

Abstract
Background
While the inflammatory response is a prerequisite for wound healing, excessive activation of the innate immune system can induce epithelial cell damage and apoptosis, which may further compromise dermal integrity. In a non infectious burn wound model, we previously demonstrated that topical inhibition of p38 MAPK, an important inflammatory signaling pathway, attenuated epithelial cell damage and apoptosis. We now questioned whether attenuating local inflammation would weaken bacterial wound resistance and compromise host defense.
Methods
Rats received 30% total body surface area burn, and the wound was treated with topical application of a p38 MAPK inhibitor or vehicle. Twenty-four hours after injury, burn wounds were inoculated with Pseudomonas aeruginosa. Forty-eight hours post-injury, animals were sacrificed and the burn wound was analyzed.
Results
Inoculating burn wounds induced a significant bacterial growth. Dermal inflammatory changes were markedly accentuated in the inoculated animals. Topical p38 MAPK inhibition reduced burn wound pro-inflammatory cytokine expression and PMN sequestration with or without bacterial inoculation. Interestingly, the bacterial wound growth was significantly attenuated in animals treated with topical p38 MAPK inhibitor.
Conclusion
Topical p38 MAPK inhibition attenuated wound inflammation without interfering with bacterial host defense. Attenuation of excessive burn wound inflammatory signaling may prevent secondary damage of the dermal barrier and reduce the growth of opportunistic pathogens.
Keywords: Inflammation, Cytokines, Signal Transduction, Infection, Burns, Wound healing, Mitogen activated protein kinase (MAPK), p38, P. aeruginosa, immunomodulation
INTRODUCTION
Therapeutic advances such as early surgical debridement and grafting as well as organ supportive measures have increased survival rates of critically injured burn patients.1, 2 However, burn wound infection and sepsis remain leading causes of death among hospitalized burn victims.3, 4 In this scenario, Pseudomonas aeruginosa - a gram negative opportunistic pathogen, is of a special relevance due to its high risk of infection in burn injured patients and its growing antibiotic resistance.5, 6 P. aeruginosa is known to be an opportunistic infection in the compromised host.7, 8 Thus, burn-specific factors, such as size and depth of the injury determine the propensity for wound infections via disruption of the dermal integrity, allowing burn wounds to become invaded by sequential waves of pathogens. Once established, P. aeruginosa wound infection and sepsis is associated with mortality rates beyond 50 percent.9, 10
Inflammation is a key component of host immune response to injurious stress. Resident cell expression of inflammatory mediators activates and recruits neutrophils, the cellular first line of defense. A key player in epithelial inflammatory and apoptotic signaling is p38 MAPK, a member of the primordial family of mitogen activated kinases11. We previously demonstrated that topical inhibition of p38 MAPK significantly reduced burn-wound expression of pro-inflammatory cytokines and chemokines.12, 13 Additionally, we found that dermal neutrophil sequestration, microvascular injury, as well as p38 MAPK associated epithelial apoptosis were reduced.
The excessive production of pro-inflammatory mediators, such as TNF-α and NO, may have deleterious effects on wound healing and can promote bacterial growth.14–17 Furthermore, inflammatory upregulation of proteinases such as matrix metalloproteinases have been shown to enhance tissue injury and support bacterial invasion.8, 18–20 One can reason that dampening the initial surge of burn wound inflammatory and apoptotic signaling could have a protective effect on dermal integrity and thereby reduce growth of opportunistic pathogens. However, inhibition of local inflammatory response may reduce chemo-attraction of immunocompetent cells, interfere with bacterial killing, and compromise host defense. Our current study was designed to investigate the effect of topical p38 MAPK inhibition on inflammatory response and bacterial growth in a burn wound inoculated with P. aeruginosa.
In this study, we demonstrated that inflammatory mediator levels and neutrophil sequestration were significantly higher in burn wounds inoculated with P. aeruginosa as compared to the non-inoculated control, indicating an inflammatory response to bacterial infection. Topical p38 MAPK inhibition reduced burn wound expression of inflammatory mediators and neutrophil sequestration, with or without bacterial inoculation. Interestingly, we found a significant reduction of P. aeruginosa growth in the inoculated wounds that received topical p38 MAPK inhibitor.
METHODS
Reagents
Unless otherwise indicated, all reagents were purchased from Sigma Aldrich (St. Louis, MO).
Experimental Animals
All experiments were performed in accordance with the guidelines set forth by the National Institutes of Health for care and use of animals. Approval for the experimental protocol was obtained from the University Committee on Use and Care of Animals (UCUCA) at the University of Michigan. Male Sprague Dawley rats (Harlan) weighing 220–240g were allowed to acclimate for at least one week prior to being used in any experiment. Animals had unrestricted access to standard rat chow and water ad libitum throughout the whole course of the study.
