

Abstract
Prostate cancer is the most common, non-dermatologic cancer in men. Since prostate cancer is highly associated with increased age, the incidence of this disease is expected to increase as the population ages. In its initial stages prostate cancer depends upon the actions of androgen, and androgen deprivation therapy induces tumor regression. Currently, androgen deprivation is achieved by either surgical or chemical androgen blockade. Unfortunately, nearly all prostate cancer patients develop tumors that grow despite androgen blockade and ultimately relapse. Many alterations in prostate cancer cells contribute to this state. Although chemotherapy induces short remissions in some patients, there are no curative therapies for metastatic disease. This review summarizes our current understanding in androgen signaling and the mechanisms that allow tumor cells to bypass androgen manipulation therapy. The identification of novel survival pathways and effector molecules that drive androgen independent growth is necessary to develop effective therapies for advanced prostate cancers.
Keywords: Prostate cancer, Androgen receptor
INTRODUCTION
In the last decade the number of newly diagnosed prostate cancer in men has increased dramatically and currently is the second leading cause of cancer deaths in men in the United States (Jemal et al., 2006). This increase is due to at least two factors. First, the introduction of prostate-specific antigen (PSA) screening has identified many men with clinically silent prostate cancer and has increased the percentage of patients identified with early stage disease. In addition, prostate cancer is strongly associated with aging and 2 out of every 3 cases are diagnosed in men older than 65 years of age (ACC, 2006). The incidence of prostate cancer varies worldwide, with the highest rates found in the United States, Canada and Scandinavia, and the lowest rates found in China, Japan and other parts of Asia (Crawford, 2003). These differences are likely due to differential genetic susceptibility (family history and race), exposure to unknown external risk factors and/or differences in health care and cancer registration.
Prostate cancer occurs when the cellular homeostasis of the prostate is disrupted. The normal human prostate is a tubular-alveolar gland composed of epithelial acini surrounded by a stromal compartment. The glandular acini contain basal and luminal cells, organized in a two-layered epithelium. The stromal compartment contains nerves, fibroblasts, infiltrating lymphocytes and macrophages, endothelial cell capillaries, and smooth muscle cells. Scattered throughout the epithelial compartment are occasional neuroendocrine cells (Marker, Donjacour, Dahiya, & Cunha, 2003). The luminal epithelial cells produce and secrete a major fraction of the seminal fluid within the prostate.
Androgens are an important growth factor for the normal prostate, signaling through the androgen receptor (AR) localized in the cytoplasm of stromal and secretory epithelial cells (Chatterjee, 2003). AR is a member of the steroid hormone receptor transcription factor family. Upon ligand binding, AR enters the nucleus and activates a panoply of target genes involved in diverse biological processes such as proliferation, differentiation, apoptosis and secretion. Androgens therefore balance proliferation and survival versus death within the prostate and control prostate homeostasis (see Figure 1).
Figure 1.
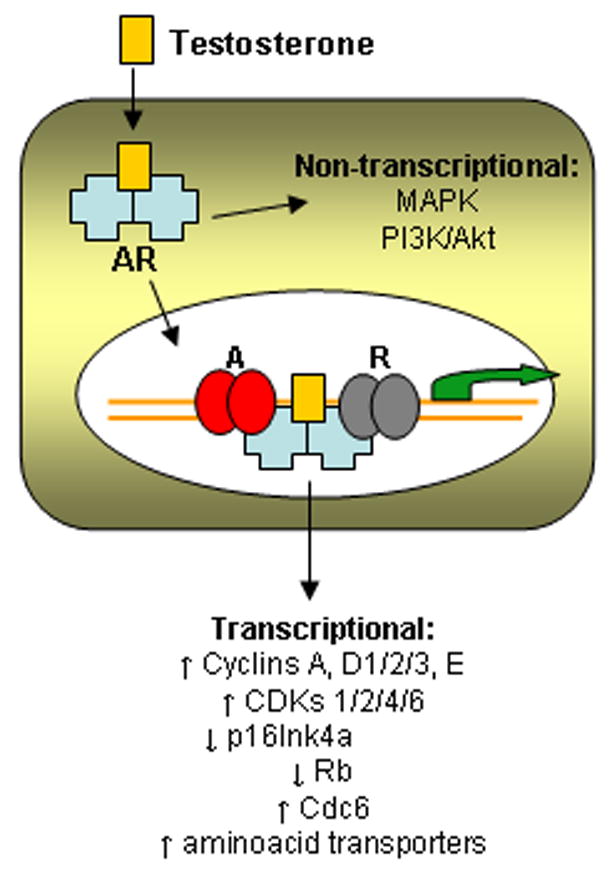
Pathways involved in proliferation induced by androgen in prostate cells. Testosterone, the main circulating androgen, binds its cytoplasmic receptor androgen receptor (AR) and translocates to the nucleus, where it activates a complex transcriptional program modulated by co-activators (A) and co-repressors (R). AR can also induce proliferation through non-transcriptional pathways that activate important mitogenic kinases.
