

Abstract
During the G1/S transition, p21 proteolysis is mediated by Skp2; however, p21 re-accumulates in G2 and is degraded again in prometaphase. How p21 degradation is controlled in mitosis remains unexplored. We found that Cdc20 (an activator of the ubiquitin ligase APC/C) binds p21 in cultured cells and identified a D-box motif in p21 necessary for APC/CCdc20-mediated ubiquitylation of p21. Overexpression of Cdc20 or Skp2 destabilized wild type p21; however, only Skp2, but not Cdc20, was able to destabilize a p21(D-box) mutant. Silencing of Cdc20 induced an accumulation of p21, increased the fraction of p21 bound to Cdk1, and inhibited Cdk1 activity in p21+/+ prometaphase cells but not in p21−/ − cells. Thus, in contrast to anaphase, during which Cdc20 induces Cdk1 inactivation by directing the proteolysis of cyclins, in prometaphase Cdc20 positively regulates Cdk1 by mediating the degradation of p21. We propose that the APC/CCdc20-mediated degradation of p21 contributes to the full activation of Cdk1 necessary for mitotic events and prevents mitotic slippage during spindle checkpoint activation.
Introduction
The cyclin-dependent kinase (CDK) inhibitor p21 is a critical negative regulator of the cell division cycle (Bloom and Pagano, 2004; Pei and Xiong, 2005). Its N-terminal domain binds to and inhibits cyclin/CDK complexes, while the C-terminal domain binds to PCNA and inhibits the elongation of DNA synthesis. In agreement with these inhibitory functions, levels of p21 increase in response to stimuli that inhibit cell proliferation or induce differentiation and senescence.
During an unperturbed cell cycle, p21 plays a role in G1 and G2 by contributing to temporal pauses that facilitate the integration of critical checkpoint signals for regulated entry into S-phase and mitosis, respectively. While p21 contributes to the maintenance of the G1 state by inhibiting the activity of Cdk2 – and likely Cdk1 and Cdk3 as well – (Bloom and Pagano, 2004; Pei and Xiong, 2005), in G2, the major target of p21 is Cdk1 (Bates et al., 1998; Bunz et al., 1998; Chan et al., 2000; Dulic et al., 1998; Medema et al., 1998; Niculescu et al., 1998). Forced expression of p21 inhibits DNA synthesis and entry into mitosis. In addition to these two well-known inhibitory effects, p21 overexpression induces polyploidy, particularly when the mitotic spindle is disrupted with nocodazole (Bates et al., 1998; Chang et al., 2000; Niculescu et al., 1998). However, the reasons for this mitotic effect are not well understood.
In non-transformed fibroblasts, p21 shows bimodal periodicity, with an initial peak during G1 and a second peak in G2, at both the mRNA and protein level (Dulic et al., 1998; Li et al., 1994). Cells in G2 (displaying visible nucleoli and no mitotic chromosome condensation) exhibit high levels of nuclear p21 associated with Cdk1 complexes. In the course of DNA condensation, p21 levels drop, and p21 remains absent during mitosis (Dulic et al., 1998). How p21 degradation is controlled in early mitosis has not been investigated. In the study presented herein, we investigated the regulation of p21 degradation in mitosis and found that the ubiquitin ligase APC/CCdc20 (Anaphase Promoting Complex/Cyclosome and its activator Cdc20) controls this process.
Results
Cdc20 physically interacts with p21
Initially, we confirmed that p21 protein levels show a bimodal distribution during the cell cycle (Dulic et al., 1998). Human T98G cells (Dorrello et al., 2006; Stein, 1979) were synchronized in G0 by serum deprivation and released from the arrest by the addition of serum. As expected, cyclin E1 levels rose in late G1, and the increase in the expression of Skp2, Cdc20, and cyclin A2 coincided with the G1/S transition, inversely correlating with p27 levels (Fig. 1A). Levels of p21 were low in G0, increased in G1, and decreased again at G1/S in parallel with p27 degradation. However, whereas p27 expression remained low for the remainder of the cell cycle, levels of p21 increased 28 hours after serum addition, when cells were exiting S-phase and approaching G2.
Figure 1. p21 is degraded in early mitosis.

(A) T98G cells were synchronized in G0 by 72 hours of serum starvation (indicated as time 0). Cells were then re-stimulated with serum and collected at the indicated time points. Protein extracts were analyzed by immunobloting with antibodies against the indicated proteins. Cell cycle phases were monitored by flow cytometry.
(B) IMR-90 fibroblasts were first synchronized in G0 by 72 hours of serum starvation and then at the G1/S boundary (indicated as time 0) by re-stimulating them with serum for 28 hours, adding aphidicolin for the last 18 hours. Cells were subsequently washed and allowed to progress thorough the cell cycle for the indicated times. Protein extracts were analyzed by immunoblotting with antibodies against the indicated proteins. Cell cycle phases were monitored by flow cytometry.
To pinpoint when p21 levels decrease again late in the cell cycle, we synchronized IMR90 cells in S-phase using aphidicolin and then released them in fresh medium to allow synchronized progression through G2 and mitosis. Levels of p21 increased and reached a peak 12 hours after aphidicolin release, then steadily decreased in coincidence with the appearance of the Ser10 phosphorylation of Histone H3 (a marker of chromosome condensation) and the degradation of cyclin A2 (Fig. 1B). Similar behavior of p21 levels was observed in multiple cell types, including T98G and U-2 OS (Fig. S1).
The SCFSkp2 ubiquitin ligase is involved in the degradation of p21 (Bornstein et al., 2003; Yu et al., 1998); therefore, we analyzed the levels of p21 in cells lacking Skp2. Both Skp2−/ − and Skp2−/ −;p27−/ − mouse embryonic fibroblasts (MEFs) displayed high levels of p21 when synchronized in S phase [as previously observed (Bornstein et al., 2003)], but not in G2 and prometaphase (Fig. 2). Moreover, the physical interaction between Skp2 and p21 was significantly decreased in early mitotic cells compared to S phase cells (data not shown). Thus, to explore the possibility that p21 is targeted for degradation by a different ubiquitin ligase, we screened 21 human F-box proteins, two activators of the APC/C, and the ubiquitin ligase Mdm2 for binding to p21. Constructs for the FLAG-tagged versions of these proteins were transfected into HEK293T cells and, after the addition of the proteasome inhibitor MG132 six hours prior to lysis, immunoprecipitations for the ubiquitin ligases were performed to evaluate their interaction with endogenous p21. We observed that, in addition to Skp2, the only other protein able to coimmunoprecipitate endogenous p21 was Cdc20 (Fig. 3A). In contrast, related proteins such as Cdh1, βTrcp1, βTrcp2, Fbxw2, Fbxw4, Fbxw5, Fbxw7α, Fbxw7γ, or Fbxw8 (all of which, like Cdc20, contain WD-40 repeats) did not bind p21 (Fig. 3A). Similarly, additional F-box proteins (Fbxl2, Fbxl3, Fbxl7, Fbxl10, Fbxl11, Fbxl15, Fbxl20, Fbxo4, Fbxo5, Fbxo7, Fbxo9, Fbxo17, Fbxo21) or Mdm2 also did not coimmunoprecipitate p21 (Fig. 3A and data not shown). Finally, the interaction of p21 with Cdc20, as well as Cdc27 (an integral subunit of APC/C), was also observed for the endogenous proteins (Fig. 3B).
Figure 2. Mitotic degradation of p21 is not dependent on Skp2.