Experimental Procedure
Animals were anesthetized by i.p. injection of 40mg/kg sodium pentobarbital (Nembutal®; Abbott Laboratories, North Chicago, IL). The dorsal area of the animals was clipped and animals were placed in a fenestrated insulating mold calculated to expose approximately 30 % total body surface area (TBSA)21. After immersion in 60° C water for 25 sec, the burn area was scrubbed using sterile gauze and rinsed with sterile 0.9 % saline. As previously demonstrated, this burn model induces epidermal necrosis, but the skin appendage epithelial cells have no necrosis with approximately 50% apoptotic rate in 24-hours, and the burn wounds remain vascular and pink12. Animals were subsequently resuscitated with 4 mL/%TBSA/kg Ringer’s Lactate i.p. and received 0.3 mg/kg BW buprenorphine s.c.(Buprenex®, Reckitt & Coleman Pharmaceuticals, Richmond, VA). Animals were housed individually and had access to water and standard chow ad libitum. Sham animals were subjected to identical procedure and resuscitation but were immersed in room temperature water.
For topical inhibition of p38 MAPK, 1 mL of 10−4M of SB202190 (Calbiochem, San Diego, CA) - a specific p38α/β inhibitor- was applied to the wound immediately after injury, and repeated twice every 8h during the first 24h. We had determined the dosing and mode of application in previous studies.12 Sham and burn controls received the drug vehicle consisting of a 4:1 mixture of acetone/olive oil (AO). Subsequently, wounds were sealed using an occlusive dressing (Tegaderm®; 3M Health Care, St. Paul, MN).
For bacterial wound inoculation, animals were anesthetized at times 24h after burn injury for topical application of 106 colony forming units (CFU) of P. aeruginosa in 100 μL of 1xPBS on sterile gauze, followed by sterile occlusive dressing. Control groups received identical burn trauma and treatment with either vehicle or SB202190 but received no bacterial inoculation. After additional 24h, animals were euthanized; skin samples were harvested, weighed and homogenized. Skin homogenates were serially diluted and plated in triplicates on blood agar plates (Becton Dickinson, Franklin Lakes, N.J.). Blinded culture plates were incubated for 24h at 37°C and CFU were counted. Pathogens were confirmed using the Oxidase method (BBL Taxo Discs, Becton Dickinson, Franklin Lakes, N.J.).22 Residual skin and organs were snap frozen in liquid nitrogen until further processing or directly processed according to protocol.
Bacterial Culture
P. aeruginosa isolates from burn patients were kindly provided the Department of Pathology, University of Michigan. Bacteria were grown overnight in 50mL Trypticase soy broth (Becton Dickinson, Franklin Lakes, N.J.) at 37°C and constant shaking at 275 rpm. An aliquot of resulting stationary-phase cultures was transferred into 30 mL of Trypticase soy broth and incubated at 37°C for 2.5h to reach log phase. Subcultures were transferred to a 50mL conical polystyrene tube and centrifugation for 10 min at 4°C to induce log phase growth. Resulting bacterial pellet was washed with sterile phosphate-buffered saline (PBS), and resuspended in 5 mL ice-cold PBS. The optical density of 100 μL of suspension was measured at 620 nm and bacterial concentrations were calculated using the formula OD620 x 2.5 x 108.
ELISA
100 mg of tissue was homogenized in 1 mL ice cold lysis buffer (50mL PBS 1X supplemented with protease inhibitor (Complete X®, Roche, Indianapolis, IN) and 50 μL Triton X). Subsequently, homogenates were centrifuged at 3000g for 5min at 4°C, supernatant were stored at −80°C until use. Rat TNF-α, IL-6, macrophage-inflammatory protein 2 (MIP-2), and matrix metalloproteinase 2 (MMP-2) were measured using prefabricated ELISA kits according to manufacturer protocol (R&D Systems, Inc. Minneapolis, MN). Plates were read at 450 and 540 nm and cytokine concentrations were calculated using a 4 point standard curve and expressed as pg/mL.
NO-Measurement
Tissue NO concentrations were measured via the stable oxidization intermediate nitrite using a prefabricated assay kit based on the Griess reaction (StressXpress, Stressgene, Ann Arbor, MI). Nitrite concentration was measured via the colored azo-dye product of the Griess reaction absorbing visible light at 540 nm via spectrophotometry according to manufacturer protocol. Briefly: 100mg tissue was homogenized in 1mL of HEPES based reaction buffer. After adding 50uL of Griess reagent to supernatants; resulting azo-dye concentration was read against nitrite standard curve and reaction buffer blanks at 540nm after 10 minutes of reaction time. Concentrations were expressed as μmol NO2−/L.
Detection of neutrophil sequestration (Myeloperoxidase (MPO) assay)
100 mg of fresh tissue was homogenized and sonicated (Branson Sonifier 250, Danbury, CT) on ice in homogenization buffer for membrane disruption and MPO release. Samples were centrifuged for 15 min. at 16,000 g and 4°C. Samples were analyzed in duplicates after addition substrate buffer (4.39 mL 1.0M monobasic, 0.615 mL 1.0M dibasic potassium phosphate, 83.3 μL 0.3% H2O2 and 0.834 mL ortho-dianisidine HCL, qs to 50 mL with sterile water). A plate reader was programmed to read kinetic OD changes every 10 seconds at 465 nm for 90 seconds total. Data was expressed as OD/mg tissue/minute.
Statistical analysis
All values are expressed as mean ± SEM. Data were analyzed using Student’s t-test and one way analysis of variance (ANOVA) followed by Bonferroni post-hoc testing (GraphPad Software, San Diego, CA). Statistical significance was set at a p-value of <0.05.