In current clinical practice, prostate cancer is usually diagnosed after an abnormal digital rectal examination (DRE) or, more commonly, by finding elevated PSA levels, a well-characterized component of the seminal fluid. Malignant cells produce aberrant PSA levels that can be measured in the blood. Levels above 4 ng/mL are considered abnormal (Crawford, 2003). Prostate cancer can also be discovered during bladder outlet obstruction surgery. The diagnosis is then confirmed by transrectal ultrasound (TRUS)-guided biopsy and examined pathologically where a Gleason score is assigned (NCCN, 2005). The Gleason score is the most frequently used grading system for prostate cancer and is based on scoring the two most predominant glandular differentiation patterns in the sample (Gleason & Mellinger, 1974). In addition, perineural invasion, glomerulations or collagenous micronodules are of prognostic relevance (DeMarzo, Nelson, Isaacs, & Epstein, 2003).
Although prostate cancer is quite common, the disease is characterized by a wide disparity in the pace of natural progression. Even when diagnosed with prostate cancer, some men will progress while others will have indolent disease. Although some factors including high PSA doubling time and Gleason grade predict a worse prognosis, a significant percentage of men progress even with apparently, early stage disease. Identifying predictive prognostic markers that permit one to distinguish prospectively between patients with aggressive and indolent disease remains an important area of active research.
PATHOGENESIS
Prostate architecture changes subtly as men age. Increased numbers of epithelial cells that retain normal cell features are often seen in the prostates of older men. These benign changes are pathologically defined as benign prostatic hyperplasia (BPH). The development of pre-invasive lesions known as prostatic intraepithelial hyperplasia (PIN) also occurs with increasing age. High-grade PIN can be clearly identified by traditional histopathology and consists of prostate acini, lined by basal cells and by luminal cells that show dysplastic features including nuclear hyperchromasia, nuclear crowding and prominent nucleoli (see Figure 2). However, a causal association between PIN lesions and the development of adenocarcinomas remains unclear as there are many early cancers that do not harbor adjacent PIN.
Figure 2.
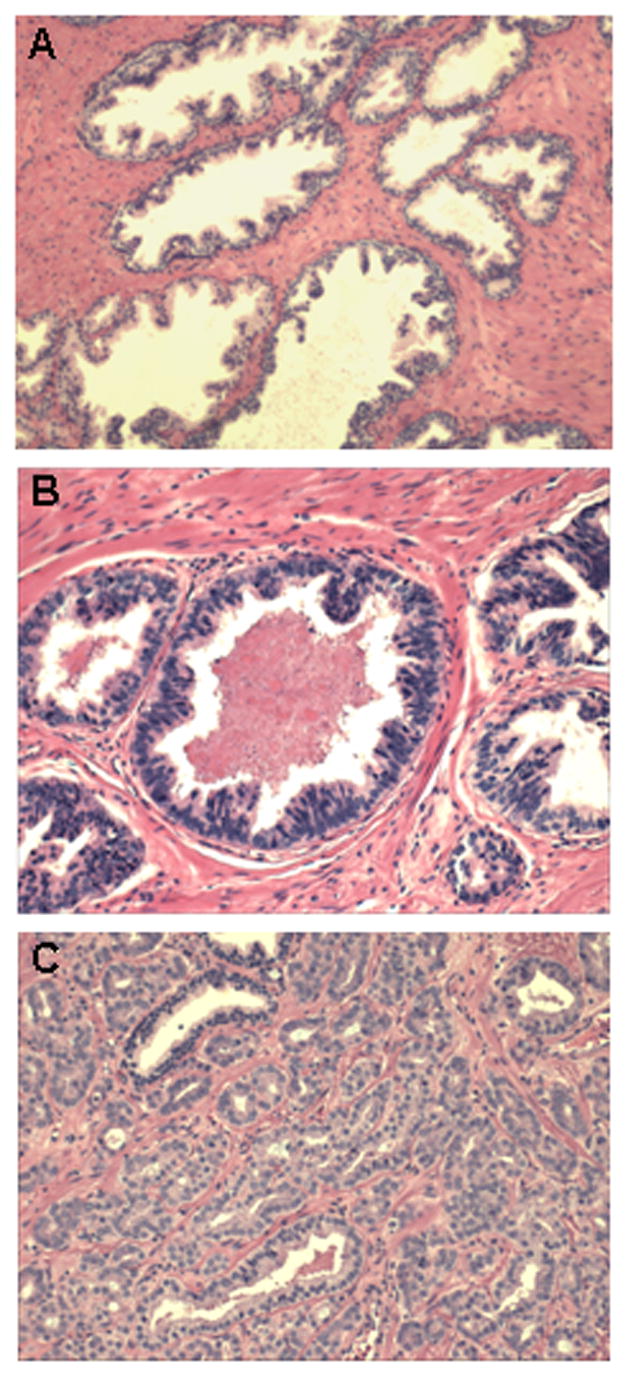
Loss of tissue architecture in prostate cancer. (A) Normal prostate: Well-defined large ducts containing luminal cells on top of a thin layer of basal cells. (B) High grade PIN: Well-defined ducts containing sparse basal cells and luminal cells that spread towards the lumen. (C) Prostate cancer: Undefined small ducts that fuse to each other and characterized by the complete absence of basal cells.
