 1:100, and for 5 min in 1x Hoechst, followed by washing in PBST. Two 5 min washes in PBST were performed between every antibody incubation. 8–10 μl of 2% N-propyl-gallate was added to each slide prior to placing and sealing the coverslip. 2) Antibody staining was also performed using a freeze-crack permeabilization/methanol fixation protocol adapted from Huang et al. (Huang et al., 2002). Triple-stained gonads were visualized using an Olympus IX70 microscope equipped with Nomarski and fluorescence optics and Deltavision Spectris deconvolution software.
Engulfment kinetics
We picked wild-type and lis-1 worms at the L4 stage and scored them 24 hours later at 20°C. dhc-1ts worms were picked at the L4 stage, placed at 25ºC for 24 hrs, and then scored in a temperature-controlled room at 25°C. In some cases engulfment took longer than a day; we reversibly arrested engulfment by shifting worms to 8°C overnight and completing the time course the following morning (Sulston and Horvitz, 1977). We used 0.2 μM levamisole to immobilize worms and used the SYTO 12 protocol of Gumienny et al. to stain corpses (Gumienny et al., 1999). We placed worms in a 2 μl drop of 10% polyvinylpyrrolidone in M9 on an agar pad, added a small amount of OP50 bacteria on the underside of the coverslip, sealed the coverslip with Vaseline, and performed time-lapse analysis using the Zeiss Axiophot microscope described above. Because the early stages of apoptosis may reflect both killing and engulfment (Reddien et al., 2001) and because very early corpses are difficult to distinguish from very late corpses (Gartner et al., 2004), we took t0 to be the peak of refractility of the corpse under Nomarski optics. We followed corpses from the refractile peak and checked them every 5 minutes or less throughout the time course of engulfment. We found no significant differences between the engulfment time course of immobilized unstained N2 worms, immobilized SYTO 12-stained N2 worms, and mobile SYTO 12-stained N2 worms (see supplemental data). These results indicate that the vital dye SYTO 12, itself, does not change engulfment kinetics and that engulfment occurs fast enough in the wild-type strain to obviate the deleterious effects of the paralytic agent levamisole. We therefore pooled our N2 results. Because lis-1 and dhc-1 mutations increase the time course of engulfment, these strains, when immobilized with levamisole, grow sick before engulfment is completed. Therefore, we only included mobile SYTO 12-stained worms in our final analysis for lis-1 and dhc-1 mutants. Furthermore, because scoring germ cell deaths may be observer-dependent (Gartner et al., 2004), lis-1 mutant worms were scored by independent observers in separate experiments. The results were indistinguishable and therefore pooled.
Statistical analysis
Data are expressed as medians or as means ± s.e.m. Statistical comparisons were performed using the unpaired, two-tailed Student’s t-test and Mann-Whitney test where appropriate. Kaplan-Meier survival curves were generated to analyze lifespans of wild-type, dpy-18 and dpy-18 lis-1 mutants and were compared using the log-rank test.
Results
Production of lis-1(lf) reagents
Analyses of the lis-1 transcript by Northern blot analysis, sequencing of lis-1 ESTs, characterization of a cDNA isolated by PCR, and 5’ RACE are consistent with a single splice form that encodes a 1309 bp ORF (data not shown). The predicted LIS-1 protein is 58% identical to the human LIS-1 protein and, like the human LIS-1 protein, contains an amino-terminal Lissencephaly type-1-like homology motif (LISH) domain (Li and Roberts, 2001) followed by seven WD40 domains (Fig. 1A).
Fig. 1.
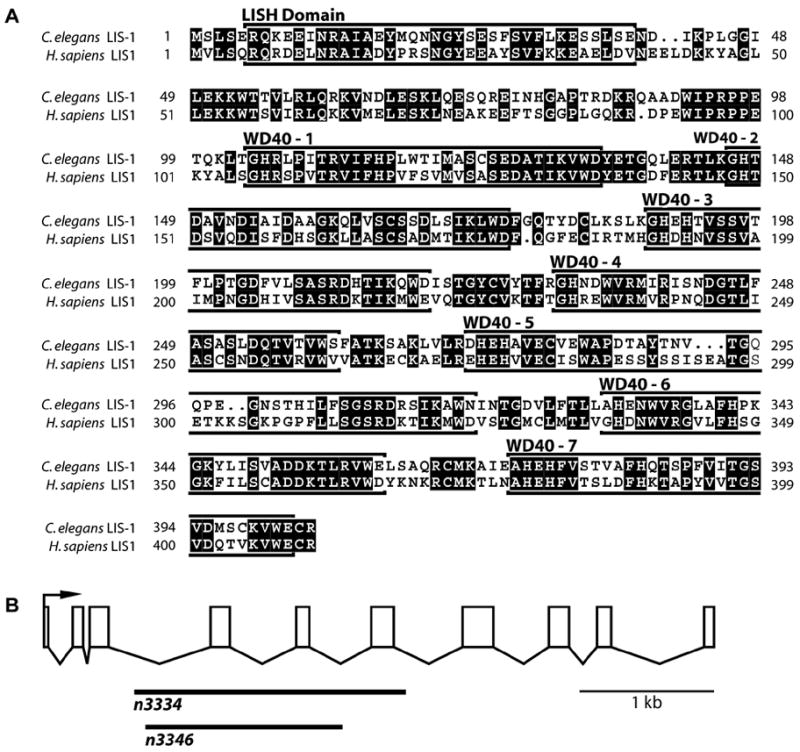
LIS-1 protein alignments and lis-1 gene structure. (A) The C. elegans LIS-1 protein is 58% identical to the human Lis1 protein. Both proteins contain an amino-terminal LISH domain followed by seven WD40 domains. The WD40 repeat domains were located using the WD40 repeat portion of Li and Roberts’ regular expression for the structure of a typical WD-repeat protein propeller blade (Li and Roberts, 2001). (B) Two deletions were obtained, both of which are completely encompassed within the lis-1 genomic sequence. n3334 is an ~2 kb out-of-frame deletion removing exons 4–6, and n3346 is an ~1.5 kb in-frame deletion removing exons 4–5. Homozygous progeny of heterozygous parents are Egl and mildly Unc. Homozygous progeny of homozygous parents display a Mel phenotype.
To determine the lis-1 phenotype we initiated RNAi by injection of lis-1 double-stranded RNA (dsRNA) into adult hermaphrodites. Progeny laid within 24 hours of injection survived and developed as mildly egg-laying defective (Egl) adults, while progeny laid more than 24 hours after injection exhibited an early embryonic arrest at the 50–100 cell stage. Two lis-1 deletion alleles, n3334 and n3346, with phenotypes similar to those observed by RNAi were isolated using a PCR-based screen (Fig. 1B). For both alleles, homozygous progeny of heterozygous parents are Egl and exhibit a subtle backing defect, while homozygous or heterozygous progeny of homozygous mothers display fully penetrant maternal-effect lethality (Mel) and arrest at the 50–100 cell stage. Injection of cosmid T03F6, which encompasses the lis-1 genomic sequence, rescues all the n3334 phenotypes.
LIS-1 is expressed throughout the germline
To study the localization and expression pattern of LIS-1, we generated a polyclonal α-LIS-1 antisera and a LIS-1::GFP (green fluorescent protein) fusion protein driven by ~3 kb of the lis-1 promoter. The GFP construct was broadly expressed throughout the animal with fluorescence strongest in the seam cells, body wall and pharyngeal muscle, vulval cells, spermatheca, the tail, and many neurons. Transgenes are rarely expressed in the germline, so we used the pie-1 promoter to drive germline expression of a full-length LIS-1::GFP fusion protein. The LIS-1::GFP was diffuse throughout the cytoplasm and strongly localized to the nuclear envelope in oocytes.