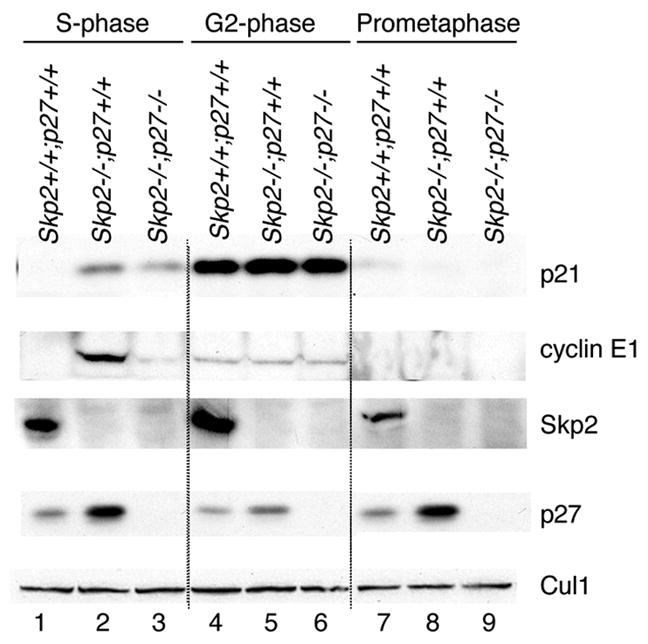
Mouse embryonic fibroblasts from Skp2−/ −;p27+/+, Skp2−/ −;p27−/ −, and Skp2+/+;p27+/+ mice were synchronized at G1/S by a double thymidine block (S-phase). Cells were subsequently washed and allowed to progress through the cell cycle for 12 hours in the presence of nocodazole. The prometaphase, round cells were collected by gentle shake-off, and finally, the remaining attached cells (G2-phase) were also harvested. Protein extracts were analyzed by immunobloting with antibodies to the indicated proteins.
Figure 3. p21 interacts with Cdc20.

(A) HEK293T cells were transfected with an empty vector (EV) or constructs encoding the indicated FLAG–tagged F-box proteins (FBPs), FLAG-Cdh1, or FLAG-Cdc20, all in the presence of a plasmid encoding His-tagged Skp1. During the last six hours before harvesting, cells were treated with the proteasome inhibitor MG132. Exogenous proteins were immunoprecipitated (IP) from whole cell extracts (WCE) with an anti-FLAG resin, and immunocomplexes were probed with antibodies to the indicated proteins.
(B) Endogenous Cdc20 and Cdc27 co-immunoprecipitates endogenous p21. Whole cell extracts (WCE) from HeLa cells were immunoprecipitated (IP) with normal IgG, anti-Cdc20, anti-Cdc27, or anti-Cdh1 antibodies. Immunocomplexes were probed with antibodies to the indicated proteins. The second lane contained molecular weight markers (MWM) that appear to cross-react with the anti-cyclin A2 antibody.
(C) HeLa cells were infected with an empty vector (EV) or constructs encoding FLAG–tagged Cdc20 or FLAG–tagged Cdc20 Δ165 mutant. Where indicated, the proteasome inhibitor MG132 was added during the last six hours in culture. Exogenous proteins were immunoprecipitated (IP) from extracts obtained from asynchronous (AS) or prometaphase (PM) cells using an anti-FLAG resin. Immunocomplexes were then probed with antibodies to the indicated proteins.
We then used seven Cdc20 mutants (Fig. S2A) to map the p21-binding domain in Cdc20. FLAG-tagged wild type Cdc20 and Cdc20 mutants were expressed in HEK293T cells and then immunoprecipitated to evaluate their interaction with endogenous p21 and other proteins. The only mutant that lost the ability to bind p21 was ΔC(471–499), which lacks the last 19 amino acids (Fig. S2B). This mutant, as well as two different mutants with N-terminal deletions (ΔN133 and ΔN165), did not bind Cdc27 [(Schwab et al., 2001; Thornton et al., 2006) and Fig. S2B].
However, in contrast to ΔC(471–499), ΔN133 and ΔN165 coimmunoprecipitated p21 much more efficiently than wild type Cdc20 (Fig. 3C and S2B).
The APC/CCdc20-mediated ubiquitylation and degradation of p21 requires an intact D-box motif in p21
Results from the immunoprecipitation experiments described above suggested that p21 may be a substrate of APC/CCdc20 for the following reasons: (i) p21 interacted with the C-terminus of Cdc20, similarly to other substrates of APC/CCdc20, such as cyclin A2 and Securin [(Ohtoshi et al., 2000) and data not shown]. (ii) Similarly to cyclin A2, p21 bound more efficiently to ΔN165 than to wild type Cdc20 (Fig. 3C). If p21 is a substrate of APC/CCdc20, this difference could be explained by the differential binding of wild type Cdc20 and ΔN165 to Cdc27. ΔN165 is unable to bind Cdc27 and, therefore, could sequester substrates without delivering them to the APC/C ligase for ubiquitylation and consequent degradation. In contrast, wild type Cdc20 binds p21, but it may also induce p21 degradation. In favor of this hypothesis, the binding between wild type Cdc20 and p21 is not immediately detectable without the addition of the proteasome inhibitor MG132 prior to harvesting the cells (Fig. 3C). (iii) Wild type Cdc20 and ΔN165 also show a second differential behavior. Whereas wild type Cdc20 interacted better with p21 and cyclin A2 in asynchronous cells than in prometaphase cells, the opposite situation was observed for ΔN165 (Fig. 3C). This result suggests that in prometaphase cells, where APC/CCdc20 is more active, wild type Cdc20 is more competent in promoting the degradation of p21 and cyclin A2, whereas ΔN165 is unable to do so, allowing increasing amounts of p21 and cyclin A2 to bind.
Based on these considerations, we investigated the involvement of APC/CCdc20 in controlling the degradation of p21. We transfected HEK293T cells with a construct expressing p21 and constructs expressing either Cdc20, Cdh1, or Skp2 (Fig. 4A). Enforced expression of Cdc20 or Skp2 resulted in a considerable decrease in the steady-state amounts of p21 (Fig. 4A, lanes 6 and 8), whereas overexpression of Cdh1 had no effect (Fig. 4A, lane 4). Notably, both the Cdc20- and the Skp2-mediated decrease in p21 levels was due to the destabilization of the CDK inhibitor, as shown by the ability of the proteasome inhibitor MG132 to prevent this downregulation (Fig. 4A, lanes 7 and 9) and by the decrease in p21 half-life (Fig. 4C, lanes 1–12). In contrast to wild type Cdc20, ΔN165 did not induce degradation of p21 (Fig. 4D), supporting the hypothesis that this mutant is inactive and unable to target p21 for degradation, regardless of its ability to bind p21.
Figure 4. Cdc20-dependent ubiquitylation and degradation of p21 depends on the D-box of p21.