Carcinoma of the prostate requires the presence of malignant acini (see Figure 2). It can be confined within the prostate (low stage, T2 or lower according to the TNM classification: www.uicc.org/tnm), spread into adjacent tissue (advanced stage, T3) or metastatic, with bone and lymph nodes being the most common colonizing sites for prostate cells. Prostate cancer, as any other type of cancer, arises from cells that accumulate genome changes affecting regulatory genes resulting in a growth or survival advantage. However, early stage prostate cancers exhibit the unique characteristic that androgens are able to regulate this aberrant tumor cell proliferation. Prostate cancer cells retain androgen responsiveness in its initial stages, and blockade of the AR pathway induces tumor regression in most patients. Indeed, the current standard of care for patients with advanced prostate cancer is androgen deprivation, either by surgical or chemical means. In late stages however, prostate cancer cells can acquire additional genetic alterations and become hormone refractory. Currently no curative therapies exist for metastatic, hormone-manipulation refractory forms of the disease.
Molecular changes associated with prostate cancer
The events associated with the initiation of prostate cancer remain poorly understood. Mutations in classic oncogenes or tumor suppressor genes are uncommon, and unlike many other cancers, few prostate cancer cell lines are available for in vitro studies. Moreover, the majority of these cell lines are derived from metastatic tumors and provide little insight about early stages of the disease (Peehl, 2005). Genomic tools such as comparative genome hybridization, spectral karyotyping and allelic imbalance analysis have provided the means to identify chromosomal alterations involved in prostate cancer. Losses at chromosomes 1p, 6q, 8p, 10q, 13q, 16q and 18q and gains at 1q, 2p, 7, 8q, 18q, and Xq have been reported [for a recent review see (Shand & Gelmann, 2006)], however only few candidate genes have been identified in these regions. A list of the genes implicated in prostate cancer is shown in Table I.
Table I.
Genetic evidences in prostate cancer progression
Over the past several years, engineered prostate epithelial cell lines and mouse models have recently become available for the study of the disease progression. The manipulation of endogenous and viral oncogenes such as the SV40 Large T and small t antigens, Ras, Myc or Akt and important tumor suppressors such as Nkx3.1, PTEN, p27Kip1, p53 or Rb have permitted the development of several new experimental models of prostate cancer [summarized in (Abate-Shen & Shen, 2002; Kasper, 2005) (Chen et al., 2005; Gao, Ouyang, Banach-Petrosky, Shen, & Abate-Shen, 2006; Zhou et al., 2006)]. These models recapitulate many aspects of prostate cancer progression and indeed, AR expression is necessary to maintain the hormonal dependency characteristic of early stage tumors (Berger et al., 2004).