RESULTS
Burn injury followed by P. aeruginosa inoculation induces p38 MAPK dependent dermal expression of pro-inflammatory cytokines
Burn animals demonstrated a significant rise of dermal inflammatory cytokines IL-6 and TNF-α compared to sham animals (Figure 1). Bacterial burn wound inoculation resulted in higher dermal pro-inflammatory cytokine expression when compared to non-inoculated burn wounds, demonstrating increased inflammatory response to bacterial inoculation. Dermal IL-6 was significantly increased in P. aeruginosa inoculated burn animals as compared to non-inoculated burn animals (Figure 1). A similar trend was seen with dermal TNF-α expression although it did not reach statistical significance. In contrast, bacterial inoculation of intact skin in sham animals led to no significant rise in either IL-6 or TNF-α expression when compared to non-inoculated sham animals.
Fig. 1. Attenuating p38 MAPK signaling reduces proinflammatory mediator expression in burn wounds.
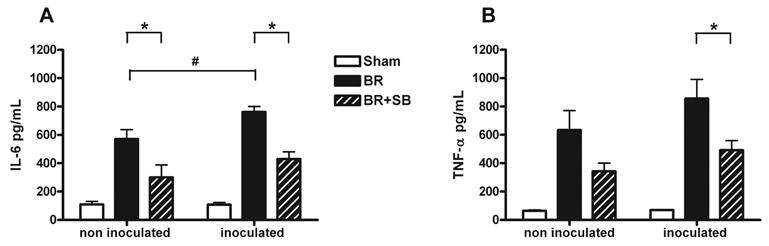
Burn wounds were treated with topical vehicle (BR) or topical SB202190 (BR+SB), a specific p38 inhibitor. After 24 hours, some of the animals were inoculated with 106 CFU P. aeruginosa (inoculated) and a matched additional group remained non-inoculated. Skin was harvested 48 hours post burn, for ELISA of IL-6 (A) and TNF-α (B). Burn injury induced a significant pro-inflammatory cytokine expression compared to sham. Dermal IL-6 expression was significantly increased in inoculated vs. non inoculated burn animals (Fig. 1A # indicates p<0.05). Inoculated groups showed significant reduction of both cytokines after topical SB202190 application (BR vs. BR+SB, * indicates p<0.05, ANOVA), whereas in non inoculated animals only dermal IL-6 expression was significantly attenuated. Graphs indicate mean ± SEM, n=12/group.
Topical p38 MAPK inhibition reduced burn wound pro-inflammatory cytokine expression, with or without bacterial inoculation. In animals receiving P. aeruginosa inoculation, topical application of SB202190 significantly (p<0.5) inhibited dermal IL-6 (Fig. 1A) and TNF- α (Fig. 1B) expression, compared to topical vehicle only. In the non-inoculated animals, TNF-α was markedly reduced, and dermal IL-6 expression was significantly reduced in burn animals receiving topical inhibitor compared to topical vehicle only.
Attenuation of inflammatory signaling in infected burn wounds reduces dermal neutrophil sequestration and expression of neutrophil chemoattractant MIP-2
Burn injury induced a significant MIP-2, a potent neutrophil chemoattractant, expression (Figure 2A, BR versus sham). In both inoculated and non-inoculated groups, p38 MAPK inhibition significantly reduced the dermal expression of MIP-2 (Figure 2A, BR versus BR+SB, * indicates p<0.05).
Fig. 2. Attenuating burn wound p38 MAPK inflammatory signaling significantly reduces dermal neutrophil sequestration and chemokine expression.
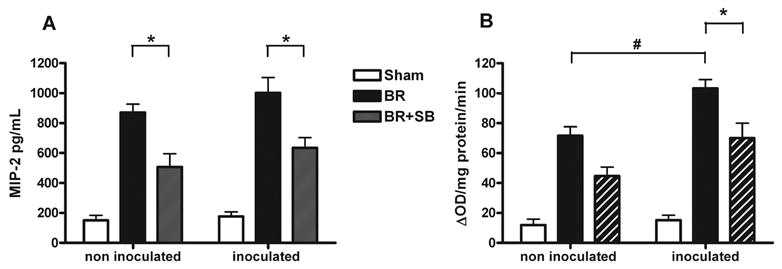
Burn wounds were treated with topical vehicle (BR) or topical SB202190 (BR+SB), a specific p38 inhibitor. After 24 hours, some of the animals were inoculated with 106 CFU P. aeruginosa (inoculated) and a matched additional group remained non-inoculated. Skin was harvested 48 hours post burn, for macrophage-inflammatory protein 2 (MIP-2) ELISA (A) and Myeloperoxidase (MPO) activity assay (B). There was significant reduction in dermal MIP-2 expression (A) in BR+SB versus BR in both inoculated and non-inoculated animals (* indicates p<0.05). Dermal MPO activity was significantly higher in the P. aeruginosa inoculated burn group compared to non-inoculated burn controls (Fig. 2A # indicates p<0.05). There was significant reduction in dermal MPO activity (A) in burn wounds receiving topical p38 MAPK inhibitor (BR+SB) versus vehicle only (BR) in the inoculated animals (* indicates p<0.05) and marked reduction for non-inoculated groups. The graphs express mean ± SEM for n=12/group.