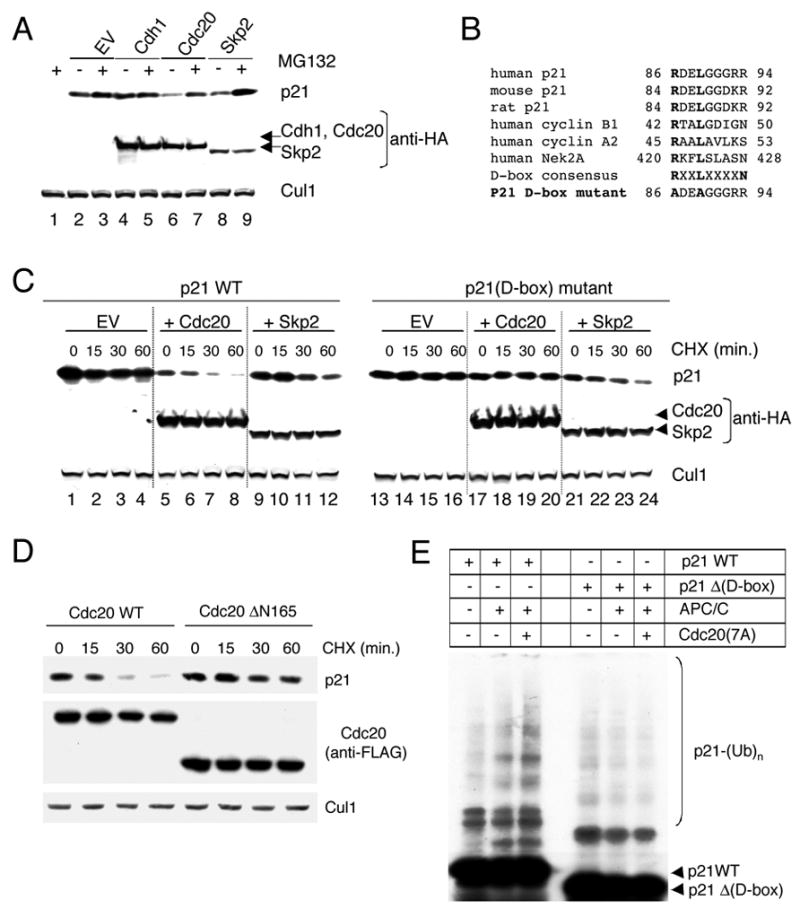
(A) Whole cell extracts were prepared from HEK293T cells transfected with p21 and either HA-tagged Cdc20, HA-tagged Cdh1, or HA-tagged Skp2. Levels of p21 were detected by immunoblotting with an anti-p21 antibody, whereas Cdc20, Cdh1, and Skp2 were analyzed using an anti-HA antibody. Cul1 was used as a loading control. Where indicated, the proteasome inhibitor MG132 was added during the last six hours in culture. No significant changes in the cell cycle phases (as monitored by flow cytometry) of transfected HEK293T cells were observed compared to controls.
(B) Alignment of the amino acid regions corresponding to the putative Destruction-box motif (D-box) in human, mouse, and rat p21 with the D-box motifs of human cyclin B1, cyclin A2 and Nek2A. A schematic representation of the Arg- and Leu-to-Ala substitutions in the putative D-box motif of human p21 is depicted at the bottom.
(C) HEK293T cells were transfected with wild type p21 or the p21(D-box) mutant in the presence of either an empty vector (EV), HA-tagged Cdc20, or HA-tagged Skp2. Twenty-four hours after transfection, cells were treated with cycloheximide (CHX) and the half-life of p21 analyzed by immunobloting.
(D) HEK293T cells were transfected with wild type p21 in the presence of either FLAG–tagged Cdc20 or FLAG–tagged Cdc20 Δ165 mutant. Twenty-four hours after transfection, cells were treated with cycloheximide (CHX), and the half-life of p21 was analyzed by immunobloting.
(E) In vitro ubiquitin ligation assay of 35S labeled in vitro transcribed/translated p21 or p21Δ (D-box) mutant was conducted in the absence or presence of immunopurified APC/C and constitutively active Cdc20(7A) mutant, as indicated. Samples were incubated at 30°C for 90 minutes. The bracket on the right side of the panels marks a ladder of bands corresponding to polyubiquitylated p21.
Substrates of APC/C are often characterized by the presence of a "destruction box" or D-box, which was first identified in cyclin B as a stretch of nine amino acids (RxxLxxIxN) that when mutated stabilized the protein (Glotzer et al., 1991). However, most substrates contain only the minimal RxxL motif in their D-boxes. We identified one conserved RxxL motif in p21 (positions 86–89 in human) that could potentially serve as a D-box (Fig. 4B), but no other potential APC/C recognition motifs (KEN-box, A-box, etc.) were identified. The RxxL domain of p21 is not present in p27, does not overlap with either the CDK- or the PCNA-binding domains, and is encompassed in a low complexity region (amino acids 81–96), similar to the D-box motifs of A and B type cyclins. We mutated the Arg and Leu residues in the putative D-box motif to Ala (Fig. 4B) and determined the stability of the p21 D-box [p21(D-box)] mutant in HEK293T cells co-expressing either Cdc20 or Skp2. Notably, whereas the half-lives of both wild type p21 and p21(D-box) mutant were shortened by Skp2 (Fig. 4C), p21(D-box) mutant was refractory to the effect of Cdc20 (Fig. 4C, lanes 17–20).
To test whether p21 is ubiquitylated via the APC/CCdc20 ubiquitin ligase and further investigate the importance of the D-box motif, we reconstituted the ubiquitylation of p21 in vitro. p21 ubiquitylation was promoted by the addition of APC/C and was further promoted by addition of both APC/C and Cdc20 (Fig. 4E). Methylated ubiquitin added to the in vitro reaction inhibited the formation of the highest molecular weight forms of p21 (Fig. S3), formally demonstrating that the high molecular weight forms of p21 are indeed polyubiquitylated species. In contrast to wild type p21, a p21 deleted in the D-box motif was not efficiently ubiquitylated, even in the presence of both APC/C and Cdc20 (Fig. 4E and S3).
Silencing of Cdc20 stabilizes p21 in early mitosis and inhibits Cdk1 activity
To confirm that Cdc20 is required for the degradation of p21, we used the small interfering RNA (siRNA) technique to reduce the expression of Cdc20. When compared to cells transfected with control double-stranded RNA (dsRNA) oligos, cells transfected with two different dsRNA oligos targeting Cdc20 [including a previously validated dsRNA oligo (Donzelli et al., 2002; Elbashir et al., 2001)] showed an upregulation of p21 levels (Fig. 5A). Since Cdc20 is mostly active in mitosis, the accumulation of p21 was presumably due to the mitotic fraction present in asynchronous cells. In agreement with this hypothesis, silencing of Cdc20 induced an increase in p21 levels only in U-2 OS cells synchronized in prometaphase but not at G1/S (Fig. 5B). We also performed a complementary experiment: U-2 OS cells arrested in prometaphase with nocodazole were collected by mitotic shake-off and then allowed to progress through mitosis and into the next cell cycle. The levels of p21 were found to be low in mitosis and to steadily increase during the G1 phase (concomitantly with an increase in p27 levels) (Fig. 5C). However, when Cdc20 was knocked-down by siRNA, a delay in the degradation of both p21 and cyclin A2 was observed (Fig. 5C). In addition, a delay in the exit from mitosis was observed, as judged by flow cytometry and the levels of phosphorylated Cdc27 and phosphorylated Histone H3.
Figure 5. Cdc20 targets p21 for degradation in prometaphase.