Androgen independent prostate cancer (AIPC)
Androgen independence defines a clinical stage in which prostate cancer cells are able to survive and proliferate without the required signals delivered normally by circulating androgens. This condition renders them insensitive to therapies that rely on androgen blockade, and it is often referred to as androgen resistant, hormone refractory, hormone resistant or castration resistant. Each of these terms is used interchangeably in the literature. Androgen insensitivity reflects the ability to grow without (or with very low) circulating androgens, but it should not be confused with an absence of the intracellular signaling activated downstream of androgen binding to AR. Indeed, most mechanisms leading to such insensitivity still rely on an active signaling through AR, which becomes able to activate its transcriptional targets independently of hormonal binding (see Figure 3).
Figure 3.
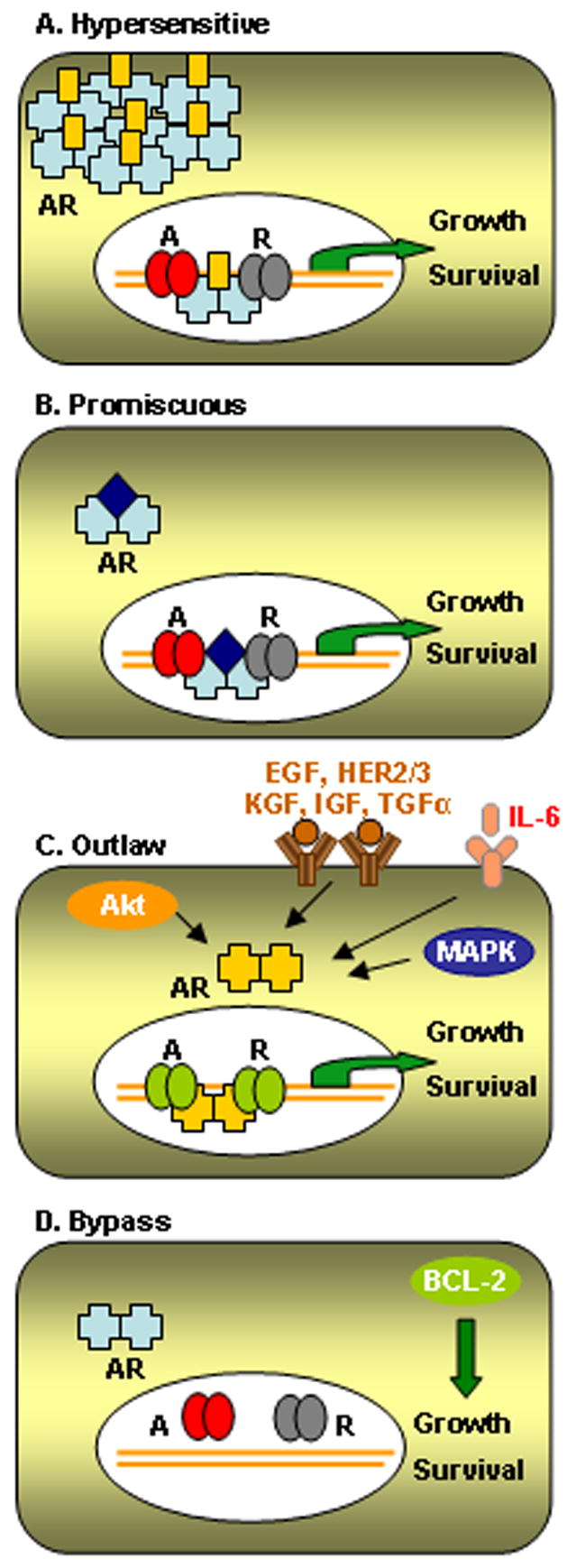
Proliferation induced by androgen independent pathways. (A) Hypersensitive pathway: Lower levels of androgen can activate signaling through the AR transcription factor. This state is usually achieved by amplification of the receptor itself, but it can also occur in the presence of activating mutations. (B) Promiscuous pathway: Mutations allow non-androgenic steroid hormones to bind and activate AR signaling. (C) Outlaw pathway: AR is activated independently of ligand binding by growth factors, cytokines, interleukins, cytoplasmic kinases or nuclear co-activators. Importantly A, B and C rely in the presence of an active AR complex. (D) Bypass pathway: Alternative survival pathways are activated and promote proliferation in the absence of androgens. The presence of an active AR is not required. Classification adapted from (Feldman and Feldman, 2001).