To confirm the LIS-1::GFP localization, we used whole-mount immunofluorescence staining of germlines. Our antisera, as well as α-LIS-1 antisera provided by Pierre Gonczy’s laboratory and commercially available α-LIS-1 antisera all showed diffuse cytoplasmic staining, but did not show the enrichment at the nuclear envelope of ooctyes seen in LIS-1::GFP. This difference may reflect instability of LIS-1 or may be a consequence of overexpression of LIS-1::GFP.
lis-1(lf) causes pleiotropic phenotypes in the germline
Our studies of the lis-1 one-cell embryo using Nomarski optics for time-lapse experiments and whole-mount immunofluorescence with 4, 6-diaminido-2-phenylindole (DAPI) and α-tubulin antibody revealed defects similar to those previously reported (Cockell et al., 2004), including defects in centrosomal migration, pronuclear migration, and bipolar spindle formation. We also observed multiple maternal pronuclei in one-cell embryos, indicating that defects in germline development may precede the one-cell stage abnormalities. This observation raised the possibility that portions of the one-cell stage phenotypes may be secondary to preexisting germline developmental defects.
To identify the earliest developmental abnormality in lis-1 mutants, we examined the germlines of both deletion alleles (n3334 and n3346) by Hoechst and found similar defects. lis-1 gonads contain fewer germline nuclei than wild-type gonads (Fig. 2A-F). Abnormalities are present in the mitotic region (polyploid nuclei), the pachytene region (large gaps devoid of nuclei), and the most proximal oocytes (extra Hoechst-positive bodies) (Figs. 2, 3). Careful examination revealed that some of the Hoechst-positive bodies in most proximal oocytes appeared to be bivalents while others appeared to be univalents, indicating that lis-1 is required for chiasmata formation or stability (Fig. S1). Similar germline phenotypes were observed in the dynein heavy chain mutant dhc-1(or195) (data not shown). Loss of dynein function phenocopies lis-1 in the C. elegans one-cell stage embryo and in other organisms (Cockell et al., 2004; Gonczy et al., 1999; Xiang et al., 1995).
Fig. 2.
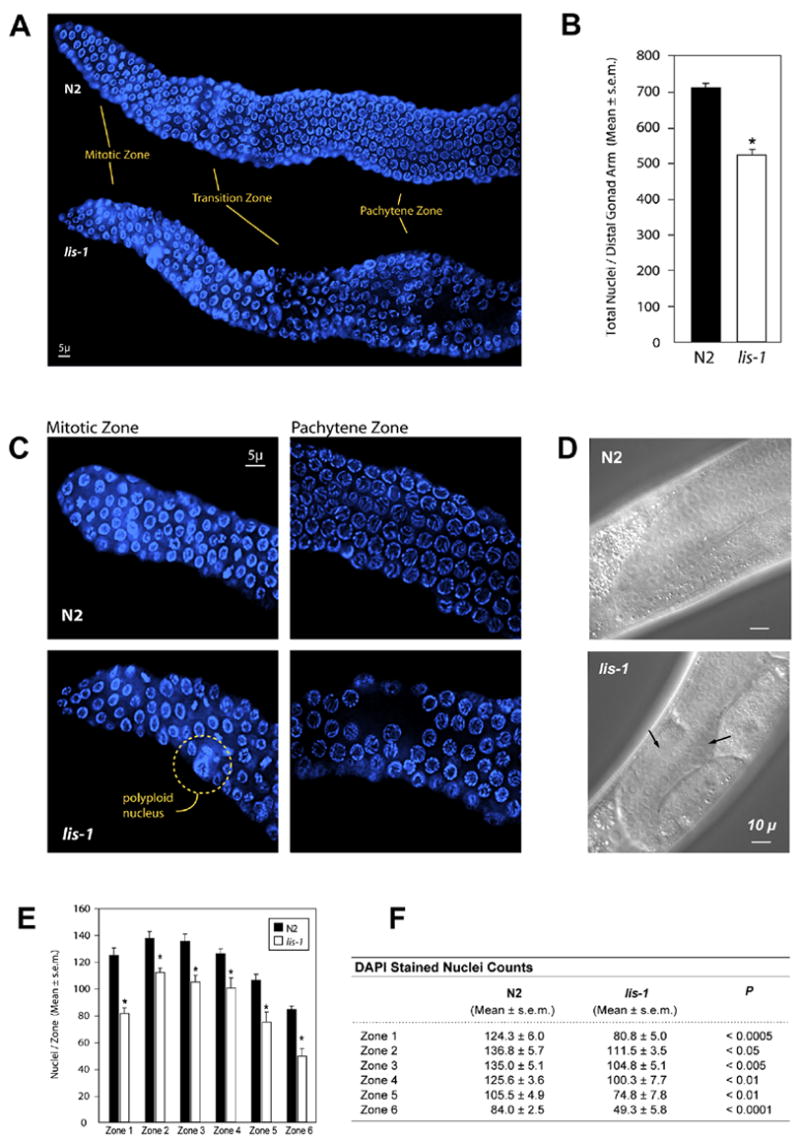
lis-1(lf) disrupts development throughout the germline. (A) Hoechst staining reveals defects in the mitotic, transition, and pachytene zones of lis-1(lf) distal gonads. (B) lis-1(n3334) mutants contain fewer germ cells per distal gonad arm than the N2 strain. lis-1 homozygotes were identified by selecting GFP-negative progeny of strain MT12272. The data are averages ± s.e.m. for N2 (n=8) and lis-1 (n=4) animals. N2 distal gonad arms contain an average of 711.1 ± 12.4 nuclei, whereas lis-1 distal gonad arms contain an average of 521.3 ± 18.1 nuclei (P<0.0001, unpaired t-test). (C) The mitotic zone of lis-1(n3334) mutants contains polyploid nuclei. The pachytene zone of lis-1(n3334) mutants contains large cell-sparse gaps. (D) Arrows point to cell-sparse gaps in the lis-1 germline visible by Nomarski DIC. (E, F) We counted germ cells in 40 μm zones from the distal tip (Zone 1) to the bend of the gonad (Zone 6). Cell counts were significantly lower in each zone of the lis-1(n3334) distal gonad than in the corresponding N2 zones.
Fig. 3.