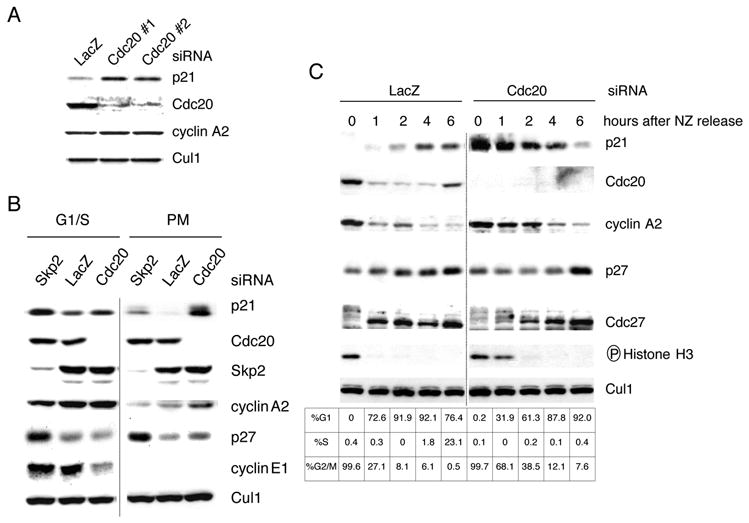
(A) HeLa cells were transfected twice with siRNA molecules to a non-relevant mRNA (Lac Z) or to Cdc20 mRNA (the latter using two different siRNA oligos: #1 and #2). Protein extracts were probed with antibodies to the indicated proteins.
(B) U-2 OS cells were transfected twice with siRNA oligos to a non-relevant mRNA (Lac Z) or to Cdc20 mRNA. Cells were then treated with aphidicolin for 24 hours to arrest cells at G1/S (first 3 lanes). In a parallel experiment, aphidicolin was washed away to allow cells to progress through the cell cycle for an additional 18 hours in the presence of nocodazole (last 3 lanes). The prometaphase (PM), round cells were collected by gentle shake-off. Cell extracts were subjected to immunobloting with antibodies to the indicated proteins.
(C) U-2 OS cells were transfected twice with siRNA oligos corresponding to a non-relevant mRNA (Lac Z) or to Cdc20 mRNA. Twelve hours after the last transfection, nocodazole (NZ) was added for an additional sixteen hours. Round, prometaphase cells were then collected by gentle shake-off and replated in fresh medium for the indicated times. Cells were harvested, and cell extracts were analyzed by immunoblotting with antibodies to the indicated proteins. Cell cycle phases were monitored by flow cytometry.
The fact that knockdown of Cdc20 is ineffective in inducing an increase in p21 levels during G1 (Fig. 5C), G1/S (Fig. 5B), and G2 (not shown) demonstrates that Cdc20 promotes the degradation of p21 only in early mitosis. The accumulation of p21 in response to Cdc20 knockdown both in unperturbed cells (Fig. 5A and data not shown) and in cells in which the spindle assembly checkpoint is activated and sustained by nocodazole (Fig. 5B and 5C), indicates that Cdc20 promotes p21 degradation irrespective of the spindle checkpoint.
Finally, we investigated whether downregulation of Cdc20 in prometaphase affects the activity of Cdk1. After downregulating Cdc20 in prometaphase HCT116 cells, we observed an accumulation of p21 (Fig. S4) and an increase of p21 bound to Cdk1, cyclin A2, and cyclin B1 (Fig. 6). Accordingly, the kinase activities associated with these CDK-cyclin complexes were strongly reduced, as assayed using Histone H1 as a substrate. However, when the same experiments were repeated in HCT116 p21−/ − cells (Waldman et al., 1996), the kinase activities associated with Cdk1 and cyclin B1 were unaffected. Cyclin A2 activity increased, since cyclin A2 is a substrate of APC/CCdc20. These experiments show that Cdc20 is involved in the activation of Cdk1 by mediating the proteolysis of p21.
Figure 6. Cdc20-mediated degradation of p21 activates Cdk1 in early prometaphase.
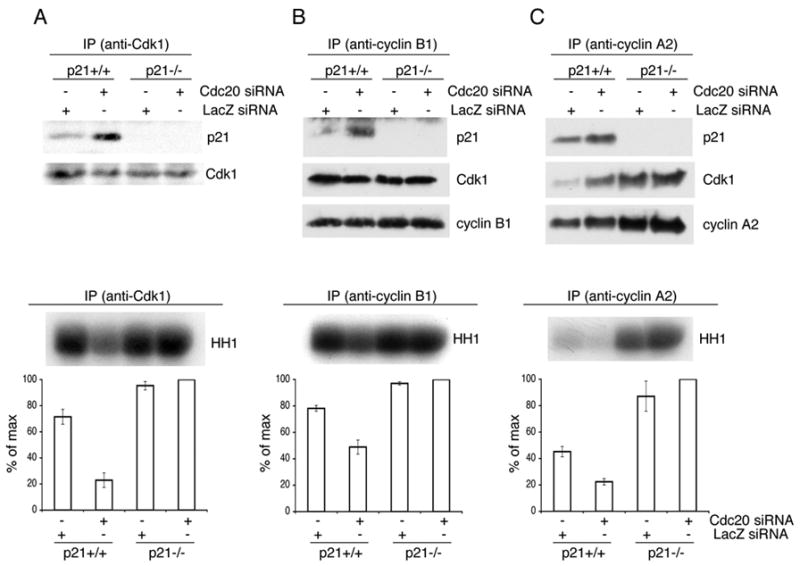
Hct116 p21 +/+ and Hct116 p21 –/−cells were transfected twice with siRNA oligos to a non-relevant mRNA (Lac Z) or to Cdc20 mRNA, as indicated. Cells were then synchronized at G1/S using a double thymidine block and then released to allow cells to progress through the cell cycle for 18 hours (adding nocodazole during the last 15 hours). Prometaphase cells were collected by gentle shake-off. Cell extracts were subjected to immunoprecipitation (IP) with antibodies against Cdk1 (A), cyclin B1 (B), or cyclin A2 (C). Immunocomplexes were probed with antibodies to the indicated proteins (upper panels) or used to perform kinase assays using Histone H1 (HH1) as a substrate (bottom panels). The error bars represent +/−SD from triplicate experiments quantified by measuring radioactivity in the HH1 band excised from the dried gel. A representative experiment showing an autoradiograph of 32P-labeled HH1 is presented on the top of the bottom panels.
Discussion
Control of p21 ubiquitylation by APC/CCdc20 and SCFSkp2 at different phases of the cell cycle
During cell cycle progression, levels of p21 increase in G1 and then decrease at G1/S to free CDK and PCNA complexes. Genetic and biochemical evidence indicates that at G1/S, the degradation of p21 (Bornstein et al., 2003; Wang et al., 2005; Yu et al., 1998) and p27 (Guardavaccaro and Pagano, 2006) is controlled by the SCFSkp2 ubiquitin ligase. However, in contrast to p27, p21 levels rise again in G2 and then decline once more in early M [(Dulic et al., 1998; Li et al., 1994) and Fig. 1A-B]. This final decrease contributes to the full activation of Cdk1 (Bates et al., 1998; Bunz et al., 1998; Chan et al., 2000; Dulic et al., 1998; Medema et al., 1998; Niculescu et al., 1998), which is necessary for mitotic events.