There are several mechanisms known to lead to androgen independence. Studies in prostate cancer derived cell lines and tumor samples have identified several genes upregulated or lost in AIPC, however their role in the pathogenesis of AIPC remains unclear. Indeed, only a few genes have been shown to drive the proliferation of prostate cells in the absence of androgens. These genes include AR and ERBB2/Her2, myc, Id-1, protocadherin-PC, IL-6, EGR1 and certain p53 mutations. Among these genes, the biochemical and genetic evidence implicating continued AR signaling is the most compelling.
The role of AR in prostate cancer has been extensively studied. AR is only rarely mutated in the early stages of the disease, however, a high percentage of tumors obtained from patients who had failed androgen ablation overexpress AR. Approximately 30% of this overexpression is associated with genetic amplification (Koivisto et al., 1997; Visakorpi et al., 1995) and 10–40% with AR mutations (Fenton et al., 1997; Taplin et al., 1995). Androgen receptor pathway can be hyperactivated through several mechanisms, which can be divided in three main categories: (1) Hypersensitivity in the androgen receptor through genomic amplification, mutations or increased systemic androgen levels, (2) Promiscuous activation of the receptor by mutation or co-regulators alterations, and (3) Receptor activation by alternative pathways such as growth-factors, receptor tyrosine-kinases or Akt (Feldman & Feldman, 2001) (see Figure 3). Taken together, the acquisition of oncogenic mutations in the AR gene appears to be responsible in many patients. Most of these mutations occur after androgen ablation therapy, indicating the strong selective pressure to maintain AR activity for the survival of prostate cells. Also the ligand-binding domain appears to be the more commonly mutated causing AR to be responsive to other molecules, such as other hormone-like ligands, growth factors or antiandrogens. To date, many mutations have been identified in the AR gene [for a detailed database of mutations consult http://www.mcgill.ca/androgendb/ (Gottlieb, Beitel, Wu, & Trifiro, 2004)]; however, only a few have been studied in any detail. Recently, two common AR mutations have been directly related to prostate cancer progression, E231G and T877A (Han et al., 2005; Sun et al., 2006).
Moreover, in the cases where AR is not hyperactivated, several investigators have shown that alternative survival pathways are activated (see Figure 3D). Bcl2 overexpression or combined loss of important tumor suppressors such as p53/Rb or PTEN/Nkx3.1 have been strongly implicated in androgen independence (Gao, Ouyang, Banach-Petrosky, Shen, & Abate-Shen, 2006; Gleave et al., 1999; Raffo et al., 1995; Zhou et al., 2006). In addition, there is increasing evidence to suggest that androgen-independent progenitors/stem cells may contribute in important ways to the initiation of prostate cancers and in the development of androgen independence (Tang et al., 2006). However, although there have been several reports of pluripotency in precursor-like populations, markers that identify the true prostate cancer stem cell have not yet been described.
CURRENT THERAPIES
Anti-androgen therapy
The first line therapy for patients with metastatic or recurrent disease is androgen deprivation, either by surgical or medical castration. The beneficial effect of androgen ablation on prostate cancer was described more that 40 years ago by Charles Huggins (Huggins, 1967). However, despite this therapy, eventually all prostate tumors adapt to hormonal ablation therapy and progress. Some evidence suggests that additional hormonal manipulations can be useful after primary castration therapy. Indeed, non-steroidal androgens such as bicalutamide, flutamide or nilutamide are able to further block androgen receptor and inhibitors of the adrenal androgen production such as ketoconazole, corticosteroids or aminoglutethiamide have been useful to inhibit testosterone production in both testes and adrenal glands. However, in the long term, prostate cells are able to grow in the absence of androgens. Although no curative therapies exist for such refractory prostate cancers, recent studies have demonstrated that treatment with regimens containing docetaxel, a cytotoxic microtubule inhibitor, modestly prolongs survival (2.5 months) in patients with metastatic, hormone refractory prostate cancer (Tannock et al., 2004); however, it is clear that more effective therapies that target late stages of the disease are required.
