

Abstract
Many different 3α-hydroxysteroids in the androstane and pregnane steroid series enhance the actions of γ-aminobutyric acid (GABA) at GABA type-A (GABAA) receptors in the mammalian central nervous system. Recent studies have shown that (3α,5α)-3-hydroxyandrostan-17-one (androsterone) is less active at these receptors than its enantiomer ent-androsterone. Further structure–activity relationship (SAR) studies are needed to explore the structural features of ent-androsterone that are important for its enhanced action at these receptors. Molecular modeling shows that 2β-hydroxysteroids are similar in three-dimensional shape to the enantiomers of 3α-hydroxysteroids. The development of synthtetic methods to gain access to C17-substituted analogues of 2β-hydroxygonanes for SAR studies is demonstrated with the synthesis of (2β,5α,13β,14β)-2-hydroxygonan-17-one.
Keywords: 2β-hydroxygonanes, abnormal Beckmann rearrangement, neurosteroids, phenanthrenes
I. Introduction
The steroid (3α,5α)-3-hydroxypregnan-20-one (allopregnanolone, Figure 1) and many analogues of it are known to be potent enhancers of γ-aminobutyric acid (GABA) at GABA Type A (GABAA) receptors.1–4 These neuroactive GABAergic steroids have activity as general anesthetics, anticonvulsants, sedative hypnotics and anxiolytics; and there is considerable current interest in the development of new analogues as pharmaceuticals having these activities.
Figure 1.
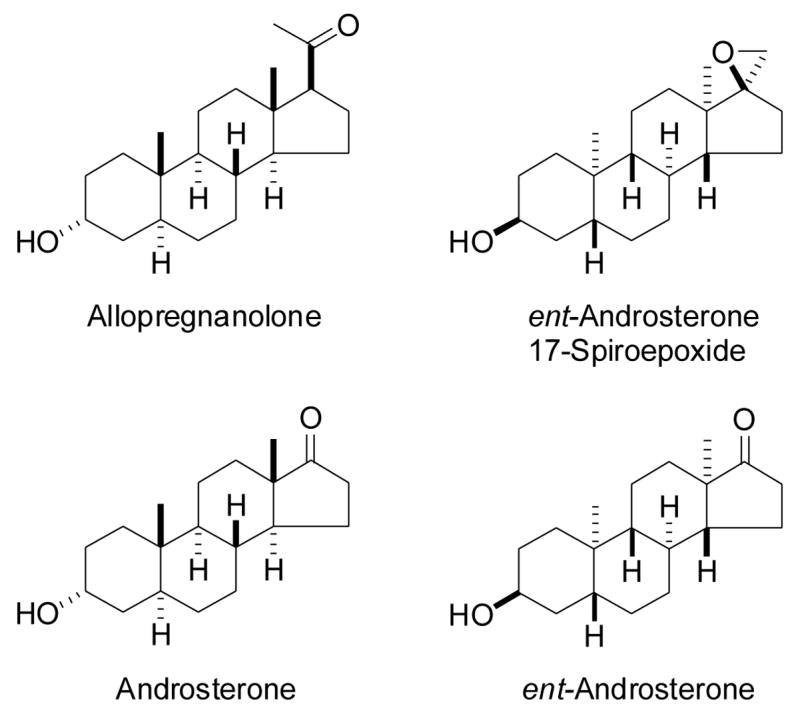
Structures of steroid modulators of GABAA receptor function. Allopregnanolone and ent-androsterone 17-spiroepoxide potently enhance the actions of GABA at GABAA receptors. Androsterone is a weak enhancer of GABA action. ent-Androsterone has activity higher than that of androsterone but less than that of the other two steroids.