During G2 and M, the SCFSkp2 ubiquitin ligase promotes the degradation of p27 and likely additional substrates. However, the mitotic phenotype present in Skp2−/ − MEFs and Skp2−/ − hepatocytes is not observed in Skp2−/ −;p27−/ − cells (Nakayama et al., 2004), strongly advocating the idea that p27 is the major G2/M substrate of Skp2 and that other substrates are degraded via different or alternative routes. Accordingly, p21 accumulates in Skp2−/ − and Skp2−/ −;p27−/ − MEFs during S-phase but not in M (Fig. 2). Herein, we show that p21 degradation in mitosis is promoted by APC/CCdc20. This process requires a conserved D-box present in p21 (Fig. 4). In contrast to APC/CCdc20, APC/CCdh1 does not play a role in the degradation of p21. In fact, by promoting degradation of Skp2 and Cdc20 (Bashir et al., 2004; Reis et al., 2006; Wei et al., 2004) APC/CCdh1 induces the stabilization of p21. Thus, APC/C exerts dual control on p21 stability: during G1, it allows p21 stabilization (via the Cdh1-mediated degradation of Skp2 and Cdc20), while in M, it induces the proteolysis of p21 (via Cdc20). Moreover, APC/CCdc20 maintains dual control over Cdk1 activity: in early M, Cdc20 promotes Cdk1 activation (by inducing the elimination of p21), whereas in late M, it induces Cdk1 inactivation (by promoting the degradation of mitotic cyclins). Notably, the positive effect of Cdc20 in the activation of Cdk1 during prometaphase is mediated by the induction of p21 proteolysis, as silencing of Cdc20 has no effect on Cdk1 activity in p21−/ − cells (Fig. 6). While both APC/CCdc20 and APC/CCdh1 are known to be involved in either the direct or indirect inactivation of CDKs (Peters, 2006), this study demonstrates that APC/C also positively controls the activity of a CDK, namely, Cdk1. Additionally, whereas all substrates of APC/CCdc20 identified so far in metazoans are also recognized by APC/CCdh1, yeast APC/CCdc20 has its own specific substrates, such as Clb5 and Kip1p (Acquaviva and Pines, 2006; Peters, 2006; Thornton and Toczyski, 2006). Thus, p21 represents the first specific substrate of metazoan APC/CCdc20.
In mitosis, the spindle assembly checkpoint inhibits the separation of sister chromatids until the microtubules radiating from the spindle poles are correctly attached to the kinetochores. At a molecular level, the spindle checkpoint ensures the inhibition of the bulk of APC/CCdc20 activity to avoid the degradation of both Securin (to prevent the separation of sister chromatids) and cyclin B (to prevent a premature exit from mitosis). However, the reason for a subpopulation of APC/CCdc20 to remain active in the presence of spindle checkpoint activity is not well understood. Since Cdk1 inhibition overcomes the prometaphase arrest mediated by the spindle checkpoint (Brito and Rieder, 2006; D'Angiolella et al., 2003), our results suggest that APC/CCdc20-mediated degradation of p21 contributes to the maintenance of the high levels of Cdk1 activity necessary to sustain the spindle checkpoint. If p21 was not degraded at this time, Cdk1 would be inhibited with consequent, rapid adaptation to the spindle checkpoint, resulting in mitotic slippage without cytokinesis and consequent aneuploidy. Interestingly, forced expression of p21 has been reported to cause polyploidy, particularly after mitotic spindle disruption (Bates et al., 1998; Chang et al., 2000; Niculescu et al., 1998).
There is extensive evidence in favor of Skp2-mediated degradation of p21. First, in vitro ubiquitylation of p21 is mediated by SCFSkp2 and its cofactor Cks1 and is promoted by the phosphorylation of p21 on Ser130 by the cyclin E-Cdk2 complex (Bornstein et al., 2003; Wang et al., 2005; Zhu et al., 2005). A conserved glutamic acid at the –2 position of Ser130 is also involved in the binding of p21 to SCFSkp2 (VA and MP, unpublished). The corresponding residues in p27 (Glu185 and Thr187) play a critical role in allowing p27 binding to the Skp2-Cks1 complex (Hao et al., 2005), suggesting that p21 and p27 are similarly recognized by SCFSkp2. Second, p21 is stabilized in Skp2−/ − cells [Fig. 2 and (Bornstein et al., 2003)], when either Skp2 or Cul1 are downregulated [Fig. 4 and (Sarmento et al., 2005; Yu et al., 1998)] , or when a Cul1 dominant-negative mutant is overexpressed (J. Bloom, R. Piva and MP, unpublished). In addition, silencing of the SCF modulator Cand1 (Y. Xiong, pers. comm.) or the inactivation of a temperature sensitive Nedd8 activating enzyme (Bloom et al., 2003), which is required for optimal SCF activity, also induce a significant accumulation of p21. Third, siRNA oligos targeting Cdh1 (a negative regulator of Skp2) or the forced expression of Skp2 induce the degradation of p21 [Fig. 4A and (Bashir et al., 2004; Wang et al., 2005)]. Despite this evidence, it has been suggested that the accumulation of p21 in Skp2−/ − MEFs is not due to a direct role of Skp2 in mediating p21 degradation but rather is a result of an accumulation of cyclin E (Chen et al., 2004), which would stabilize p21 by a unknown mechanism. However, several data conflict with this explanation: (i) p21 stabilization is also observed under conditions in which cyclin E accumulation is not observed, such as in Skp2−/ −;p27−/ − MEFs [Fig. 4 of (Bornstein et al., 2003), Fig. 3 of (Kossatz et al., 2004) and Fig. 2 of this paper] and after Skp2 silencing (Fig. 5B). (ii) p21 is destabilized in response to the forced expression of cyclin E (Wang et al., 2005). (iii) A p21 mutant deficient for interaction with cyclin-CDK complexes displayed enhanced stability (Cayrol and Ducommun, 1998). (iv) Phosphorylation of p21 by cyclin E-Cdk2 promotes its ubiquitylation and degradation (Bornstein et al., 2003; Wang et al., 2005; Zhu et al., 2005). (v) At G1/S, p21 is degraded in concomitance with an increase in cyclin E levels (see, for example, Fig. 1A).
p21 is targeted by APC/C and SCF at different phases of the cell cycle, similar to other cell cycle regulatory proteins such as Cdc25A (Guardavaccaro and Pagano, 2006). Although our data indicate a role for Skp2 in the proteolysis of p21 during S phase, we cannot exclude that a minor fraction of p21 is also degraded via Skp2 in mitosis, since silencing of Skp2 induces a slight accumulation of p21 in prometaphase cells (Fig. 5B). The lack of p21 accumulation in mitotic Skp2−/ − MEFs (Fig. 2) could be in fact due to compensation by related proteins during development, which cannot occur during acute silencing of Skp2 in somatic cells.
Ubiquitin-mediated and ubiquitin-independent degradation of p21
p21 is a short-lived protein and is degraded by the proteasome. Ubiquitylated forms of both endogenously and exogenously expressed p21 have been detected in cultured cells, and these ubiquitylated forms increase after proteasome inhibition (Bloom et al., 2003; Cayrol and Ducommun, 1998; Coulombe et al., 2004; Fukuchi et al., 2002; Maki and Howley, 1997; Maki et al., 1996; Rousseau et al., 1999; Sheaff et al., 2000; Sugimoto et al., 2006), suggesting that the proteasome degrades p21 marked with ubiquitin chains. In spite of these reports, a ubiquitin-independent, proteasome-dependent mechanism for p21 degradation has been proposed as the sole route for proteolysis (Chen et al., 2004; Sheaff et al., 2000). Initially, this theory was based on the observation that a lysine-less p21 mutant [p21(K0)] is degraded with kinetics similar to those of p21 wild type. Since ubiquitin chains are usually covalently bound to lysine residues of the substrate, there was a potential that this mutant could not be ubiquitylated. In contrast to this idea, two groups have independently shown that the p21(K0) mutant is indeed N-terminal ubiquitylated both in vitro and in vivo (Bloom et al., 2003; Coulombe et al., 2004). In addition, two temperature-sensitive cell lines carrying mutations in the ubiquitin- and Nedd8-activating enzymes, respectively, accumulated p21 at the restrictive temperature [(Bloom et al., 2003; Coulombe et al., 2004) and F. Bernassola and G. Melino, pers. comm.]. Inhibition of p21 ubiquitylation both in vitro (using methylated ubiquitin) and in vivo (using a lysine-less ubiquitin mutant) stabilized endogenous p21, as well as exogenous wild type p21 and a p21(K0) mutant (Bloom et al., 2003; Coulombe et al., 2004). These experiments strongly indicated that the degradation of p21 largely requires its ubquitylation. The regulating function exerted by the APC/CCdc20 and SCFSkp2 ubiquitin ligases on p21 stability further confirms the role of the ubiquitin system in this important biological process.