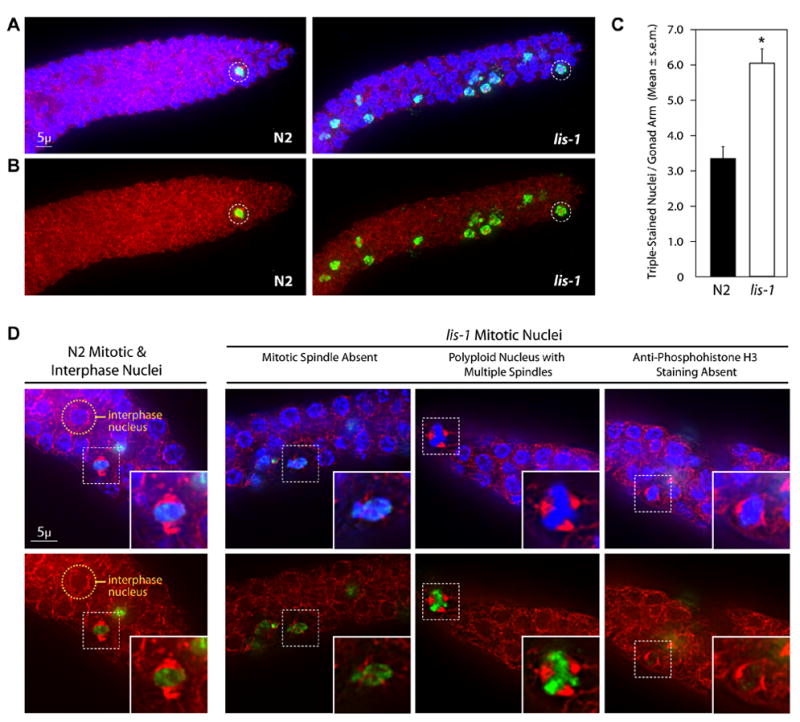
lis-1(lf) disrupts the mitotic spindle and produces cell cycle arrest. (A) lis-1(n3334) and N2 gonads stained with Hoechst, α-tubulin antibody, and α-phosphohistone H3 antibody (α-PH3). lis-1 gonads contain an increased number of α-PH3-positive nuclei compared to N2 gonads. Representative mitotic nuclei are circled for both strains. Photographs represent a z-stack through the width of the gonad, flattened using deconvolution microscopy. (B) Photographs of the same animals presented in (a), showing only α-tubulin antibody and α-PH3 staining. (C) The mean number of triple-stained nuclei per distal gonad arm is significantly greater in lis-1(n3334) than in the N2 control (P<0.0001, unpaired t-test). The data are averages ± s.e.m. of N2 (n=47) and lis-1 (n=61). (D) N2 mitotic nuclei were easily distinguished by their symmetrical bipolar spindles and distinct centralized Hoechst and α-PH3 staining. An N2 interphase nuclei is circled above for comparison. lis-1 nuclei, on the other hand, display a range of defects including spindle abnormalities, polyploidy, and lack of α-PH3 staining. Representative nuclei are indicated by dashed-squares and are magnified in the inset images.
To quantify the cell-sparse germline phenotype, we divided the gonad into six 40 μ zones and counted nuclei from the distal tip to the bend of the gonad in wild type and lis-1 (n3334). A statistically significant reduction in nuclei was seen in all six zones from the mitotic region through the pachytene region of the lis-1 mutant (Fig. 2E,F), suggesting that lis-1(lf) produces a proliferation defect.
lis-1(lf) causes mitotic arrest
Germline proliferation occurs exclusively in the mitotically active distal tip region of the gonad, thus to characterize the lis-1 proliferation defect we counted mitotic nuclei in this region. To unambiguously and quantitatively count mitotic nuclei, we triple-stained gonads with Hoechst to identify DNA, with α-tubulin antibody to identify spindles, and with α-phosphohistone H3 antibody to identify mitotic cells. lis-1 gonads exhibited a significant increase in triple-stained nuclei in the mitotic region (Fig. 3A–C). On average the mitotic region of a wild-type gonad contained 3.3 ± 0.4 triple-stained nuclei, whereas the mitotic region of a lis-1 gonad contained 6.1 ± 0.4 triple-stained nuclei (P<0.0001, unpaired t-test). In addition to the increased number of triple-stained mitotic nuclei, many nuclei with abnormal spindles, DNA or α-phosphohistone H3 staining were apparent when cells were inspected at higher resolution (see below). These observations suggest that depletion of lis-1 causes delayed exit from mitosis or mitotic arrest among a portion of the germline nuclei, a mechanism that could account for the observed depletion of nuclei throughout the germline (Fig. 2). Defects in germline proliferation appear to be less penetrant than defects seen in the one-cell embryo. Microtubule defects in the embryo are likely more severe than those in the germline, because the cell cycle is faster in the embryo and because the one-cell embryo is a much larger cell. These differences likely make the embryonic microtubules more sensitive to depletion of the LIS-1 microtubule-associated protein.
To investigate the requirement for lis-1 in mitosis we used deconvolution microscopy to characterize the mitotic defects. First, lis-1(lf) disrupts the mitotic spindle (Fig. 3D). To quantify this defect, we took nuclei to be in mitosis if they displayed at least two of the following: a condensed metaphase plate by Hoechst staining, strong nuclear staining by α-phosphohistone H3 antibody, or a bipolar spindle by α-tubulin antibody staining. Many mitotic cells in lis-1(n3334) lack a spindle altogether or have abnormal spindle architecture, abnormal spindle position, or multipolar spindles (Fig. 3D). We also found nuclei in lis-1(n3334) that display absent or very weak α-phosphohistone H3 antibody staining (Fig. 3D) despite the presence of both a bipolar spindle and a condensed metaphase plate. Since phosphorylation of histone H3 by the AIR-2 kinase is required for mitosis (Hsu et al., 2000), this result suggests that the dynein motor complex may be required for proper trafficking and/or localization of AIR-2 kinase. Alternatively, nuclei may arrest at two different stages, one that is early and α-phosphohistone H3 antibody-negative and one that is late and α-phosphohistone H3 antibody-positive. Finally, we also noted an increased number of nuclei with abnormal staining of condensed DNA. In many cases, nuclei with abnormal spindles displayed nuclear morphology defects suggestive of altered DNA content (ploidy) (Fig. 3D). We were unable to reliably distinguish whether some nuclei also had a defect in chromosome condensation independent of abnormalities in α-tubulin antibody and α-phosphohistone H3 antibody staining.
Since lis-1(lf) disrupts the mitotic spindle, we reasoned that lis-1(lf)-induced cell cycle arrest might depend on mdf-1, the C. elegans homolog of the spindle checkpoint gene MAD1 (Encalada et al., 2005; Kitagawa and Rose, 1999). To determine whether mdf-1 function is required for the lis-1(lf)-induced mitotic arrest we soaked wild-type and mdf-1 mutant worms in lis-1 dsRNA and then counted α-phosphohistone H3-positive nuclei in the mitotic region. lis-1 (RNAi) causes a significant increase in α-phosphohistone H3-positive nuclei in the mitotic region compared to the soaking buffer control (P<0.005, Tukey test), whereas mdf-1; lis-1 (RNAi) is not significantly different than mdf-1 in soaking buffer alone (P>0.24). As a control we asked whether mutation of the p53 DNA-damage checkpoint homolog, cep-1, also blocked lis-1(lf) induced mitotic arrest. As expected, an increase in α-phosphohistone H3-positive nuclei in the mitotic region of lis-1(n3334) was not blocked in cep-1(gk138); lis-1(n3334) double mutants (data not shown). Because lis-1(lf)-induced cell cycle arrest is reduced in the absence of mdf-1 (Fig. 4), we conclude that lis-1(lf) leads to cell cycle arrest by disrupting the mitotic spindle and activating the spindle checkpoint.
Fig. 4.