As part of our ongoing studies of the enantioselectivity of neurosteroid action at GABAA receptors, we recently investigated the enantioselectivity for (3α,5α)-3-hydroxyandrostan-17-one (androsterone, Figure 1) effects at these receptors.5 Androsterone has only weak actions on GABAA receptor function6,7 and, based on our previous results from enantioselectivity studies of allopregnanolone,8 we expected that ent-androsterone (Figure 1) would have even weaker actions than androsterone. Unexpectedly, we found that ent-androsterone was more active than androsterone. Moreover, a 17-spiroepoxide derivative of ent-androsterone (Figure 1) was shown to have actions comparable to those of allopregnanolone.5
Additional steroid analogues are needed for future structure–activity relationship (SAR) studies of ent-androgen action at GABAA receptors. In this regard, we were intrigued by the structural similarity between 2β-hydroxysteroids and the enantiomers of 3α-hydroxysteroids. Figure 2A shows the structure of (2β,5α,13β,14β)-2-hydroxygonan-17-one (1) and ent-androsterone. Figure 2B shows a three-dimensional overlay of molecular models of steroid 1 and ent-androsterone. Both steroids are shown with their β face down because in this orientation the compounds most closely resemble the highly active 3α-hydroxysteroid allopregnanolone. As presented in Figure 2, each of the four steroid rings of one compound is proximate to the corresponding ring of the other compound. The O2 and O3 atoms of the two molecules are 0.68 Å apart and the O17 atoms are 0.54 Å from each other in this overlay.
Figure 2.
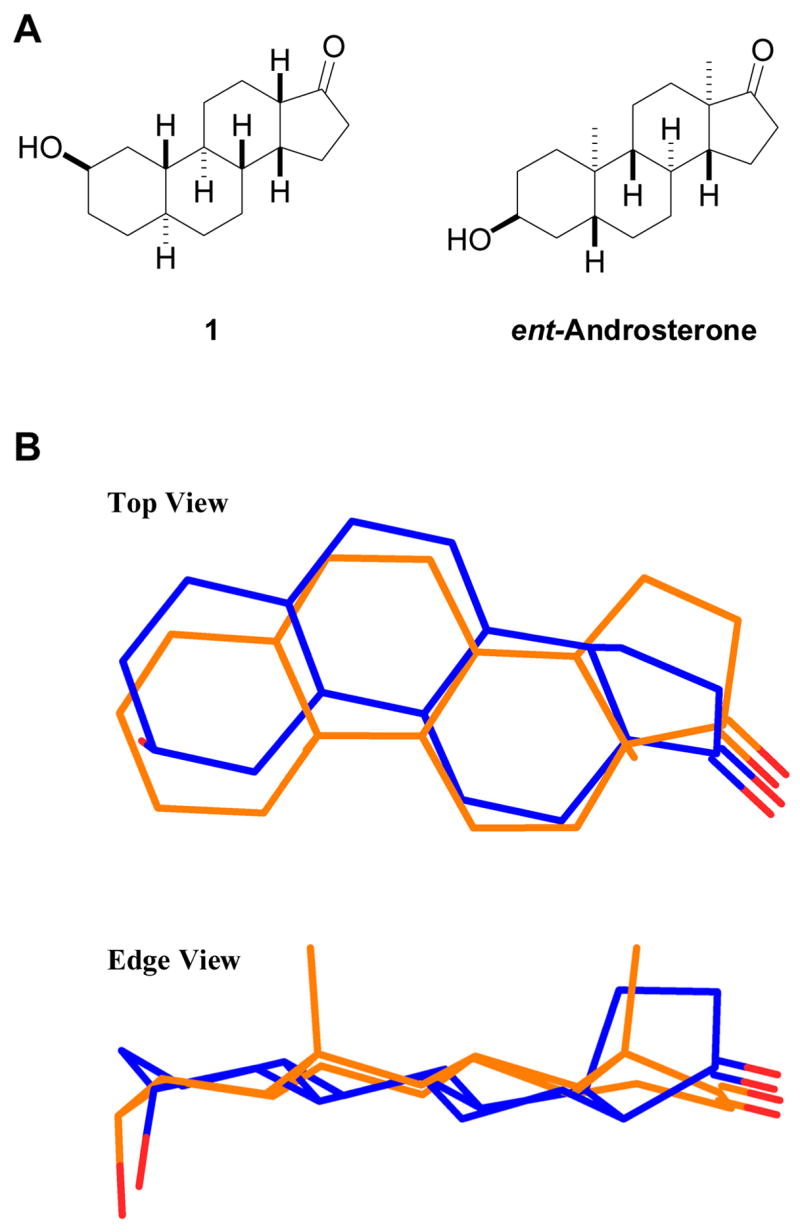
Panel A: Structures of (2β,5α,13β,14β)-2-hydroxygonan-17-one (1) and ent-androsterone. Panel B: An overlay of steroid 1 (blue structure) and ent-androsterone (orange structure). The structures were overlaid using a RMS fit of atoms C9, C11, C12, C14, O3 and O17 in each structure for the alignment. The O2–O17 (9.37 Å) distance in steroid 1 is shorter than the O3–O17 distance (9.67 Å) in ent-androsterone.