It is possible, however, that a fraction of cellular p21 is degraded in a ubiquitin-independent manner. For example, free recombinant p21, but not p21 bound to a cyclin-CDK complex is degraded in vitro by the proteasome in the absence of ubiquitylation (Bloom et al., 2003; Liu et al., 2003). This effect is due to the fact that p21 is natively unfolded and, as such, can enter the proteasome pore by free diffusion. Although we have been unable to detect free p21 in extracts from human fibroblasts (Bloom et al., 2003), it is conceivable that some free p21 exists, at least transiently and under certain cellular conditions, and that this fraction is degraded in a ubiquitin-independent manner. Together, these views would reconcile the ubiquitin-independent and the ubiquitin-dependent theories of p21 degradation. In contrast, a hypothesis proposing the ubiquitin-independent degradation of p21 as the only mechanism cannot explain how the proteasome degrades, in a temporally regulated fashion, p21 that is present in different protein complexes in the absence of ubiquitylation, and why, if p21 is ubiquitylated in vivo, its degradation should not require ubiquitylation.
Ubiquitinylation on the N-terminus is sufficient for p21 degradation
It has been reported that endogenous p21 is acetylated at the N-terminus [Fig. 1 of (Chen et al., 2004)]. Based on the assumption that mass spectrometry can quantitatively attest that 100% of a protein is acetylated and that acetylation is irreversible, it has been inferred that endogenous p21 cannot be ubiquitylated at the N-terminus as N-terminal acetylation would block N-terminal ubiquitylation. However, in this experiment the p21 band for the mass spectrometry analysis was excised from a gel, and therefore, slower-migrating, ubiquitylated species of p21 (which potentially may not be acetylated) were not accounted for. Moreover, N-terminal acetylation has been proposed as corroboration for the ubiquitin-independent degradation of p21, based on the belief that ubiquitylation of internal lysines, although present, is not necessary for p21 degradation. However, there is no evidence that ubiquitylation of internal lysines is not required to target endogenous p21 for degradation. Yet, when a p21(K0) mutant is expressed in mammalian cells, N-terminal ubiquitylation appears as one possible way of p21 degradation. Accordingly, a p21(K0) mutant tagged at the C-terminus (and therefore not interfering with N-terminal acetylation) is robustly ubiquitylated in vivo [Figure 5 in (Bloom et al., 2003)]. Thus, ubiquitylation at the N-terminus is sufficient for degradation, though it may not be essential for endogenous p21. Interestingly, Cdc20 and Skp2 are both able to induce the degradation of the p21(K0) mutant (Fig. S5), suggesting that Cdc20 and Skp2 can use N-terminal ubiquitylation of p21 to induce its degradation, at least under these particular circumstances.
Conclusions
p21 contributes to the maintenance of the G2 state by inhibiting Cdk1 activity (Bates et al., 1998; Bunz et al., 1998; Chan et al., 2000; Dulic et al., 1998; Medema et al., 1998; Niculescu et al., 1998). It is well established that Cdk1 promotes its own full activation through two amplification loops, one involving the activation or stabilization of its activators (the Cdc25 phosphatases) and a second promoting the degradation of its inhibitor (the Wee1 kinase) (Guardavaccaro and Pagano, 2006). Our results suggest a positive feedback model where Cdk1, by phosphorylating certain subunits of APC/CCdc20, promotes the activity of APC/CCdc20 (Peters, 2006), consequently triggering the degradation of p21, resulting in a further activation of Cdk1 (Fig. 7). We propose that the degradation of p21 via APC/CCdc20 contributes to the full activation of Cdk1 in early M and prevents mitotic slippage during activation of the spindle assembly checkpoint.
Figure 7. Model of Cdk1 autoactivation via three feedback loops, including the activation of the APC/CCdc20-mediated degradation of p21 occurring in early mitosis.
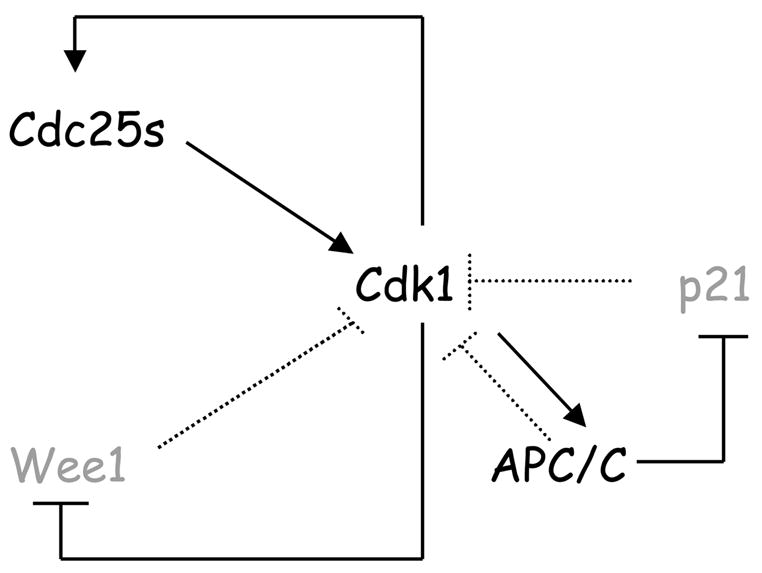
Black signifies activated forms of the respective proteins, gray indicates inactive forms or degraded proteins. Solid lines indicate activating (→) or inhibitory (--I) activities in prometaphase, dotted lines indicate activating or inhibitory activities in different phase of the cell cycle. See text for details.
Experimental Procedures
Cell culture, synchronization and treatment with drugs
T98G cells are revertants from T98 glioblastoma cells that acquired the ability to arrest in G0/G1 in low serum (Dorrello et al., 2006; Lisztwan et al., 1998; Stein, 1979). Normal human fetal lung fibroblasts (IMR-90) were obtained from the American Type Culture Collection. HEK293T, IMR90, U-2 OS, HeLa, and T98G cells and mouse embryonic fibroblasts (MEFs) from Skp2−/ −;p27+/+, Skp2−/ −;p27−/ −, and wild type littermate control mice (Skp2+/+;p27+/+) were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum (FCS). Skp2−/ −;p27+/+, Skp2−/ −;p27−/ − and wild type MEFs were a gift from K. Nakayama (Kyushu University). HCT116 p21+/+ and HCT116 p21−/ − colon cancer cells (Waldman et al., 1996) were a gift from B. Vogelstein (Johns Hopkins University). T98G and IMR90 cells were serum starved in the presence of 0.02% and 0.2% FCS, respectively, as previously described (Dorrello et al., 2006; Lisztwan et al., 1998). Cells were synchronized at G1/S using aphidicolin (2 μg/ml) or a double thymidine block (2.5 mM), as described (Peschiaroli et al., 2006). Synchronization in prometaphase with nocodazole and mitotic shake-off was performed as described (Guardavaccaro et al., 2003). To test the in vivo interaction between different proteins and endogenous p21 the proteasome inhibitor MG132 (10 μM final concentration) was added for 6 hours prior to harvesting the cells. To measure protein half-lives, cells were incubated in the presence of 100 μg/ml cycloheximide (CHX) (Sigma-Aldrich) diluted in 100% ethanol. Cell cycle phases were monitored by flow cytometry, as described (Carrano and Pagano, 2001).