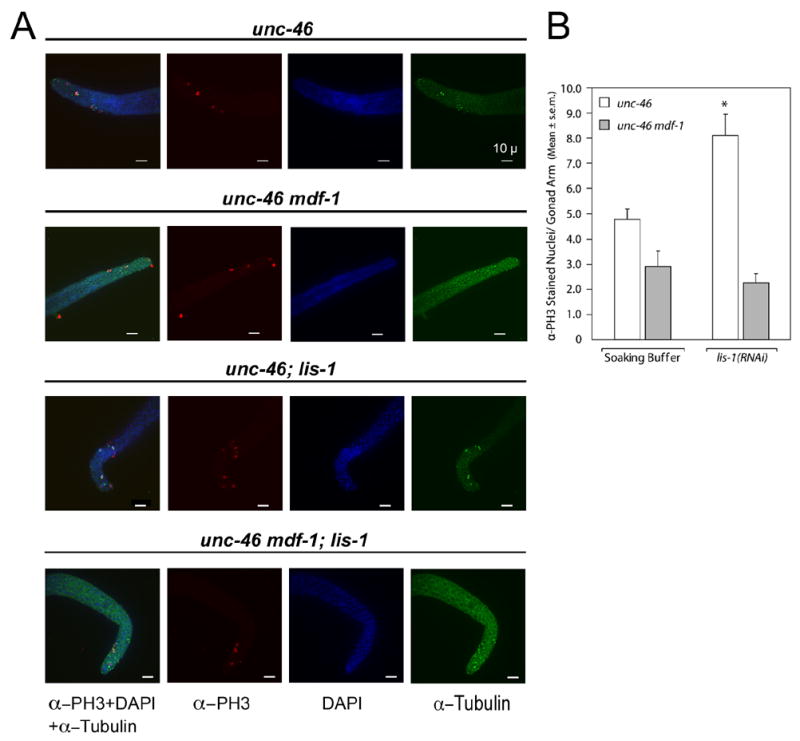
mdf-1(gk2) blocks lis-1(RNAi)-induced cell cycle arrest. (A) Triple-staining of dissected gonads with α-tubulin antibody, α-PH3 antibody, and DAPI. Gonads are from four groups: unc-46 in soaking buffer alone, unc-46 mdf-1 in soaking buffer alone, unc-46; lis-1(RNAi), unc-46 mdf-1;lis-1(RNAi). unc-46; lis-1(RNAi) gonads display an increased number of mitotic nuclei compared to unc-46 gonads in soaking buffer. Staining of unc-46 mdf-1;lis-1(RNAi) gonads displays no such increase in the number of mitotic nuclei when compared to unc-46 mdf-1 gonads in soaking buffer. Photographs represent z-stacks through the width of the gonad, flattened using deconvolution microscopy. (B) The average number of α-PH3-stained nuclei per gonad arm in unc-46 gonads in soaking buffer (4.8 ± 0.4, n=21) is significantly less than in unc-46;lis-1(RNAi) gonads (8.2. ±0.9, n=25) (P<0.005, Tukey test). There is no difference between the average number of α-PH3 stained nuclei per gonad arm in unc-46 mdf-1 gonads in soaking buffer (2.9 ± 0.6, n=17) and unc-46 mdf-1;lis-1(RNAi) gonads (2.3 ± 0.4, n=30).
lis-1(lf) causes an increase in germline apoptotic cells
Mitotic arrested nuclei are only observed in the proliferative zone, yet lis-1(lf) gonads are nuclei sparse throughout (Fig. 2). Nuclei that exit the mitotic zone enter meiosis and traverse to the pachytene zone, which is where germline apoptosis occurs. If mitotic arrested abnormal nuclei are cleared by apoptosis, as occurs in other systems (Bhalla 2003), then to account for the nuclei sparse lis-1(lf) phenotype we may see an increase in apoptotic cells. To quantify apoptotic nuclei we used the vital dye SYTO 12 (Gumienny et al., 1999). We compared dpy-18(e364) lis-1(n3334) to dpy-18(e364) worms and found an increased number of SYTO 12-positive germline corpses in the lis-1 mutants (P<0.001, Mann-Whitney test). A similar increase is seen in dhc-1ts mutants at the restrictive temperature of 25°C compared to wild-type animals at 25°C (P<0.05, Mann-Whitney test). These SYTO 12-positive corpses display the anatomic and histologic characteristics of apoptotic corpses: they cluster in the distal arm near the bend of the gonad; they contain condensed DNA by Hoechst staining; they appear refractile by Nomarski optics; and they stain with SYTO 12 dye. To confirm that these are normal ced-3-dependent cell deaths we introduced a ced-3 mutation into the lis-1(lf) background and found that this significantly reduced the number of SYTO 12-positive corpses (Fig. 5A,B; Table 1). We also visualized dying cells using a CED-1::GFP reporter under the control of the lim-7 promoter (Plim-7ced-1::gfp). CED-1 is a cell surface phagocytic receptor that recognizes cell corpses (Zhou et al., 2001). CED-1::GFP marks dying cells earlier in the dying process than SYTO 12 (Zhou et al., 2001) and consequently marked significantly more germ cell corpses in the pachytene region than did SYTO 12, dramatically increasing the difference between wild type and lis-1(n3334) (Fig. 5C).
Fig. 5.
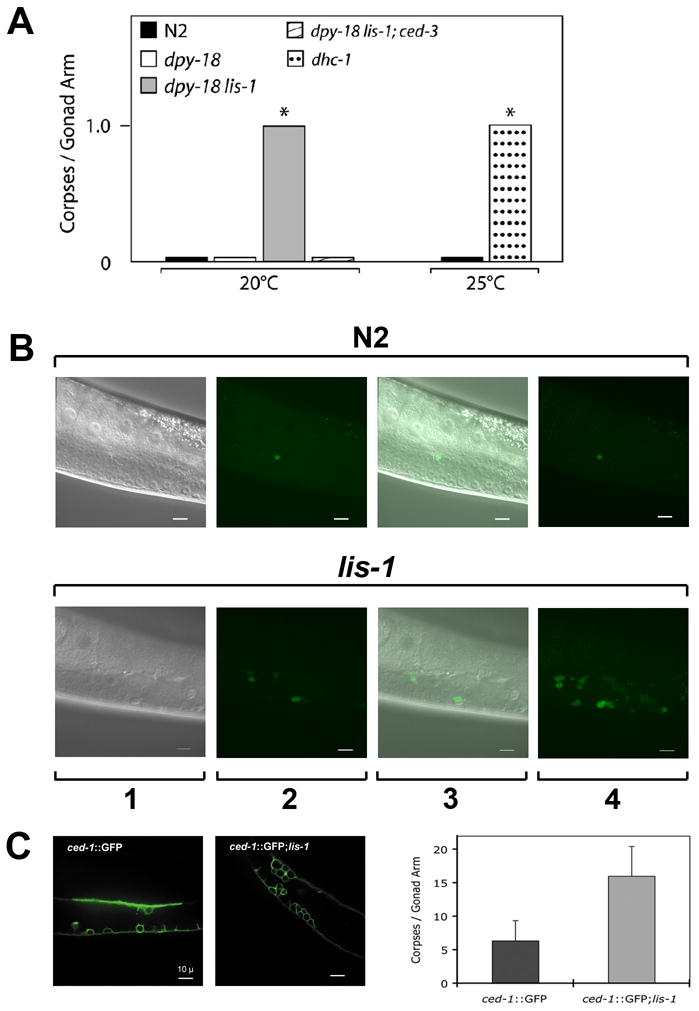
lis-1(lf) triggers apoptosis in the C. elegans germline. (A) dpy-18 lis-1 animals contain significantly more corpses per gonad arm than dpy-18 control animals (P<0.001, Mann-Whitney test). ced-3 blocks the lis-1(lf)-induced increase in apoptosis, as evidenced by the reduced number of corpses per gonad arm in dpy-18 lis-1; ced-3 animals. At the restrictive temperature dhc-1ts mutants exhibit a similar increase compared to N2 (P<0.05, Mann-Whitney test). dpy-18 data and dpy-18 lis-1 data were pooled from 2 datasets, one with and one without a lin-11::GFP marker used for strain construction. The data presented are medians. (B) Corpse number is increased in the lis-1 germline compared to N2. 1) Nomarski DIC alone. 2) SYTO-12-positive corpses in a single focal plane. 3) Nomarski DIC and SYTO-12 images together in a single focal plane. 4) SYTO-12 positive corpses visible in a flattened z-stack. (C) A CED-1::GFP reporter reveals a marked increase in corpse number in lis-1(n3334) gonads.