Two additional design criteria were considered in the decision to select steroid 1 as an initial potential structural mimic of ent-androsterone. Previous SAR studies have shown that having methyl groups on the steroid α face decreases the activity of 3α-hydroxysteroid modulators of GABAA receptors.9,10 When aligned as shown in Figure 2B, the C18 and/or C19 methyl groups of steroids in the androstane, estrane (19-norandrostane) and 18-norandrostane series would be located below the plane of the steroid rings (i.e. they would occupy positions similar to those occupied by methyl groups on the α face of 3α-hydroxysteroids, and therefore could be expected to negatively affect pharmacological activity). By choosing a steroid in the gonane class as a synthetic target, worries about unfavorable steric effects of these methyl groups is avoided.
The decision to have the 13β,14β cis C,D-ring fusion present in steroid 1 was made because this cis ring fusion places the C17 carbonyl group further above the plane of the steroid rings than it would be in the corresponding 13β,14α trans ring fusion. This was considered to be desirable since previous SAR studies indicate that the D-ring hydrogen bonding groups needs to be above the plane of the steroid rings for high pharmacological activity.1,6,7
2. Results and Discussion
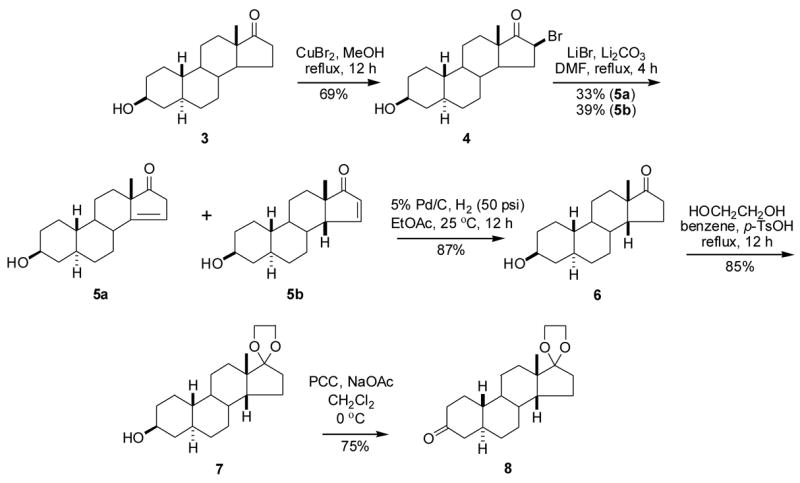
The starting material, (3β,5α)-3-hydroxyestran-17-one (3), was prepared in three steps by known methodology11,12 from commercially available 19-nortestosterone (2) in 85% total yield (Scheme 1). Steroid 3 was brominated selectively in the C16 position using CuBr213,14 to give compound 4 in 69% yield (Scheme 2). HBr was eliminated from steroid 4 using LiBr/Li2CO315 to give compounds 5a (33%) and 5b (39%). For characterization purposes, some spectroscopic data was collected on each of these products after their separation by chromatograpy. For synthetic purposes, products 5a and 5b were not separated and the mixture was subjected to catalytic hydrogenation to give compound 6 in 87% yield. The 17-keto group was ketalized in the standard way16 to afford compound 7 in 85% yield. Oxidation of the 3β-hydroxyl group of compound 7 using PCC in the presence of NaOAc17 gave compound 8 (75%).
Scheme 1.

Scheme 2.