Burn injury induced a significant neutrophil sequestration, which was further amplified with subsequent bacterial inoculation (Figure 2B, comparing inoculated versus non-inoculated burn wounds, # indicates p<0.05). In the inoculated group, topical p38 MAPK inhibition significantly inhibited dermal wound MPO activity when compared to burn controls receiving the vehicle only (Figure 2B, BR versus BR+SB, * indicates p<0.05). In non-inoculated group, there was a lower MPO activity after topical p38 MAPK inhibition, but it did not reach statistical significance.
Topical p38 MAPK inhibition reduced bacterial colony formation in burn wounds
We next examined the effect of topical p38 MAPK inhibition on bacterial growth after P.aeruginosa inoculation. Skin samples obtained from inoculated sham animals showed minimal growth (Figure 3). In contrast, abundant P. aeruginosa growth was seen in the inoculated burn animals treated with vehicle only (Figure 3, BR versus shame, p<0.05). There was a significant reduction in colony forming units in plated skin homogenates from burn animals treated with topical SB202190 versus vehicle only (Figure 3B, BR 1.8x108±1x107 versus BR+SB 3.1x107±1.2x107; p<0.001, ANOVA).
Fig. 3. Topical attenuation of burn activated p38 MAPK reduces P. aeruginosa growth on burn wounds.
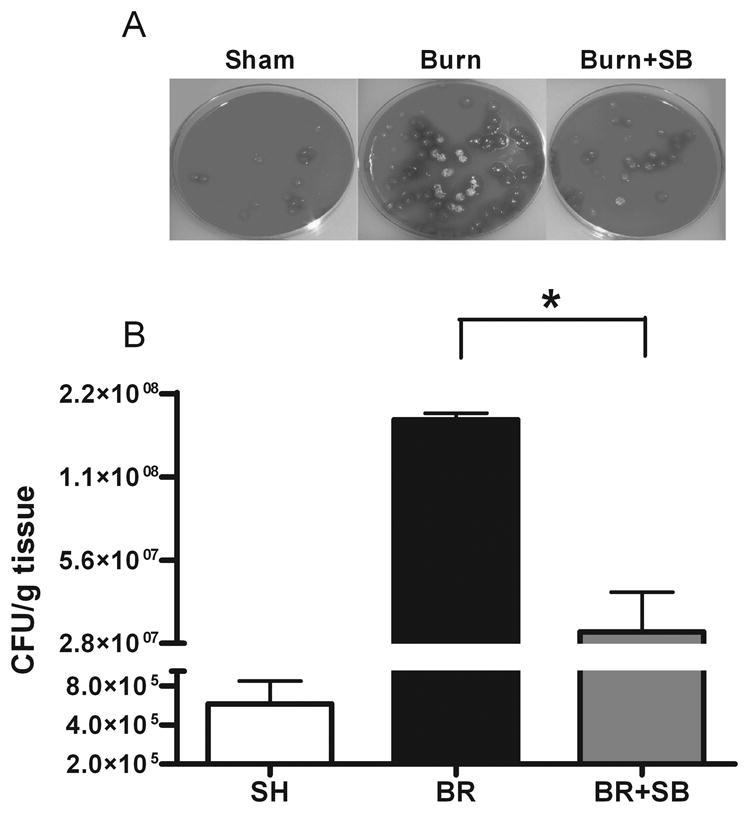
Male Sprague Dawley rats received burn or sham (SH) treatment, and burn wounds were treated with topical vehicle (BR) or topical SB202190 (BR+SB). Twenty-four hours post-injury, animals were inoculated with 106 CFU P. aeruginosa. After additional 24 hours, animals were euthanized; skin samples were homogenized, plated (A), and CFU were counted (B). There was only minimal pathogen growth in the SH animals. Animals with burn injury showed a robust growth of P. aeruginosa, which was significantly reduced in the BR+SB group versus BR. Graph B demonstrates mean ± SEM (Units: CFU/g tissue, * indicates p<0.01, BR vs. BR+SB) for n=10/group.
Topical p38 MAPK inhibition reduces burn wound NO production
Nitric oxide production by iNOS has been implicated in host responses to bacterial wound colonization.23 We, therefore, sought to examine the effect of topical p38 MAPK inhibition on NO production by measuring local nitrate concentrations using the Griess reaction. There was an upregulation of nitrite in burn wounds, which was significantly attenuated in response to application of topical p38 MAPK inhibitor (Fig. 4A) (BR 52±5 vs. BR+SB 31±3; p<0.01).
Fig. 4. Topical p38 MAPK inhibition reduces burn induced dermal NO production but has no effect on burn induced upregulation of dermal MMP-2.