Biochemical methods
Extract preparation, immunoprecipitation, and immunoblotting were previously described (Dorrello et al., 2006; Busino et al., 2007). Rabbit polyclonal antibodies were from Santa Cruz Biotechnology (against p21, Cdc20 and HA-tag). Mouse monoclonal antibodies were from Sigma-Aldrich (anti-Cdc27, FLAG), Lab Vision (anti-Cdh1), Invitrogen (anti- Cul1, Skp1, and Skp2), Covance (anti-HA) and BD Transduction Laboratories (anti- p21 and p27). The rabbit polyclonal antibodies against Cdk1, cyclin A2, cyclin B1, cyclin E1, and phospho-Histone H3 were previously described (Carrano et al., 1999; Peschiaroli et al., 2006). Histone H1 kinase reactions were performed as described (Carrano and Pagano, 2001).
Plasmids
Cdc20 and p21 point mutants were generated using the QuickChange Site-directed Mutagenesis kit (Stratagene). Deletion mutants were generated by a PCR-based approach. All constructs were sequenced prior to use, and primer sequences are available upon request.
Transient Transfections, and Retroviral-mediated Gene Transfer
HEK293T cells were transfected using the calcium phosphate method as described (Bashir et al., 2004). U-2 OS cells were transiently transfected using ExGen 500 reagent (Fermentas), according to the manufacturer's instructions. For experiments investigating the ability of Cdc20 to induce the degradation of p21 (Fig. 4), Cdc20 and p21 were cotransfected in a 3:1 ratio. For retrovirus production, packaging GP-293 cells (Clontech) were transfected with FuGENE transfection reagent (Roche) according to the manufacturer’s instructions. Forty-eight hours after transfection, the virus-containing medium was collected and supplemented with 8 μg/ml polybrene (Sigma). Cells were then infected by replacing the cell culture medium with the viral supernatant for six hours.
In vitro ubiquitylation assay
Ubiquitylation assays were carried out using a procedure based on a combination of previously described protocols (Fang et al., 1998; Kramer et al., 1998). Briefly, prometaphase HeLa cells were washed with PBS and lysed for 30 minutes on ice using Triton buffer (50 mM Tris pH 7.5, 250 mM NaCl, 0.1% Triton X-100, 1 mM EDTA, 1 mM DTT, and phosphatase and protease inhibitors). An anti-Cdc27 antibody (15 μg) was added to 10 mg of cell extract and incubated for approximately 3 hours at 4ºC. Protein G-agarose was then added and incubated for 45 minutes at 4ºC on a rotating wheel. The beads were washed 4 times in Triton buffer and 4 times in QA buffer (10 mM Tris-HCl pH 7.5, 100 mM KCl, 1 mM MgCl2, 0.1 mM CaCl2, 1 mM DTT). In vitro transcribed/translated, unlabelled, constitutively active Cdc20(7A) mutant (Yudkovsky et al., 2000) was added and incubated 30 minutes at 37ºC. The beads were subsequently washed 3 times with QA buffer. The resulting beads were used for two reactions of in vitro ubiquitylation. Ubiquitylation assays were performed in a volume of 10 μl containing 40 mM Tris pH 7.6, 5 mM MgCl2, 1 mM DTT, 2 mM ATP, 1.5 μg/ml E1 (Boston Biochem), 20 μg/ml UbcH10 (Boston Biochem), 2.5 mg/ml ubiquitin (Sigma), 1μM ubiquitin aldehyde, approx. 0.25 μg/ml cyclin A2/Cdk2 complex, 20 μM Staurosporine, and 1 μl of in vitro transcribed/translated [35S] p21 as a substrate. 0.2 mg/ml methyl-ubiquitin was added where indicated. The reactions were incubated at 30ºC for the indicated times and analyzed by SDS-PAGE.
Gene Silencing by Small Interfering RNA
siRNA duplexes were transfected into cells using Oligofectamine (Invitrogen) according to manufacturer’s instructions and as described (Dorrello et al., 2006; Peschiaroli et al., 2006). Forty-eight hours after transfection, cells were collected after further treatments. The siRNA oligonucleotide sequence for Cdh1 was 5′-AAUGAGAAGUCUCCCAGUCAGdTdT-3′ (corresponding to nt 199–219 of human Cdh1 cDNA). The siRNA oligonucleotide sequences for Cdc20 were: 5′-AACGGCAGGACUCCGGGCCGAdTdT-3′ (oligo #1, corresponding to nt 156–170 of human Cdc20 cDNA) and 5′-AAUGGCCAGUGGUGGUAAUGAdTdT-3′ (oligo #2, corresponding to nt 969–989 of human Cdc20 cDNA). The siRNA oligonucleotide sequence for Skp2 was 5′-AAGGGAGUGACAAAGACUUUGdTdT-3′ (corresponding to nt 218–238 of human Skp2 cDNA). A 21 nt siRNA duplex corresponding to a nonrelevant gene (LacZ) was used as control.
Supplementary Material
Acknowledgments
We thank A. Hershko for discussion and suggestions; T. Bashir, F. Bassermann, M. Brandeis, D. Frescas, P. Kaldis, J. Pines, and J. Skaar for critically reading the manuscript; M. Brandeis, G. Draetta, K. Helin, J. Lukas, K. Nakayama, and B. Vogelstein for reagents. MP is grateful to T. M. Thor for continuous support. This work was supported by a fellowship from the Human Frontiers Science Program fellowship to VA, a Spanish Ministry of Culture grant and an American Lung Association fellowship to PGS, an Emerald Foundation grant to DG, and grants from the NIH to MP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquaviva C, Pines J. The anaphase-promoting complex/cyclosome: APC/C. J Cell Sci. 2006;119:2401–2404. doi: 10.1242/jcs.02937. [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 2004;428:190–193. doi: 10.1038/nature02330. [DOI] [PubMed] [Google Scholar]
- Bates S, Ryan KM, Phillips AC, Vousden KH. Cell cycle arrest and DNA endoreduplication following p21Waf1/Cip1 expression. Oncogene. 1998;17:1691–1703. doi: 10.1038/sj.onc.1202104. [DOI] [PubMed] [Google Scholar]
- Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-Mediated Degradation of p21 via N-Terminal Ubiquitinylation. Cell. 2003;115:71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- Bloom J, Pagano M. To be or not to be ubiquitinated? Cell Cycle. 2004;3:138–140. [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. doi: 10.1016/j.cub.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Busino L, Bassermann F, Maiolica A, Lee C, Godinho S, Nolan P, Draetta G, Pagano M. SCFFbxl3 Controls the Oscillation of the Circadian Clock by Directing the Degradation of Cryptochrome Proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M. Skp2 is required for the ubiquitin-mediated degradation of the Cdk-inhibitor p27. Nat Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Carrano AC, Pagano M. Role of the F-box protein Skp2 in adhesion-dependent cell cycle progression. J Cell Biol. 2001;153:1381–1389. doi: 10.1083/jcb.153.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene. 1998;17:2437–2444. doi: 10.1038/sj.onc.1202189. [DOI] [PubMed] [Google Scholar]
- Chan TA, Hwang PM, Hermeking H, Kinzler KW, Vogelstein B. Cooperative effects of genes controlling the G(2)/M checkpoint. Genes Dev. 2000;14:1584–1588. [PMC free article] [PubMed] [Google Scholar]
- Chang BD, Broude EV, Fang J, Kalinichenko TV, Abdryashitov R, Poole JC, Roninson IB. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene. 2000;19:2165–2170. doi: 10.1038/sj.onc.1203573. [DOI] [PubMed] [Google Scholar]