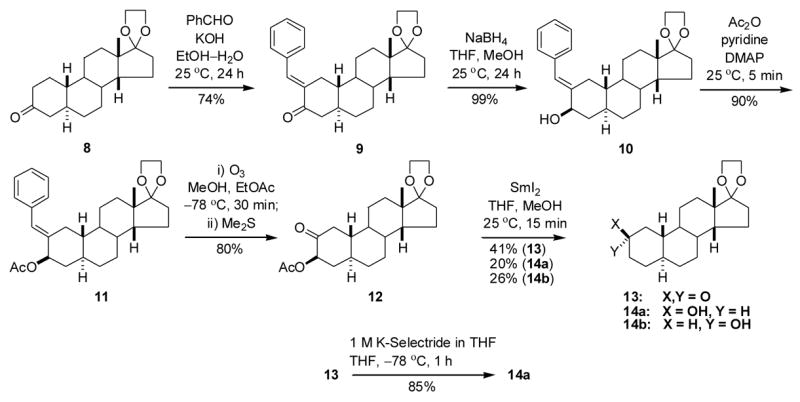
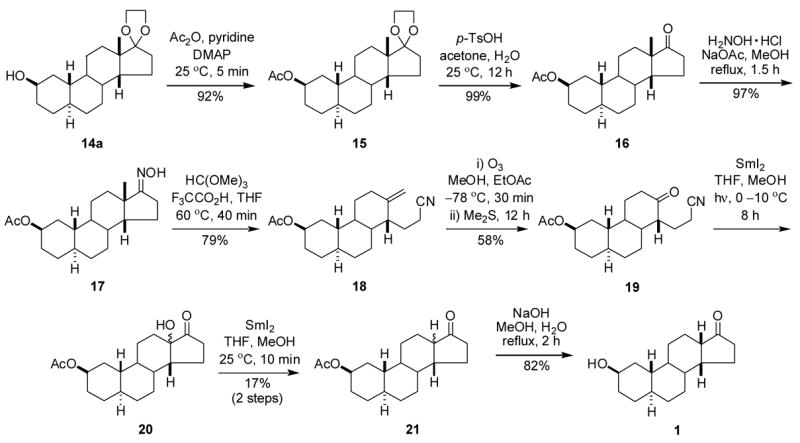
When treated with 10% ethanolic KOH at room temperature for 24 h, compound 8 underwent an aldol condensation18 to give compound 9 in 74% yield (Scheme 3). Reduction of the 3-ketone group with NaBH4 at room temperature led to the 3β-hydroxysteroid 10 (99%), and acetylation of the hydroxyl group gave the acetate derivative 11 in 90% yield. Compound 11 was treated with ozone in the usual manner18 to give steroid 12 (80%). Samarium(II) iodide-THF mediated reductive removal of the 3-acetyloxy group from compound 12 gave 2-ketosteroid 13 (41%), 2β-hydroxysteroid 14a (20%) and 2α-hydroxysteroid 14b (26%). Reduction of compound 13 with K-Selectride in THF at −78 ºC gave additional amounts of compound 14a (85%).
Scheme 3.
The hydroxyl group of 2β-hydroxysteroid 14a was reacted with acetic anhydride and pyridine in the presence of a catalytic amount of 4-dimethylaminopyridine to afford 2-(acetyloxy)steroid 15 (92%) after 5 min at room temperature. The 17-ketal of compound 15 was readily removed using p-TsOH in acetone at room temperature for 12 h to give, in quantitative yield, steroid 16. The reaction of compound 16 with NH2OH·HCl and NaOAc gave oxime 17 (97%). Oxime 17 underwent an abnormal Beckmann rearrangement19 upon treatment with CH(OCH3)3/TFA in THF to give carbonitrile 18 (79%). Ozonolysis of compound 18 at −78 °C yielded ketone-nitrile 19 (58%). Treatment of compound 19 with SmI2 in THF and irradiation with a 500 W lamp20 at 0–10 °C for 8 h gave crude 13-hydroxysteroid 20. After isolation, crude product 20 was again treated with SmI2 in THF at room temperature for 10 min to give compound 21 (17%, from compound 19). Finally, base-catalyzed hydrolysis of the 3-acetyloxy group of compound 21 and column chromatography to remove the presumed 13α,14β stereoisomer gave compound 1 (82%). The structure of steroid 1 was confirmed by single crystal X-ray diffraction analysis (Figure 3).21
Figure 3.