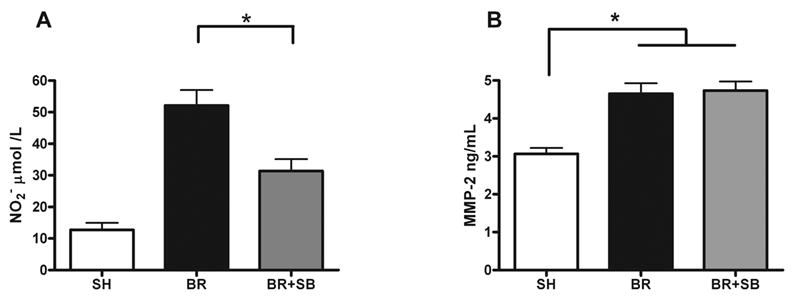
Male Sprague Dawley rats received burn or sham (SH) treatment, and burn wounds were treated with topical vehicle (BR) or topical SB202190 (BR+SB). Twenty-four hours post-injury, animals were inoculated with 106 CFU P. aeruginosa. After additional 24 hours, skin was harvested for NO concentration measurement (A) and matrix metalloproteinase 2 (MMP-2) ELISA (B). There was a significant increase in dermal NO production in burn animals receiving topical vehicle versus sham (A). Topical application of a p38 MAPK inhibitor significantly reduced dermal concentration of NO when compared to burn controls receiving vehicle only (A) (* indicates p<0.01, BR vs. BR+SB). Burn injury resulted in significant upregulation of dermal MMP-2 expression (B) (* indicates p<0.01, BR vs. SH and BR+SB vs. SH). Topical treatment with p38 inhibitor had no effect on this burn induced dermal MMP-2 expression (B). Graphs indicate mean ± SEM for n=8/group.
Burn injury induces p38 MAPK independent dermal MMP-2 expression
Inflammatory activation of matrix metalloproteinases is known to play an integral role in wound physiology, and there is a subtle balance that exists between necessary bed digestion for healing and injurious wound breakdown.18 There was a significant upregulation of dermal MMP-2 expression in inoculated burn animals when compared to sham controls (Figure 4B). This surge in dermal MMP-2 expression was independent of topical p38 inhibitor treatment. As such neither vehicle nor topical p38 MAPK inhibitor application reduced dermal MMP-2 expression.
DISCUSSION
Burn injury dramatically alters host immune responses.24, 25 Additionally, thermal injury results in a burn wound micro milieu rich in degraded proteins, providing for excellent pathogen growth.10, 26 These conditions are ideally suited for opportunistic pathogens such as P. aeruginosa which pose considerable clinical problems.3 P. aeruginosa is equipped with multiple virulence factors such as endo- and exotoxins, proteinases and pili which induce inflammation, tissue destruction, and provide for pathogen-motility. All of these factors facilitate wound invasion and challenge treatment once infection and systemic sepsis is established.27, 28 Given increasing pathogen resistance to antibiotic treatment, alternate antimicrobial strategies and prophylactic measures to reduce host receptiveness for bacterial invasion after burn injury are needed.29, 30
We previously demonstrated that topical p38 MAPK inhibition reduced burn wound inflammation and hair follicle cell apoptosis.12, 13 Several potentially beneficial aspects can be derived from this strategy, such as attenuating dermal injury from excessive inflammatory response and attenuating epithelial tissue loss mediated by proapoptotic p38 MAPK signaling. Dampening excessive inflammation and epithelial apoptosis may preserve skin appendages - structures that are required for wound reepithelialization and maintenance of dermal barrier function- and decrease the ultimate burn wound depth.
It is well known that p38 MAPK is central to the innate immune response. This pathway up-regulates pro-inflammatory cytokine production via enhanced target gene expression, as well as posttranscriptional mRNA stabilization, enhancing efficiency of cytokine expression.31–34 Neutrophil attraction, activation, and degranulation are p38 MAPK dependent processes.35–37 Additionally, p38 MAPK signaling pathway can inactivate neutrophil apoptosis via caspase 3 and 8 phosphorylation and thereby extend neutrophil half life.38, 39 Since p38 MAPK plays an important role in the innate immune response, inhibiting this pathway may weaken the first line in cellular immunity and increase the risk of burn wound bacterial infection. Therefore, we used a burn wound infection model to study the effect of topical p38 MAPK inhibition on wound response to bacterial challenge. In our rat burn model, we induced a dermal wound with epidermal necrosis, but the skin appendage-epithelial cells remained viable with approximately 50% apoptotic rate in 24-hours. Since rats heal by burn wound contraction and not reepithelialization, this may not be a good model to study wound healing; however, the model is appropriate for investigating innate immune response to burn wound infection.
Similar to our previous studies,12, 13 the current data affirms that attenuating dermal p38 MAPK signaling via application of topical inhibitor had pronounced effects on dermal inflammatory responses. In both inoculated and non-inoculated wounds, there was inhibition of dermal IL-6, TNF-α, MIP-2 chemokine expression, and neutrophil sequestration.
Bacterial inoculation of burn wounds accentuated dermal inflammation and cytokine production, suggesting that bacterial inoculation poses an additional inflammatory challenge exceeding the ordinary burn wound response. Thus, in this model the bacterial inoculation and growth was not merely colonization but represented an infectious challenge. Some of the increased host inflammatory response may be due to the bacterial quorum-sensing system. In many Gram-negative bacteria, such as Pseudomonas aeruginosa, virulence factor production is linked to the quorum-sensing, a system of bacterial communication that uses diffusible N-acyl-L-homoserine lactones to co-ordinate gene expression within a population.40 The quorum sensing molecules are not only active in the communication among bacteria, but also induce a proinflammatory phenotype in the host innate immune cells, such as macrophages.41 Quorum sensing was found to be responsible for a surge in the inflammatory cytokine production following thermal injury and P. aeruginosa infection.8 Recent findings have demonstrated that quorum sensing molecules directly activate macrophages via a p38 MAPK dependent pathway.41 Inhibition of this pathway with topical p38 MAPK inhibitor may be another potential mechanism for attenuation of inflammatory response in our burn-infection model.