Table 1.
SYTO 12 Experimental Data
| lis-1(lf) Increases the Average Number of Corpses per Gonad Arm | |||
|---|---|---|---|
| Strain | Treatment | Mean ± s.e.m. | N |
| N2 | 0.5 ± 0.1 | 154 | |
| dpy-18 | 0.2 ± 0.1 | 107 | |
| cep-1; dpy-18 | 0.2 ± 0.1 | 35 | |
| dpy-18 lis-1 | 1.6 ± 0.2 | 81 | |
| cep-1; dpy-18 lis-1 | 1.3 ± 0.1 | 26 | |
| dpy-18 lis-1; ced-3 | 0.5 ± 0.2 | 24 | |
|
| |||
| dhc-1(lf) Increases the Average Number of Corpses per Gonad Arm | |||
|
| |||
| Strain | Treatment | Mean ± s.e.m. | N |
|
| |||
| N2 | 25°C | 0.6 ± 0.2 | 39 |
| dhc-1 | 25°C | 2.0 ± 0.5 | 28 |
|
| |||
| mdf-1 Blocks the lis-1(lf)-induced Increase in Average Number of Corpses per Gonad Arm | |||
|
| |||
| Strain | Treatment | Mean ± s.e.m. | N |
|
| |||
| N2 | Soaking Buffer | 1.2 ± 0.2 | 31 |
| unc-46 | Soaking Buffer | 1.3 ± 0.3 | 37 |
| unc-46 mdf-1 | Soaking Buffer | 1.3 ± 0.2 | 33 |
| N2 | lis-1(RNAi) | 2.9 ± 0.4 | 26 |
| unc-46 | lis-1(RNAi) | 2.8 ± 0.3 | 48 |
| unc-46 mdf-1 | lis-1(RNAi) | 1.3 ± 0.2 | 27 |
Germline corpse number increases with animal age, and single-gene mutants, including dhc-1 and dli-1, can accelerate aging (Gumienny et al., 1999; Hsu et al., 2003; (Cockell et al., 2004). Therefore we asked whether lis-1 mutants also accelerate aging. Our initial results were confounded by the observation that dpy-18, the genetically-linked marker used to identify lis-1 homozygotes, extended lifespan (Fig. 6B,C). dpy-18 lis-1 worms were not as long-lived as dpy-18 worms but lived as long as the wild-type strain (Fig. 6B,C). Since dpy-18 lis-1(lf) mutants contain more germline corpses than the wild-type strain but age at the same rate as wild type, lis-1-induced cell death is not a consequence of rapid aging. In fact, lis-1 probably shortens lifespan non-specifically by disrupting essential cellular functions rather than by accelerating aging, since we noted changes in lis-1(n3334) characteristic of sickness and not characteristic of rapid aging (Garigan et al., 2002).
Fig. 6.
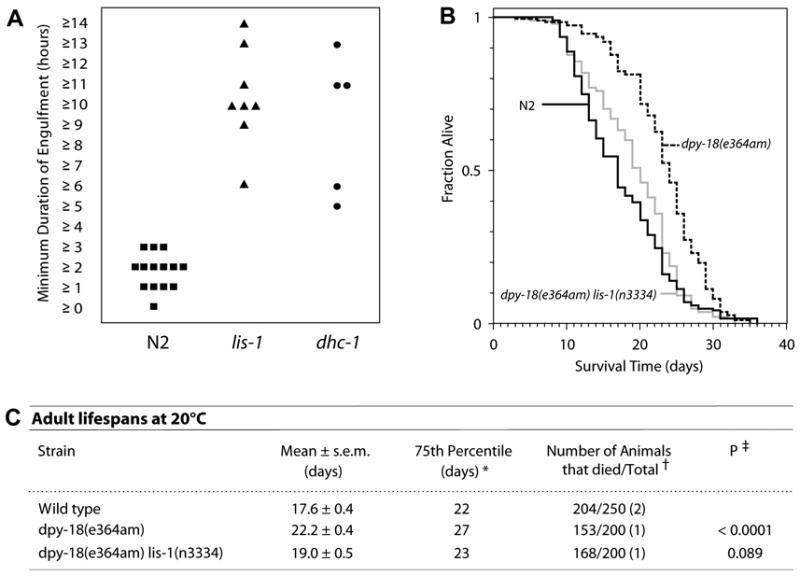
lis-1 and dhc-1 mutants are defective in engulfment but not in aging. (A) lis-1(lf) and dhc-1 exhibit significantly increased engulfment kinetics compared to N2 (P<0.0005 and P<0.005 respectively, Mann-Whitney test). The average minimum engulfment times for lis-1(n3334) (10.9 hours ± 0.9, n=8) and dhc-1 (9.4 hours ± 1.7, n=5) were greater than the N2 average (2.3 hr ± 0.2, n=14). Statistical comparisons using the Mann-Whitney test were conducted between pooled N2 data (unstained mobile, stained mobile and stained immobile worms), and stained mobile lis-1(lf) and dhc-1 worms (see Supplementary data, Fig. S1). (B) Kaplan-Meier lifespan curve analysis confirms that the increase in apoptosis seen in lis-1(n3334) is not a result of accelerated aging. (c) There was no significant difference between the lifespans of wild-type and dpy-18 lis-1 animals; they exhibited similar average lifespans, 75th percentiles, and number of deaths relative to the number of animals observed. For unknown reasons, dpy-18 animals exhibited a longer than wild-type average lifespan (P<0.0001, log-rank test). * The 75th percentile is the age when the fraction of animals alive reaches 0.25. † The total number of observations equals the number of animals that died plus the number censored. The number of independent times the lifespan was determined is in parentheses. Animals that crawled off the plate, exploded or bagged were censored at the time of the event.‡ Compared with wild-type control in all experiments.
lis-1 and dhc-1 mutants are defective in engulfment
The increase in corpse number observed in lis-1 and dhc-1 mutants may be caused by either an increased killing rate, perhaps associated with mitotic arrest, or by slowed engulfment of dead cells. Therefore, we measured the time course of germline corpse engulfment. We found that lis-1 and dhc-1 mutations slow engulfment kinetics dramatically, such that corpses require as long as ~14 hours to vanish, whereas engulfment is completed within ~1–3 hours in the wild-type strain (Fig. 6A). Because paralyzed worms grow sick after ~3 hours, we performed our experiments on freely moving animals (Kimble and Hirsh, 1979; Sulston and Horvitz, 1977), a requirement that presented two technical obstacles. First, germline deaths occur within a syncytium and often cluster tightly along the lateral margin of the pachytene region (Gartner et al., 2004), such that we often observed deaths immediately overlying one another along the edge of the gonad. Second, slowed engulfment requires constant monitoring for many hours to days. To ensure that each time course represented the engulfment of a single cell, we developed a new protocol in which we followed engulfment in unrestrained worms both by SYTO 12 fluorescence and by refractility under Nomarski optics (see Materials andMethods, Fig. S2). This protocol offered the added benefit of ensuring that we scored the same population of cells when counting corpses and scoring engulfment. Quantitative temporal analysis of the corpse engulfment process has not been previously reported. We found that in the N2 strain, the time for corpse engulfment ranged between 1.0 – 3.7 hours. The mean duration of engulfment was 2.3 ± 0.2 hours. In contrast, the range for lis-1 was 6.2 – 14.2 hours with a mean of 10.9 ± 0.9 hours (P<0.0005, Mann-Whitney test). The range for dhc-1 was similar, 5.0 – 13.8 hours with a mean of 9.4 ± 1.7 hours (P<0.005, Mann-Whitney test). These results suggest that engulfment is a dynein motor-driven cellular motility-dependent event that requires lis-1 and dhc-1 for changes in cytoskeletal dynamics and/or cargo trafficking.