New approaches in drug development: Re-focusing on the AR axis
Over 200 compounds have entered clinical trial for use in advanced prostate cancer, alone or in combination with cytotoxic agents (Armstrong & Carducci, 2006). The identification of new pathways important for the development of prostate cancer provides several potential therapeutic targets. Several compounds are currently being tested in clinical trials in order to explore their efficacy targeting prostate cancer cells. They include inhibition of survival pathways through essential kinases (phosphatidylinosytol 3-kinase (PI3K), Akt, mTOR) and growth factors (epithermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), insulin growth factor receptor (IGFR), Her2/Her3), immunological approaches, novel cytotoxic compounds and targeting of important cellular processes such as angiogenesis, apoptosis, vitamin D metabolism, differentiation and stem cell biology [for a recent review see (Mimeault & Batra, 2006)]. However, although efforts to develop new therapies are promising, clinical efficacy of these compounds is yet to be proven.
On the other hand, the AR pathway is a well-established target for treating prostate cancer. The currently available treatments targeting AR fail to fully block this signaling pathway. Further strategies targeting AR signaling thus are likely to provide additional clinical benefit while maintaining unique specificity for prostate tumors. Accomplishing this goal will require us to more fully understand the pathways involved in AR signaling and how such pathways become activated during the androgen insensitive state. Targeting such effector proteins alone or in combination with current anti-androgen drugs may improve our ability to treat advanced prostate cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abate-Shen C, Shen MM. Mouse models of prostate carcinogenesis. Trends Genet. 2002;18(5):S1–5. doi: 10.1016/s0168-9525(02)02683-5. [DOI] [PubMed] [Google Scholar]
- ACC. Prostate Cancer: Risk Factors and Prevention. 2006 (from http://www.cancer.org)
- Armstrong AJ, Carducci MA. New drugs in prostate cancer. Curr Opin Urol. 2006;16(3):138–145. doi: 10.1097/01.mou.0000193390.69845.bb. [DOI] [PubMed] [Google Scholar]
- Asatiani E, Huang WX, Wang A, Rodriguez Ortner E, Cavalli LR, Haddad BR, et al. Deletion, methylation, and expression of the NKX3.1 suppressor gene in primary human prostate cancer. Cancer Res. 2005;65(4):1164–1173. doi: 10.1158/0008-5472.CAN-04-2688. [DOI] [PubMed] [Google Scholar]
- Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S, et al. Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res. 2004;64(24):8867–8875. doi: 10.1158/0008-5472.CAN-04-2938. [DOI] [PubMed] [Google Scholar]
- Bookstein R, MacGrogan D, Hilsenbeck SG, Sharkey F, Allred DC. p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 1993;53 (14):3369–3373. [PubMed] [Google Scholar]
- Bubendorf L, Kononen J, Koivisto P, Schraml P, Moch H, Gasser TC, et al. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999;59 (4):803–806. [PubMed] [Google Scholar]
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57(22):4997–5000. [PubMed] [Google Scholar]
- Chatterjee B. The role of the androgen receptor in the development of prostatic hyperplasia and prostate cancer. Mol Cell Biochem. 2003;253(1–2):89–101. doi: 10.1023/a:1026057402945. [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436(7051):725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombel M, Symmans F, Gil S, O'Toole KM, Chopin D, Benson M, et al. Detection of the apoptosis-suppressing oncoprotein bc1-2 in hormone-refractory human prostate cancers. Am J Pathol. 1993;143(2):390–400. [PMC free article] [PubMed] [Google Scholar]
- Cooney KA, Wetzel JC, Merajver SD, Macoska JA, Singleton TP, Wojno KJ. Distinct regions of allelic loss on 13q in prostate cancer. Cancer Res. 1996;56 (5):1142–1145. [PubMed] [Google Scholar]