Interestingly, topical application of p38 MAPK inhibitor not only attenuated wound inflammation, as expected, but also significantly reduced wound bacterial growth. This finding was contrary to our initial hypothesis in favor of increased bacterial growth based on the elemental role that p38 MAPK plays in recruitment of immunocompetent cells. While we currently do not know the mechanism, several explanatory avenues can be discussed based on our data and on findings of other investigators.
Preservation of skin barrier is the most effective mean of avoiding dermal infection. Our study demonstrates that burn injury leads to tremendous changes in growth patterns of P.aeruginosa on skin. While intact skin is resistant to pathogen growth, thermal injury profoundly disturbs this dermal shield, and there was abundant growth of P.aeruginosa in burned skin. Secondary tissue destruction due to an excessive inflammatory response may be an important factor that compromises the physical dermal barrier. In addition, based on our previous work,12 attenuating p38 MAPK mediated epithelial apoptosis may decrease the ultimate burn wound depth. Therefore, attenuation p38 MAPK may preserve the dermal barrier and decrease the risk for wound infection.
It is known that acute and chronic inflammatory activation of protein degrading enzymes such as the matrix metalloproteinases leads to ongoing wound bed degradation, further destabilizing the skin barrier and favoring invasion of opportunistic pathogens.18, 42 Although we found a significant wound expression of MMP-2 – a metalloproteinase heavily involved in inflammatory matrix degradation - this expression appeared to be p38 MAPK independent. Matrix metalloproteinase are regulated at transcription and post-translational modification, pro-MMP to active form. There is data to suggest that in skin inflammatory diseases cytokine-induced activation of pro-MMP is an important regulatory mechanism.43, 44 We only looked at the total expression of MMP-2 and not the activation process. Whether p38 MAPK is an important regulatory mechanism for MMP-2 activation in our model remains to be elucidated. We also did not look at other matrix metalloproteinase, which may be important.
Recent findings, demonstrating increased bacterial growth in response to high cytokine concentration, suggest another potential mechanism that may explain our data. In vitro studies have shown a U-shaped bacterial growth pattern upon exposure to varying concentrations of inflammatory cytokines IL-6, IL-1β and TNF-α.17, 45 Moderate increases in these cytokines improved bacterial clearance, but bacterial growth was found to be increased under high cytokine concentrations. It appears that pathogens such as P. aeruginosa may acquire the phenotypic ability to use cytokines as growth factors.45 While these in vitro data may not clearly translate to our in vivo model, it is interesting to note that for P. aeruginosa, IL-6 and TNF-α concentration conducive for bacterial growth was in the 800–1200 pg range, a concentration which matches dermal cytokine levels in our infected burn wounds (Figure 1). Application of topical p38 MAPK inhibitors reduced IL-6 and TNF-α concentration to less than 600 pg/ml, which may explain the decreased growth of P. aeruginosa in our treatment group.
In summary, blunting the inflammatory signaling in burn wounds via topical p38 MAPK inhibition attenuates burn wound growth of the opportunistic pathogen P. aeruginosa. While we currently do not know the exact pathways responsible for this decreased bacterial growth, the mechanism is probably a combination of factors already discussed. Topical inhibition of p38 MAPK may preserve the dermal barrier by attenuating excessive inflammation and/or directly inhibiting apoptotic signaling in the epithelial cells. Inhibition of inflammatory signaling may also directly reduce bacterial growth by attenuating cytokine production. Emerging knowledge that excessive host inflammatory signaling and cytokine expression may directly increase the virulence factor and the growth of invading pathogens highlights the importance of immunomodulatory strategies in the early management of burn injuries.
Acknowledgments
This work was supported by grants from the NIH: K08-GM069437, (PI: S.A.), the Surgical Infection Society Foundation and the American Association for the Surgery of Trauma.