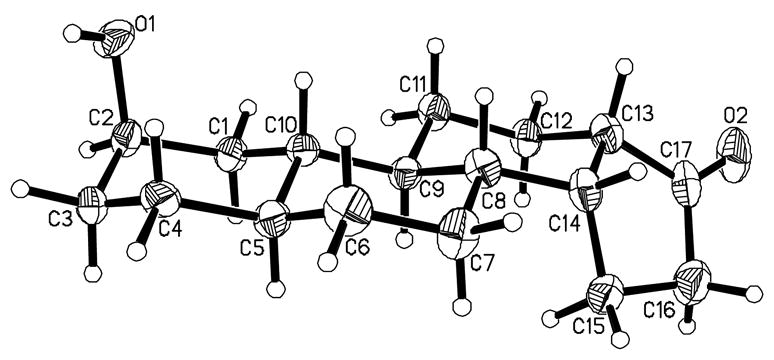
Projection view of one of the two unique molecules of steroid 1 shown with 50% thermal ellipsoids for non-hydrogen atoms.
Using methods reported previously,22 the actions of compound 1 as a modulator of GABAA receptor function were compared to those of ent-androsterone. Whereas ent-androsterone allosterically displaced 50% of [35S]-t-butylbicyclophosphorthionate bound to the picrotoxin binding site on GABAA receptors at a concentration of 0.31 μM,5 no displacement of [35S]-t-butylbicyclophosphorthionate occurred at concentrations up to 30 μM of compound 1. ent-Androsterone (10 μM) enhances 2 μM GABA-mediated chloride currents at rat α1β2γ2L GABA receptors expressed in Xenopus laevis oocytes.5 Compound 1 at concentrations up to 10 μM does not enhance 2 μM GABA-mediated chloride currents at these expressed receptors. Finally, ent-androsterone causes 50% loss of righting reflex for tadpoles at a concentration of 3.38 μM.5 By contrast, compound 1 did not cause loss of righting reflex in tadpoles at a concentration as high as 10 μM. Thus, despite the similarities in the overall shapes of the two steroids, only ent-androsterone is effective as a positive modulator of GABAA receptors at concentrations below 10 μM.
3. Conclusion
We have described the synthesis and crystal structure of (2β, 5α,13β,14β)-2-hydroxygonan-17-one (1) from commercially available 19-nortestosterone. Novel features of the synthetic route developed include the first use of a SmI2-promoted reaction to remove a 3-acetyloxy group from a 2-keto-3-(acetyloxy)steroid, the first report of the abnormal Beckmann rearrangement on steroids having the 13β,14β cis C,D-ring fusion and the first use of SmI2-promoted reactions to prepare 18-norsteroids having a 13β,14β cis C,D-ring fusion. Omission of the part of the reported synthetic sequence that was used to construct the 13β,14β cis C,D-ring fusion would also allow other 2β-hydroxygonane analogues with the 13β,14α trans C,D-ring fusion to be prepared using methods reported previously for the synthesis of (13β,14α)-18-norsteroids.20,23
4. Experimental
4.1 General Methods
Melting points were determined on a Kofler micro hot stage and are uncorrected. NMR spectra were recorded in CDCl3 at 300 MHz (1H) or 75 MHz (13C). IR spectra were recorded as films on a NaCl plate. Elemental analyses were carried out by M-H-W Laboratories, Phoenix, AZ.
4.1.1 (3β,5α,16ββ))-16-Bromo-3-hydroxyestran-17-one (4)
A mixture of known compound 324 (1.40 g, 5.07 mmol) and copper bromide (3.06 g, 13.7 mmol) in methanol (35 ml) was stirred under reflux for 12 h. Then the solvent was removed under reduced pressure to give a residue and water (25 mL) was added. The product was extracted with CH2Cl2 (25 mL), the organic layer was washed with water (2 × 10 mL), brine (20 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; CH2Cl2/EtOAc, 10:1) to give compound 4 (1.24 g, 69%) as white crystals: mp 108–110 °C (EtOAc-hexanes); [α] 20D = +59.1 (c = 0.30, CHCl3); 1H NMR δ 4.56 (m, 1H), 3.56 (brs, 1H), 3.05 (s, 1H), 2.18 (m, 2H), 0.91 (s, 3H); 13C NMR δ 13.6, 24.4, 27.7, 29.0, 31.7, 32.5, 33.3, 34.9, 39.2, 40.4, 42.4, 45.4, 46.0, 46.5, 47.2, 47.3, 69.3, 213.0; IR νmax 3400, 2920, 2853, 1748, 1448, 1023 cm−1. Anal. Calcd. For C18H27BrO2: C 60.85, H 7.66; Found C 60.78, H 7.87.