These results indicate that the observed increase in corpse number in lis-1(lf) animals may be due entirely to slowed kinetics and not to an increase in mitotically arrested nuclei entering the cell death pathway. If so, then removing mdf-1 should not affect either the engulfment rate or the observed number of corpses. Therefore, we compared lis-1(RNAi)-induced cell deaths between mdf-1(+) and mdf-1(−) strains. We found that mdf-1 function is required for the lis-1(RNAi)-induced increase in corpse number per gonad arm (Fig. 7). However engulfment of corpses is still slowed (n=7). Furthermore, an mdf-1 mutation eliminated the lis-1(RNAi)-induced nuclei sparse appearance throughout the gonad. These results demonstrate that activation of the spindle checkpoint is necessary to trigger apoptosis in lis-1(lf) mutants. As a control, we again confirmed that the effect was specific to the mdf-1-mediated spindle checkpoint by asking whether the p53 homolog, cep-1, was also required for lis-1(lf)-induced cell death. We found no significant difference in the number of corpses per gonad arm between dpy-18 lis-1 and cep-1; dpy-18 lis-1 mutants (Fig. 7A; Table 1). This result suggests that the mdf-1-mediated spindle checkpoint pathway links cell cycle arrest to apoptosis.
Fig. 7.
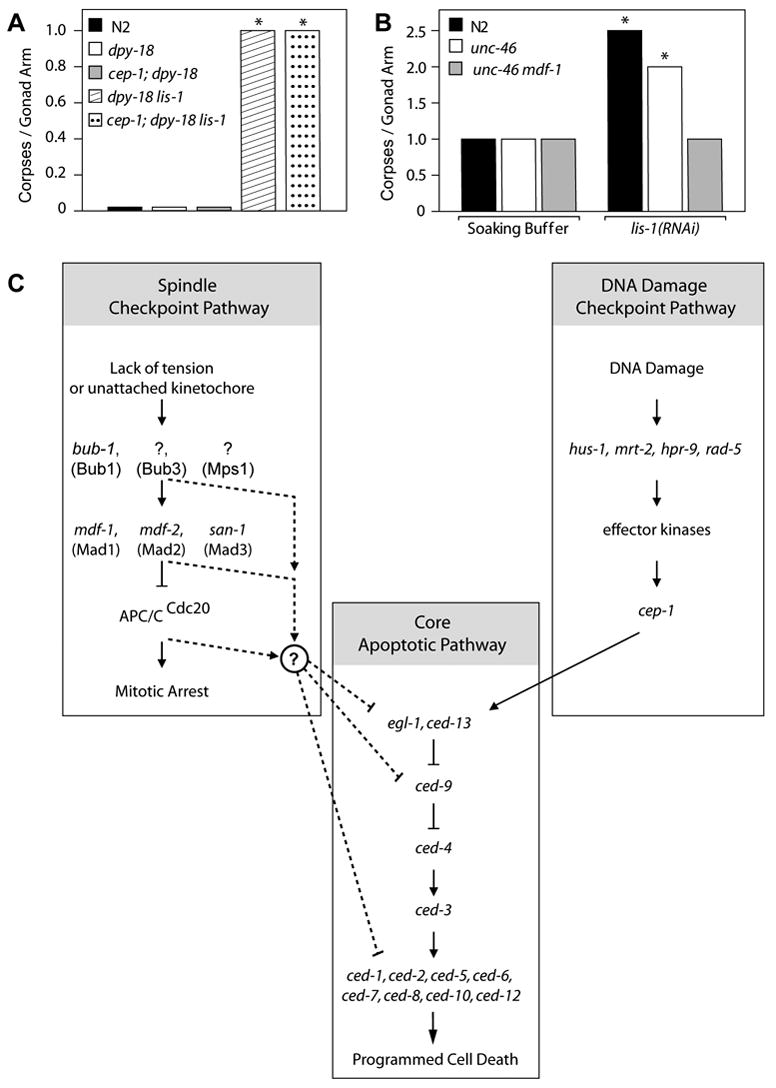
The spindle checkpoint lies upstream of the programmed cell death pathway. (A) dpy-18 lis-1 and cep-1; dpy-18 lis-1 mutants exhibit no significant difference in the average number of corpses per gonad arm when stained with SYTO 12. On the other hand, they both contain a significantly greater average number of corpses per gonad arm than N2, dpy-18 and cep-1; dpy-18 (P<0.001, for all comparisons, Mann-Whitney test). The data presented are medians. (B) Treatment of N2 and unc-46 with lis-1 dsRNA produces a significant increase in the average number of corpses per gonad arm, compared to soaking buffer alone (P<0.05 and P<0.001 respectively, Mann-Whitney test). The unc-46 mdf-1 double mutant contains a similar average number of corpses per gonad arm as N2 and unc-46 in soaking buffer alone but does not show an increase in corpse number upon treatment with lis-1 dsRNA. The data are medians, while the averages ± the s.e.m. and are presented in Table 1. Because mdf-1 homozygotes display variable phenotypes (Kitagawa and Rose, 1999), we excluded animals exhibiting tumerous gonads (tum phenotype) or abnormal gonad development (gon phenotype) from our analysis. (C) The spindle checkpoint gene mdf-1 is required for lis-1(lf)-induced apoptosis, suggesting that the spindle checkpoint lies upstream of the programmed cell death pathway and in parallel to the DNA damage checkpoint. Genes linking the spindle checkpoint to the programmed cell death pathway remain unknown.
Discussion
We show that lis-1(lf) induces a spindle checkpoint-dependent mitotic arrest in the germline and an increase in germ-cell apoptosis. That both of these events are abrogated by null alleles of the spindle checkpoint gene mdf-1 implies, but does not demonstrate, that these two events are causally related. This observation may be relevant to understanding lissencephalic brains as well as providing insight into the mechanism of action of anti-cancer drugs that target microtubule-dependent functions. Furthermore, we have identified other previously unreported germline biological processes that require lis-1 function.
Loss of lis-1 function leads to a cell-sparse phenotype in the C. elegans germline reminiscent of the cell-sparse cortex seen in human lissencephaly. Our analysis correlated the germ cell sparseness to spindle checkpoint-dependent cell cycle arrest among proliferating nuclei. The relatively small increase in mitotic arrested nuclei compared to the large decrease in germline nuclei is inconsistent with simple retention in the proliferation zone. Thus either the mitotic arrested nuclei are resolved as they migrate along the proximal-distal axis, or they are cleared by apoptosis. Consistent with the latter possibility, we observed an increase in germline cell corpses. A portion of the increase in accumulated corpses is likely caused by slowed engulfment kinetics due to loss of lis-1 function. However, because an mdf-1(null) allele abrogates the accumulation of lis-1(lf)-associated corpses without suppressing the lis-1(lf) engulfment defect, it is probable that lis-1(lf) also leads to an increase in the rate of programmed cell death in the gonad. As expected, mutation of mdf-1 alone did not affect cell death or engulfment. The picture that emerges from these observations is that loss of lis-1 function leads to spindle defects that cause spindle checkpoint mitotic arrest; those nuclei that do not resolve the spindle checkpoint are then cleared by programmed cell death. However, because we observed mitotic arrest in fixed samples we cannot follow the fate of individual nuclei. Consistent with this scenario, Tao et al. (2005) reported that an inhibitor of the mitotic kinesin KSP induces apoptosis of a HeLa-derived cell line via activation of the spindle checkpoint. At this time we cannot distinguish whether increased apoptosis results directly from activation of the spindle checkpoint or whether other events occurring during mitotic arrest affect apoptosis with the spindle checkpoint playing a permissive role.