Footnotes
This manuscript was presented, in part, at the Society of University Surgeons Sixty-six annual meeting at Nashville, Tennessee, February 2005, and Surgical Infection Society 25th annual meeting, Miami Beach, Florida, May 5 2005.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shirani KZ, Vaughan GM, Mason AD, Jr, Pruitt BA., Jr Update on current therapeutic approaches in burns. Shock. 1996;5(1):4–16. [PubMed] [Google Scholar]
- 2.Ipaktchi K, Arbabi S. Advances in burn critical care. Crit Care Med. 2006;34(9 Suppl):S239–44. doi: 10.1097/01.CCM.0000232625.63460.D4. [DOI] [PubMed] [Google Scholar]
- 3.Tredget EE, Shankowsky HA, Rennie R, Burrell RE, Logsetty S. Pseudomonas infections in the thermally injured patient. Burns. 2004;30(1):3–26. doi: 10.1016/j.burns.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 4.Shankowsky HA, Callioux LS, Tredget EE. North American survey of hydrotherapy in modern burn care. J Burn Care Rehabil. 1994;15(2):143–6. doi: 10.1097/00004630-199403000-00007. [DOI] [PubMed] [Google Scholar]
- 5.McManus AT, Mason AD, Jr, McManus WF, Pruitt BA., Jr Twenty-five year review of Pseudomonas aeruginosa bacteremia in a burn center. Eur J Clin Microbiol. 1985;4(2):219–23. doi: 10.1007/BF02013601. [DOI] [PubMed] [Google Scholar]
- 6.Santucci SG, Gobara S, Santos CR, Fontana C, Levin AS. Infections in a burn intensive care unit: experience of seven years. J Hosp Infect. 2003;53(1):6–13. doi: 10.1053/jhin.2002.1340. [DOI] [PubMed] [Google Scholar]
- 7.Carney SA, Dyster RE, Jones RJ. The invasion of burned skin by Pseudomonas aeruginosa. Br J Dermatol. 1973;88(6):539–45. doi: 10.1111/j.1365-2133.1973.tb08016.x. [DOI] [PubMed] [Google Scholar]
- 8.Rumbaugh KP, Hamood AN, Griswold JA. Cytokine induction by the P. aeruginosa quorum sensing system during thermal injury. J Surg Res. 2004;116(1):137–44. doi: 10.1016/j.jss.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 9.Wurtz R, Karajovic M, Dacumos E, Jovanovic B, Hanumadass M. Nosocomial infections in a burn intensive care unit. Burns. 1995;21(3):181–4. doi: 10.1016/0305-4179(95)80005-9. [DOI] [PubMed] [Google Scholar]
- 10.Pruitt BA, Jr, McManus AT, Kim SH, Goodwin CW. Burn wound infections: current status. World J Surg. 1998;22(2):135–45. doi: 10.1007/s002689900361. [DOI] [PubMed] [Google Scholar]
- 11.Dillman JF, 3rd, McGary KL, Schlager JJ. An inhibitor of p38 MAP kinase downregulates cytokine release induced by sulfur mustard exposure in human epidermal keratinocytes. Toxicol In Vitro. 2004;18(5):593–9. doi: 10.1016/j.tiv.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Hemmila MR, Su GL, et al. Topical p38MAPK Inhibition Reduces Dermal Inflammation and Epithelial Apoptosis in Burn Wounds. Shock. 2006;26(2):201–209. doi: 10.1097/01.shk.0000225739.13796.f2. [DOI] [PubMed] [Google Scholar]
- 13.Ipaktchi K, Mattar A, Niederbichler AD, Hoesel LM, Vollmannshauser S, Hemmila MR, et al. Attenuating burn wound inflammatory signaling reduces systemic inflammation and acute lung injury. J Immunol. 2006;177(11):8065–71. doi: 10.4049/jimmunol.177.11.8065. [DOI] [PubMed] [Google Scholar]
- 14.Henson PM. Dampening inflammation. Nat Immunol. 2005;6(12):1179–81. doi: 10.1038/ni1205-1179. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Soejima K, Nozaki M, Tanabe Y, Sakurai H. Effect of early wound excision on changes in plasma nitric oxide and endothelin-1 level after burn injury: an experimental study in rats. Burns. 2004;30(8):793–7. doi: 10.1016/j.burns.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Ruckert R, Lindner G, Bulfone-Paus S, Paus R. High-dose proinflammatory cytokines induce apoptosis of hair bulb keratinocytes in vivo. Br J Dermatol. 2000;143(5):1036–9. doi: 10.1046/j.1365-2133.2000.03784.x. [DOI] [PubMed] [Google Scholar]
- 17.Meduri GU, Kanangat S, Stefan J, Tolley E, Schaberg D. Cytokines IL-1beta, IL-6, and TNF-alpha enhance in vitro growth of bacteria. Am J Respir Crit Care Med. 1999;160(3):961–7. doi: 10.1164/ajrccm.160.3.9807080. [DOI] [PubMed] [Google Scholar]
- 18.Elkington PT, O'Kane CM, Friedland JS. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol. 2005;142(1):12–20. doi: 10.1111/j.1365-2249.2005.02840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sparkes BG. Immunological responses to thermal injury. Burns. 1997;23(2):106–13. doi: 10.1016/s0305-4179(96)00089-7. [DOI] [PubMed] [Google Scholar]
- 20.Ravage ZB, Gomez HF, Czermak BJ, Watkins SA, Till GO. Mediators of microvascular injury in dermal burn wounds. Inflammation. 1998;22(6):619–29. doi: 10.1023/a:1022366514847. [DOI] [PubMed] [Google Scholar]
- 21.Gilpin DA. Calculation of a new Meeh constant and experimental determination of burn size. Burns. 1996;22(8):607–11. doi: 10.1016/s0305-4179(96)00064-2. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs N. Identification of Pseudomonas pyocyanea by the oxidase reaction. Nature. 1956;178(4535):703. doi: 10.1038/178703a0. [DOI] [PubMed] [Google Scholar]
- 23.Mahoney E, Reichner J, Bostom LR, Mastrofrancesco B, Henry W, Albina J. Bacterial colonization and the expression of inducible nitric oxide synthase in murine wounds. Am J Pathol. 2002;161(6):2143–52. doi: 10.1016/s0002-9440(10)64492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sparkes BG. Mechanisms of immune failure in burn injury. Vaccine. 1993;11(5):504–10. doi: 10.1016/0264-410x(93)90218-m. [DOI] [PubMed] [Google Scholar]