- Crawford ED. Epidemiology of prostate cancer. Urology. 2003;62(6 Suppl 1):3–12. doi: 10.1016/j.urology.2003.10.013. [DOI] [PubMed] [Google Scholar]
- DeMarzo AM, Nelson WG, Isaacs WB, Epstein JI. Pathological and molecular aspects of prostate cancer. Lancet. 2003;361(9361):955–964. doi: 10.1016/S0140-6736(03)12779-1. [DOI] [PubMed] [Google Scholar]
- Downing SR, Russell PJ, Jackson P. Alterations of p53 are common in early stage prostate cancer. Can J Urol. 2003;10(4):1924–1933. [PubMed] [Google Scholar]
- Effert PJ, McCoy RH, Walther PJ, Liu ET. p53 gene alterations in human prostate carcinoma. J Urol. 1993;150(1):257–261. doi: 10.1016/s0022-5347(17)35458-7. [DOI] [PubMed] [Google Scholar]
- Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998;16(13):1743–1748. doi: 10.1038/sj.onc.1200205. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1(1):34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997;3(8):1383–1388. [PubMed] [Google Scholar]
- Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clin Cancer Res. 1996;2(2):389–398. [PubMed] [Google Scholar]
- Gao H, Ouyang X, Banach-Petrosky WA, Shen MM, Abate-Shen C. Emergence of androgen independence at early stages of prostate cancer progression in nkx3.1; pten mice. Cancer Res. 2006;66(16):7929–7933. doi: 10.1158/0008-5472.CAN-06-1637. [DOI] [PubMed] [Google Scholar]
- Gleason DF, Mellinger GT. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J Urol. 1974;111(1):58–64. doi: 10.1016/s0022-5347(17)59889-4. [DOI] [PubMed] [Google Scholar]
- Gleave M, Tolcher A, Miyake H, Nelson C, Brown B, Beraldi E, et al. Progression to androgen independence is delayed by adjuvant treatment with antisense Bcl-2 oligodeoxynucleotides after castration in the LNCaP prostate tumor model. Clin Cancer Res. 1999;5(10):2891–2898. [PubMed] [Google Scholar]
- Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat. 2004;23(6):527–533. doi: 10.1002/humu.20044. [DOI] [PubMed] [Google Scholar]
- Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ, et al. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci U S A. 2005;102(4):1151–1156. doi: 10.1073/pnas.0408925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WW, Sciavolino PJ, Wing J, Augustus M, Hudson P, Meissner PS, et al. A novel human prostate-specific, androgen-regulated homeobox gene (NKX3.1) that maps to 8p21, a region frequently deleted in prostate cancer. Genomics. 1997;43(1):69–77. doi: 10.1006/geno.1997.4715. [DOI] [PubMed] [Google Scholar]
- Heidenberg HB, Sesterhenn IA, Gaddipati JP, Weghorst CM, Buzard GS, Moul JW, et al. Alteration of the tumor suppressor gene p53 in a high fraction of hormone refractory prostate cancer. J Urol. 1995;154(2 Pt 1):414–421. doi: 10.1097/00005392-199508000-00024. [DOI] [PubMed] [Google Scholar]
- Huggins C. Endocrine-induced regression of cancers. Cancer Res. 1967;27(11):1925–1930. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, et al. Cancer statistics, 2006. CA Cancer J Clin. 2006;56(2):106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Kasper S. Survey of genetically engineered mouse models for prostate cancer: analyzing the molecular basis of prostate cancer development, progression, and metastasis. J Cell Biochem. 2005;94(2):279–297. doi: 10.1002/jcb.20339. [DOI] [PubMed] [Google Scholar]
- Kibel AS, Freije D, Isaacs WB, Bova GS. Deletion mapping at 12p12-13 in metastatic prostate cancer. Genes Chromosomes Cancer. 1999;25(3):270–276. [PubMed] [Google Scholar]
- Kibel AS, Schutte M, Kern SE, Isaacs WB, Bova GS. Identification of 12p as a region of frequent deletion in advanced prostate cancer. Cancer Res. 1998;58 (24):5652–5655. [PubMed] [Google Scholar]
- Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57(2):314–319. [PubMed] [Google Scholar]