Microtubule-dependent processes likely underlie all these phenotypes, since, as reported in other systems (Cockell et al., 2004; Gonczy et al., 1999; Xiang et al., 1995), we find that dhc-1 mutants phenocopy the full spectrum of germline phenotypes of multiple lis-1 mutants and lis-1 RNAi. lis-1(lf) may activate the spindle checkpoint through multiple mechanisms. First, LIS-1 is a MAP that affects microtubule polymerization, so lis-1(lf) may compromise spindle integrity resulting in decreased tension at kinetochores (Sapir et al., 1997; Stern and Murray, 2001). Second, since lis-1 plays a role in dynein-mediated transport of spindle checkpoint proteins away from the kinetochore to the spindle pole(Howell et al., 2001; Schmidt et al., 2005; Wojcik et al., 2001; Yan et al., 2003), lis-1(lf) may interfere with kinetochore disassembly.
Much of the original characterization of the spindle checkpoint has been carried out in yeast, indeed, specifically in dynein loss-of-function (DYN1) mutants (Eshel et al., 1993). However, yeast cells do not undergo apoptosis, and the role of the spindle checkpoint in mediating cell death in metazoans has been controversial. The mouse spindle checkpoint protein Bub1 may be required for nocodazole-induced apoptosis (Taylor and McKeon, 1997), but this study was limited by a gain-of-function approach and nonspecific markers of apoptotic death. Subsequent studies of mutations in Drosophila bub1 (Basu et al., 1999) and mouse Mad2 (Dobles et al., 2000) found that spindle checkpoint gene mutations trigger, rather than block, cell death. These results may reflect the terminal phenotypes of cells that cycled through multiple aberrant mitoses, death being triggered by secondary processes such as chromosomal instability rather than by the spindle checkpoint directly. The simplest model to explain why lis-1(lf)-induced apoptosis requires a functional spindle checkpoint but not a functional DNA damage checkpoint is that the spindle checkpoint pathway, like the DNA damage checkpoint pathway (Hofmann et al., 2002), lies upstream of the programmed cell death pathway (Fig. 7C). Such a model may explain why mammalian Mad2 haploinsufficiency causes papillary lung adenocarcinoma (Michel et al., 2001) and may explain the mechanism of action of anticancer drugs such as paclitaxel (Taxol) and vinca alkaloids that act directly on spindle microtubules (Rieder and Maiato, 2004).
lis-1(lf) increases germ cell corpse number by both activating killing and impairing engulfment. Our time-lapse observations indicate that mutations in lis-1 and dhc-1 disrupt engulfment but do not abolish it, since dead cells are eventually cleared from the germline. This, and the fact that both genes are essential, probably explains why mutations in these genes were not recovered in screens for ced genes. lis-1 and dhc-1 could mediate engulfment directly by modulating microtubule- or actin-dependent processes (Kinchen et al., 2005; Rehberg et al., 2005) or indirectly by trafficking molecules such as an eat-me signal (Wang et al., 2003) or organelles such as phagosomes (Blocker et al., 1997; Koushika et al., 2004) that mediate engulfment. However, lis-1(lf) does not increase corpse number solely via an engulfment defect; in the lis-1(RNAi) mdf-1(lf) double mutants engulfment was still substantially slowed, yet corpse number was nearly normal and the nuclei sparse appearance of the germline was suppressed.
Mutations in human Lis1 or its genetic interactors have been associated with increased apoptosis. Gambello et al. (2003) reported a role for apoptosis in mouse Lis1 mutants, while Feng and Walsh (2004) found changes in cell death insufficient to account for progenitor reduction in mouse Nde1 mutants. Both studies assayed apoptosis using only TUNEL staining, which is neither quantitative, specific, nor sufficient to identify apoptotic cells (Prochazkova et al., 2003). Moreover, absent TUNEL staining alone does not rule out apoptosis, because rapid clearance of dying cells may hinder detection of apoptosis in human diseases. In C. elegans, for example, thousands of female germ cells undergo physiological programmed cell death, but, since clearance occurs rapidly (Fig. 6A), only 1–3 female germ cells are usually visualized by SYTO 12 at a given moment (Gumienny et al., 1999). Our results offer a more rigorous demonstration of Lis1-induced apoptosis than has been reported in other systems. Our results also offer genetic evidence implicating apoptosis in the pathogenesis of human lissencephaly. lis-1(lf)-induced cell killing not only displays the anatomical and morphological features of germline apoptosis but also requires ced-3, a killer gene within the core programmed cell death pathway (Fig. 5A). Since C. elegans lis-1 and mammalian LIS-1 are orthologs, common genetic pathways likely underlie the cell-sparse phenotype of the C. elegans lis-1 germline and the cell-sparse phenotype of the human lissencephalic cortex. If so, the following sequence of events may contribute to lissencephaly: loss of MAP function damages the mitotic spindle, in turn activating the spindle checkpoint and causing cell cycle arrest among proliferating neuroblasts (Feng and Walsh, 2004; Gambello et al., 2003), a portion of which die by apoptosis. Many additional hypotheses have been forwarded to explain why lissencephalic cortex is cell-sparse, including impaired neuronal migration due to a defect in nuclear translocation (Hirotsune et al., 1998; Xiang et al., 1995) or due to a defect in platelet-activating factor metabolism (Hattori et al., 1994) and including altered spindle position and altered cell fate of cortical progenitors (Feng and Walsh, 2004; Sasaki et al., 2000). The striking pleiotropy seen with mutation or silencing of C. elegans lis-1 suggests that multiple mechanisms likely contribute to the cell-sparse cortex seen in human lissencephaly (Cockell et al., 2004; Williams et al., 2004).
Our findings hold implications not only for the pathogenesis of lissencephaly, but also for the relationship between apoptosis and disease more generally. The discovery that the C. elegans cell survival gene ced-9 is orthologous to the human proto-oncogene BCL-2 first implicated inhibition of the programmed cell death pathway in oncogenesis (Hengartner and Horvitz, 1994). In contrast, the only definitive genetic evidence for a role of apoptosis in diseases arising from excessive cell death is the gain-of-function disease retinitis pigmentosa (Davidson and Steller, 1998). If confirmed in lissencephalic brains, our findings would constitute the first definitive genetic evidence of a role for apoptosis in the pathogenesis of a loss-of-function human disease. Moreover, our studies offer a specific cell biological and genetic model through which loss-of-function mutations in a disease gene homolog activate the core programmed cell death pathway.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at 10.1016/j.ydbio.2007.02.024.