- 25.Alexander JW. Mechanism of immunologic suppression in burn injury. J Trauma. 1990;30(12 Suppl):S70–5. doi: 10.1097/00005373-199012001-00017. [DOI] [PubMed] [Google Scholar]
- 26.Gibran NS, Heimbach DM. Current status of burn wound pathophysiology. Clin Plast Surg. 2000;27(1):11–22. [PubMed] [Google Scholar]
- 27.Wretlind B, Pavlovskis OR. Pseudomonas aeruginosa elastase and its role in pseudomonas infections. Rev Infect Dis. 1983;5 (Suppl 5):S998–1004. doi: 10.1093/clinids/5.supplement_5.s998. [DOI] [PubMed] [Google Scholar]
- 28.Baltch AL, Franke M, Smith RP, Asperilla M, Griffin P, Michelsen P, et al. Serum antibody concentrations of cytotoxin, exotoxin, A, lipopolysaccharide, protease, and elastase and survival of patients with Pseudomonas aeruginosa bacteremia. Clin Infect Dis. 1996;23(5):1109–16. doi: 10.1093/clinids/23.5.1109. [DOI] [PubMed] [Google Scholar]
- 29.McDonald LC. Trends in antimicrobial resistance in health care-associated pathogens and effect on treatment. Clin Infect Dis. 2006;42 (Suppl 2):S65–71. doi: 10.1086/499404. [DOI] [PubMed] [Google Scholar]
- 30.Haynes A, 3rd, Rumbaugh KP, Park PW, Hamood AN, Griswold JA. Protamine sulfate reduces the susceptibility of thermally injured mice to Pseudomonas aeruginosa infection. J Surg Res. 2005;123(1):109–17. doi: 10.1016/j.jss.2004.07.251. [DOI] [PubMed] [Google Scholar]
- 31.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, et al. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. Embo J. 1999;18(18):4969–80. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274(1):264–9. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 33.Arbabi S, Garcia I, Bauer GJ, Maier RV. Alcohol (ethanol) inhibits IL-8 and TNF: role of the p38 pathway. J Immunol. 1999;162(12):7441–5. [PubMed] [Google Scholar]
- 34.Arbabi S, Rosengart MR, Garcia I, Jelacic S, Maier RV. Priming interleukin 8 production: role of platelet-activating factor and p38. Arch Surg. 1999;134(12):1348–53. doi: 10.1001/archsurg.134.12.1348. [DOI] [PubMed] [Google Scholar]
- 35.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM, et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A. 2005;102(2):431–6. doi: 10.1073/pnas.0405193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher CA, Manns JM, Young ED, Kannan S. Molecular mechanism of extracellular nucleotide-induced degranulation from human polymorphonuclear leukocytes: Role of leukotriene B4 and p38 MAP kinase as essential intermediates. J Biol Chem. 2003 doi: 10.1074/jbc.M306830200. [DOI] [PubMed] [Google Scholar]
- 37.Partrick DA, Moore EE, Offner PJ, Meldrum DR, Tamura DY, Johnson JL, et al. Maximal human neutrophil priming for superoxide production and elastase release requires p38 mitogen-activated protein kinase activation. Arch Surg. 2000;135(2):219–25. doi: 10.1001/archsurg.135.2.219. [DOI] [PubMed] [Google Scholar]
- 38.Alvarado-Kristensson M, Melander F, Leandersson K, Ronnstrand L, Wernstedt C, Andersson T. p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J Exp Med. 2004;199(4):449–58. doi: 10.1084/jem.20031771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu M, Du Q, Vancurova I, Lin X, Miller EJ, Simms HH, et al. Proapoptotic effect of curcumin on human neutrophils: activation of the p38 mitogen-activated protein kinase pathway. Crit Care Med. 2005;33(11):2571–8. doi: 10.1097/01.ccm.0000186760.20502.c7. [DOI] [PubMed] [Google Scholar]
- 40.Kim MH, Choi WC, Kang HO, Lee JS, Kang BS, Kim KJ, et al. The molecular structure and catalytic mechanism of a quorum-quenching N-acyl-L-homoserine lactone hydrolase. Proc Natl Acad Sci U S A. 2005;102(49):17606–11. doi: 10.1073/pnas.0504996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vikstrom E, Magnusson KE, Pivoriunas A. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone stimulates phagocytic activity in human macrophages through the p38 MAPK pathway. Microbes Infect. 2005;7(15):1512–8. doi: 10.1016/j.micinf.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 42.Han YP, Tuan TL, Wu H, Hughes M, Garner WL. TNF-alpha stimulates activation of pro-MMP2 in human skin through NF-(kappa)B mediated induction of MT1-MMP. J Cell Sci. 2001;114(Pt 1):131–139. doi: 10.1242/jcs.114.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han YP, Downey S, Garner WL. Interleukin-1alpha-induced proteolytic activation of metalloproteinase-9 by human skin. Surgery. 2005;138(5):932–9. doi: 10.1016/j.surg.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han YP, Zhou L, Wang J, Xiong S, Garner WL, French SW, et al. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem. 2004;279(6):4820–8. doi: 10.1074/jbc.M310999200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meduri GU. Clinical review: a paradigm shift: the bidirectional effect of inflammation on bacterial growth. Clinical implications for patients with acute respiratory distress syndrome. Crit Care. 2002;6(1):24–9. doi: 10.1186/cc1450. [DOI] [PMC free article] [PubMed] [Google Scholar]