- Li C, Larsson C, Futreal A, Lancaster J, Phelan C, Aspenblad U, et al. Identification of two distinct deleted regions on chromosome 13 in prostate cancer. Oncogene. 1998;16(4):481–487. doi: 10.1038/sj.onc.1201554. [DOI] [PubMed] [Google Scholar]
- Marker PC, Donjacour AA, Dahiya R, Cunha GR. Hormonal, cellular, and molecular control of prostatic development. Dev Biol. 2003;253(2):165–174. doi: 10.1016/s0012-1606(02)00031-3. [DOI] [PubMed] [Google Scholar]
- Melamed J, Einhorn JM, Ittmann MM. Allelic loss on chromosome 13q in human prostate carcinoma. Clin Cancer Res. 1997;3(10):1867–1872. [PubMed] [Google Scholar]
- Mimeault M, Batra SK. Recent advances on multiple tumorigenic cascades involved in prostatic cancer progression and targeting therapies. Carcinogenesis. 2006;27 (1):1–22. doi: 10.1093/carcin/bgi229. [DOI] [PubMed] [Google Scholar]
- NCCN. National Comprehensive Cancer Network. 2005. Clinical Practice Guidelines in Oncology: Prostate Cancer. [DOI] [PubMed] [Google Scholar]
- Peehl DM. Primary cell cultures as models of prostate cancer development. Endocr Relat Cancer. 2005;12(1):19–47. doi: 10.1677/erc.1.00795. [DOI] [PubMed] [Google Scholar]
- Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995;55(19):4438–4445. [PubMed] [Google Scholar]
- Sarkar FH, Sakr W, Li YW, Macoska J, Ball DE, Crissman JD. Analysis of retinoblastoma (RB) gene deletion in human prostatic carcinomas. Prostate. 1992;21(2):145–152. doi: 10.1002/pros.2990210207. [DOI] [PubMed] [Google Scholar]
- Shand RL, Gelmann EP. Molecular biology of prostate-cancer pathogenesis. Curr Opin Urol. 2006;16(3):123–131. doi: 10.1097/01.mou.0000193384.39351.64. [DOI] [PubMed] [Google Scholar]
- Sommerfeld HJ, Meeker AK, Piatyszek MA, Bova GS, Shay JW, Coffey DS. Telomerase activity: a prevalent marker of malignant human prostate tissue. Cancer Res. 1996;56(1):218–222. [PubMed] [Google Scholar]
- Sun C, Shi Y, Xu LL, Nageswararao C, Davis LD, Segawa T, et al. Androgen receptor mutation (T877A) promotes prostate cancer cell growth and cell survival. Oncogene. 2006 doi: 10.1038/sj.onc.1209424. [DOI] [PubMed] [Google Scholar]
- Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, et al. Prostate cancer stem/progenitor cells: Identification, characterization, and implications. Mol Carcinog. 2006 Aug 18; doi: 10.1002/mc.20255. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351(15):1502–1512. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332(21):1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310(5748):644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- Van Den Berg C, Guan XY, Von Hoff D, Jenkins R, Bittner, Griffin C, et al. DNA sequence amplification in human prostate cancer identified by chromosome microdissection: potential prognostic implications. Clin Cancer Res. 1995;1(1):11–18. [PubMed] [Google Scholar]
- Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 1995;9(4):401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- Voeller HJ, Augustus M, Madike V, Bova GS, Carter KC, Gelmann EP. Coding region of NKX3.1, a prostate-specific homeobox gene on 8p21, is not mutated in human prostate cancers. Cancer Res. 1997;57(20):4455–4459. [PubMed] [Google Scholar]
- Zhang W, Kapusta LR, Slingerland JM, Klotz LH. Telomerase activity in prostate cancer, prostatic intraepithelial neoplasia, and benign prostatic epithelium. Cancer Res. 1998;58(4):619–621. [PubMed] [Google Scholar]
- Zheng SL, Ju JH, Chang BL, Ortner E, Sun J, Isaacs SD, et al. Germ-line mutation of NKX3.1 cosegregates with hereditary prostate cancer and alters the homeodomain structure and function. Cancer Res. 2006;66(1):69–77. doi: 10.1158/0008-5472.CAN-05-1550. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, et al. Synergy of p53 and Rb Deficiency in a Conditional Mouse Model for Metastatic Prostate Cancer. Cancer Res. 2006;66(16):7889–7898. doi: 10.1158/0008-5472.CAN-06-0486. [DOI] [PubMed] [Google Scholar]