- Chen X, Chi Y, Bloecher A, Aebersold R, Clurman BE, Roberts JM. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1) Mol Cell. 2004;16:839–847. doi: 10.1016/j.molcel.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol. 2004;24:6140–6150. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angiolella V, Mari C, Nocera D, Rametti L, Grieco D. The spindle checkpoint requires cyclin-dependent kinase activity. Genes Dev. 2003;17:2520–2525. doi: 10.1101/gad.267603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M, Squatrito M, Ganoth D, Hershko A, Pagano M, Draetta GF. Dual mode of degradation of Cdc25 A phosphatase. Embo J. 2002;21:4875–4884. doi: 10.1093/emboj/cdf491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–471. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol. 1998;18:546–557. doi: 10.1128/mcb.18.1.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell. 1998;2:163–171. doi: 10.1016/s1097-2765(00)80126-4. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Hagiwara T, Nakamura K, Ichimura S, Tatsumi K, Gomi K. Identification of the regulatory region required for ubiquitination of the cyclin kinase inhibitor, p21. Biochem Biophys Res Commun. 2002;293:120–125. doi: 10.1016/S0006-291X(02)00198-5. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray A, Kirschner M. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, Kudo Y, Boulaire J, Barchi M, Busino L, Donzelli M, Margottin-Goguet F, Jackson PK, Yamasaki L, Pagano M. Control of Meiotic and Mitotic Progression by the F Box Protein beta-Trcp1 In Vivo. Dev Cell. 2003;4:799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D, Pagano M. Stabilizers and destabilizers controlling cell cycle oscillators. Mol Cell. 2006;22:1–4. doi: 10.1016/j.molcel.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell. 2005;20:9–19. doi: 10.1016/j.molcel.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev. 2004;18:2602–2607. doi: 10.1101/gad.321004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ER, Gieffers C, Holzl G, Hengstschlager M, Peters JM. Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr Biol. 1998;8:1207–1210. doi: 10.1016/s0960-9822(07)00510-6. [DOI] [PubMed] [Google Scholar]
- Li Y, Jenkins C, Nichols M, Xiong Y. Cell cycle expression and p53 regulation of the cyclin-dependent kinase inhibitor p21. Oncogene. 1994;9:2261–2268. [PubMed] [Google Scholar]
- Lisztwan J, Marti A, Sutterluty H, Gstaiger M, Wirbelauer C, Krek W. Association of human CUL-1 and ubiquitin-conjugating enzyme CDC34 with the F-box protein p45(SKP2): evidence for evolutionary conservation in the subunit composition of the CDC34-SCF pathway. Embo J. 1998;17:368–383. doi: 10.1093/emboj/17.2.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki C, Howley P. Ubiquitination of p53 and p21 are differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki CG, Huibregtse JM, Howley PM. In vivo ubiquitination and proteasome-mediated degradation of p53(1) Cancer Res. 1996;56:2649–2654. [PubMed] [Google Scholar]
- Medema RH, Klompmaker R, Smits VA, Rijksen G. p21waf1 can block cells at two points in the cell cycle, but does not interfere with processive DNA-replication or stress-activated kinases. Oncogene. 1998;16:431–441. doi: 10.1038/sj.onc.1201558. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, Kitagawa M, Iemura S, Natsume T, Nakayama KI. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell. 2004;6:661–672. doi: 10.1016/s1534-5807(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Niculescu AB, 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtoshi A, Maeda T, Higashi H, Ashizawa S, Hatakeyama M. Human p55(CDC)/Cdc20 associates with cyclin A and is phosphorylated by the cyclin A-Cdk2 complex. Biochem Biophys Res Commun. 2000;268:530–534. doi: 10.1006/bbrc.2000.2167. [DOI] [PubMed] [Google Scholar]
- Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene. 2005;24:2787–2795. doi: 10.1038/sj.onc.1208611. [DOI] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Peters JM. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Reis A, Levasseur M, Chang HY, Elliott DJ, Jones KT. The CRY box: a second APCcdh1-dependent degron in mammalian cdc20. EMBO Rep. 2006;7:1040–1045. doi: 10.1038/sj.embor.7400772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau D, Cannella D, Boulaire J, Fitzgerald P, Fotedar A, Fotedar R. Growth inhibition by CDK-cyclin and PCNA binding domains of p21 occurs by distinct mechanisms and is regulated by ubiquitin-proteasome pathway. Oncogene. 1999;18:4313–4325. doi: 10.1038/sj.onc.1202686. [DOI] [PubMed] [Google Scholar]
- Sarmento LM, Huang H, Limon A, Gordon W, Fernandes J, Tavares MJ, Miele L, Cardoso AA, Classon M, Carlesso N. Notch1 modulates timing of G1-S progression by inducing SKP2 transcription and p27 Kip1 degradation. J Exp Med. 2005;202:157–168. doi: 10.1084/jem.20050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Neutzner M, Mocker D, Seufert W. Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. Embo J. 2001;20:5165–5175. doi: 10.1093/emboj/20.18.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell. 2000;5:403–410. doi: 10.1016/s1097-2765(00)80435-9. [DOI] [PubMed] [Google Scholar]
- Stein GH. T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979;99:43–54. doi: 10.1002/jcp.1040990107. [DOI] [PubMed] [Google Scholar]
- Sugimoto M, Gromley A, Sherr CJ. Hzf, a p53-responsive gene, regulates maintenance of the G2 phase checkpoint induced by DNA damage. Mol Cell Biol. 2006;26:502–512. doi: 10.1128/MCB.26.2.502-512.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, Ng TM, Matyskiela ME, Carroll CW, Morgan DO, Toczyski DP. An architectural map of the anaphase-promoting complex. Genes Dev. 2006;20:449–460. doi: 10.1101/gad.1396906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton BR, Toczyski DP. Precise destruction: an emerging picture of the APC. Genes Dev. 2006;20:3069–3078. doi: 10.1101/gad.1478306. [DOI] [PubMed] [Google Scholar]
- Waldman T, Lengauer C, Kinzler KW, Vogelstein B. Uncoupling of S phase and mitosis induced by anticancer agents in cells lacking p21. Nature. 1996;381:713–716. doi: 10.1038/381713a0. [DOI] [PubMed] [Google Scholar]
- Wang W, Nacusi L, Sheaff RJ, Liu X. Ubiquitination of p21Cip1/WAF1 by SCFSkp2: substrate requirement and ubiquitination site selection. Biochemistry. 2005;44:14553–14564. doi: 10.1021/bi051071j. [DOI] [PubMed] [Google Scholar]
- Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG., Jr Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428:194–198. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- Yu ZK, Gervais J, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkovsky Y, Shteinberg M, Listovsky T, Brandeis M, Hershko A. Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem Biophys Res Commun. 2000;271:299–304. doi: 10.1006/bbrc.2000.2622. [DOI] [PubMed] [Google Scholar]
- Zhu H, Nie L, Maki CG. Cdk2-dependent Inhibition of p21 stability via a C-terminal cyclin-binding motif. J Biol Chem. 2005;280:29282–29288. doi: 10.1074/jbc.M407352200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.