Acknowledgments
We would like to thank Matt Michael and members of the Hunter laboratory for comments on the manuscripts. This work was supported by the NINDS (#NS02083-02) and the Bernhard Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Basu J, Bousbaa H, Logarinho E, Li Z, Williams BC, Lopes C, Sunkel CE, Goldberg ML. Mutations in the essential spindle checkpoint gene bub1 cause chromosome missegregation and fail to block apoptosis in Drosophila. J Cell Biol. 1999;146:13–28. doi: 10.1083/jcb.146.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla KN. Microtubule-targeted anticancer agents and apoptosis. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- Blocker A, Severin FF, Burkhardt JK, Bingham JB, Yu H, Olivo JC, Schroer TA, Hyman AA, Griffiths G. Molecular requirements for bi-directional movement of phagosomes along microtubules. J Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockell MM, Baumer K, Gonczy P. lis-1 is required for dynein-dependent cell division processes in C. elegans embryos. J Cell Sci. 2004;117:4571–4582. doi: 10.1242/jcs.01344. [DOI] [PubMed] [Google Scholar]
- Davidson FF, Steller H. Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature. 1998;391:587–591. doi: 10.1038/35385. [DOI] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell. 2000;101:635–645. doi: 10.1016/s0092-8674(00)80875-2. [DOI] [PubMed] [Google Scholar]
- Encalada SE, Willis J, Lyczak R, Bowerman B. A Spindle Checkpoint Functions during Mitosis in the Early Caenorhabditis elegans Embryo. Mol Biol Cell. 2005;16:1056–1070. doi: 10.1091/mbc.E04-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshel D, Urrestarazu LA, Vissers S, Jauniaux JC, van Vliet-Reedijk JC, Planta RJ, Gibbons IR. Cytoplasmic dynein is required for normal nuclear segregation in yeast. Proc Natl Acad Sci USA. 1993;90:11172–11176. doi: 10.1073/pnas.90.23.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O'Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA. Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron. 2004;44:279–293. doi: 10.1016/j.neuron.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Gambello MJ, Darling DL, Yingling J, Tanaka T, Gleeson JG, Wynshaw-Boris A. Multiple dose-dependent effects of Lis1 on cerebral cortical development. J Neurosci. 2003;23:1719–1729. doi: 10.1523/JNEUROSCI.23-05-01719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–1112. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner A, MacQueen AJ, Villeneuve AM. Methods for Analyzing Checkpoint Responses in Caenorhabditis elegans. Methods Mol Biol. 2004;280:257–274. doi: 10.1385/1-59259-788-2:257. [DOI] [PubMed] [Google Scholar]
- Gonczy P, Pichler S, Kirkham M, Hyman AA. Cytoplasmic dynein is required for distinct aspects of MTOC positioning, including centrosome separation, in the one cell stage Caenorhabditis elegans embryo. J Cell Biol. 1999;147:135–150. doi: 10.1083/jcb.147.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumienny TL, Lambie E, Hartwieg E, Horvitz HR, Hengartner MO. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development. 1999;126:1011–1022. doi: 10.1242/dev.126.5.1011. [DOI] [PubMed] [Google Scholar]
- Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M, Hengartner MO. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol. 2002;12:1908–1918. doi: 10.1016/s0960-9822(02)01262-9. [DOI] [PubMed] [Google Scholar]
- Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155:1159–1172. doi: 10.1083/jcb.200105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, Grushcow JM, Brame CJ, Caldwell JA, Hunt DF, Lin R, Smith MM, Allis CD. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- Huang NN, Mootz DE, Walhout AJ, Vidal M, Hunter CP. MEX-3 interacting proteins link cell polarity to asymmetric gene expression in Caenorhabditis elegans. Development. 2002;129:747–759. doi: 10.1242/dev.129.3.747. [DOI] [PubMed] [Google Scholar]
- Kimble J, Hirsh D. The postembryonic cell lineages of the hermaphrodite and male gonads in Caenorhabditis elegans. Dev Biol. 1979;70:396–417. doi: 10.1016/0012-1606(79)90035-6. [DOI] [PubMed] [Google Scholar]
- Kinchen JM, Cabello J, Klingele D, Wong K, Feichtinger R, Schnabel H, Schnabel R, Hengartner MO. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Rose AM. Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol. 1999;1:514–521. doi: 10.1038/70309. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML. Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci. 2004;24:3907–3916. doi: 10.1523/JNEUROSCI.5039-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Steward R, Luo L. Drosophila Lis1 is required for neuroblast proliferation, dendritic elaboration and axonal transport. Nat Cell Biol. 2000;2:776–783. doi: 10.1038/35041011. [DOI] [PubMed] [Google Scholar]
- Liu Z, Xie T, Steward R. Lis1, the Drosophila homolog of a human lissencephaly disease gene, is required for germline cell division and oocyte differentiation. Development. 1999;126:4477–4488. doi: 10.1242/dev.126.20.4477. [DOI] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr Biol. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty V, Benezra R. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- Prochazkova J, Kylarova D, Vranka P, Lichnovsky V. Comparative study of apoptosis-detecting techniques: TUNEL, apostain, and lamin B. Biotechniques. 2003;35:528–534. doi: 10.2144/03353st06. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- Rehberg M, Kleylein-Sohn J, Faix J, Ho TH, Schulz I, Graf R. Dictyostelium LIS1 Is a Centrosomal Protein Required for Microtubule/Cell Cortex Interactions, Nucleus/Centrosome Linkage, and Actin Dynamics. Mol Biol Cell. 2005;16:2759–2571. doi: 10.1091/mbc.E05-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Sapir T, Elbaum M, Reiner O. Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. Embo J. 1997;16:6977–6984. doi: 10.1093/emboj/16.23.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Schmidt DJ, Rose DJ, Saxton WM, Strome S. Functional analysis of cytoplasmic dynein heavy chain in Caenorhabditis elegans with fast-acting temperature-sensitive mutations. Mol Biol Cell. 2005;16:1200–1212. doi: 10.1091/mbc.E04-06-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 Operates in a Common Pathway with LIS1 and Cytoplasmic Dynein to Regulate Cortical Neuronal Positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Siller KH, Serr M, Steward R, Hays TS, Doe CQ. Live Imaging of Drosophila Brain Neuroblasts Reveals a Role for Lis1/Dynactin in Spindle Assembly and Mitotic Checkpoint Control. Mol Biol Cell. 2005;16:5127–5140. doi: 10.1091/mbc.E05-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- Stern BM, Murray AW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. Curr Biol. 2001;11:1462–1467. doi: 10.1016/s0960-9822(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Swan A, Nguyen T, Suter B. Drosophila Lissencephaly-1 functions with Bic-D and dynein in oocyte determination and nuclear positioning. Nat Cell Biol. 1999;1:444–449. doi: 10.1038/15680. [DOI] [PubMed] [Google Scholar]
- Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005;8:49–59. doi: 10.1016/j.ccr.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Wang X, Wu YC, Fadok VA, Lee MC, Gengyo-Ando K, Cheng LC, Ledwich D, Hsu PK, Chen JY, Chou BK, Henson P, Mitani S, Xue D. Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science. 2003;302:1563–1566. doi: 10.1126/science.1087641. [DOI] [PubMed] [Google Scholar]
- Williams SN, Locke CJ, Braden AL, Caldwell KA, Caldwell GA. Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in C. elegans. Hum Mol Genet. 2004;13:2043–2059. doi: 10.1093/hmg/ddh209. [DOI] [PubMed] [Google Scholar]
- Wojcik E, Basto R, Serr M, Scaerou F, Karess R, Hays T. Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat Cell Biol. 2001;3:1001–1007. doi: 10.1038/ncb1101-1001. [DOI] [PubMed] [Google Scholar]
- Xiang X, Osmani AH, Osmani SA, Xin M, Morris NR. NudF, a nuclear migration gene in Aspergillus nidulans, is similar to the human LIS-1 gene required for neuronal migration. Mol Biol Cell. 1995;6:297–310. doi: 10.1091/mbc.6.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Li F, Liang Y, Shen Y, Zhao X, Huang Q, Zhu X. Human Nudel and NudE as regulators of cytoplasmic dynein in poleward protein transport along the mitotic spindle. Mol Cell Biol. 2003;23:1239–1250. doi: 10.1128/MCB.23.4.1239-1250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at 10.1016/j.ydbio.2007.02.024.