4.1.2 (3β,5α)-3-Hydroxyestr-14-en-17-one (5a) and (3β,5α,14ββ))-3-Hydroxyestr -15-en-17- one (5b)
The mixture of compound 4 (0.65 g, 1.8 mmol), LiBr (0.42 g, 5.0 mmol) and Li2CO3 (0.35 g, 5.0 mmol) in DMF (30 mL) was stirred under reflux for 4 h. Afterwards, the reaction mixture was chilled to room temperature and was poured into water (50 mL), the mixture was extracted with EtOAc (2 ×3 0 mL), the combined organic extract was washed with saturated NaHCO3 (10 mL), water (10 mL), brine (10 mL) and dried over Na2SO4. The solvent was removed under reduced pressure. The products were purified by column chromatography (silica gel; CH2Cl2/EtOAc, 10:1) to give compound 5a (0.17 g, 33%; Rf = 0.47, CH2Cl2/EtOAc, 10:1) and compound 5b (0.20 g, 39%; Rf = 0.33, CH2Cl2/EtOAc, 10:1). Compound 5a was obtained as an oil: 1H NMR δ 5.47 (d, J = 1.8 Hz, 1H), 3.59 (m, 1H), 2.86 (m, 2H), 2.29 (s, 1H), 1.12 (s, 3H); 13C NMR δ 19.7, 25.2, 27.8, 28.0, 32.9, 33.0, 35.4, 40.8, 41.0, 41.3, 43.0, 46.5, 48.6, 50.7, 70.0, 112.3, 152.8, 222.7; IR νmax 3400, 3059, 2921, 2855, 1744, 1641, 1449, 1046 cm−1.
Compound 5b was obtained as an oil: 1H NMR δ 7.64 (dd, J = 6.0, 2.4 Hz, 1H), 6.15 (dd, J = 6.0, 2.4 Hz, 1H), 3.57 (m, 1H), 2.63 (m, 1H), 1.11 (s, 3H); 13C NMR δ 21.3, 24.8, 28.0, 30.1, 31.7, 33.4, 35.5, 39.7, 39.8, 40.8, 42.9, 47.3, 47.5, 54.4, 70.2, 132.2, 163.3, 215.2. IR νmax 3400, 2921, 2854, 1705, 1585, 1449, 1052 cm−1. Compound 5a and 5b were not purified further and were used in the next step directly.
4.1.3 (3β,5α,14β )-3-Hydroxyestran-17-one (6)
A mixture of compounds 5a and 5b (3.90 g, 14.2 mmol) was dissolved in EtOAc (50 mL) and hydrogenated under H2 (50 psi) in the presence of 5% Pd-C (400 mg) at room temperature overnight. The reaction mixture was filtered through a pad of Celite 545® to remove catalyst and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography (silica gel; EtOAc/hexanes, 1:1) to give compound 6 (3.81 g, 97%) as white crystals: mp 116–118 ºC (EtOAc); [α] 20D = +92.0 (c = 0.27, CHCl3); 1H NMR δ 3.56 (m, 1H), 3.27 (brs, 1H), 2.45 (dd, J = 19.2, 8.1 Hz, 1H), 2.13 (dd, J = 19.2, 9.9 Hz, 1H), 1.07 (s, 3H); 13C NMR δ 18.1, 19.3, 24.6, 27.6, 27.7, 30.0, 33.4, 35.1, 35.6, 39.1, 40.0, 40.6, 42.7, 46.2, 47.5, 47.8, 69.6, 222.8; IR νmax 3401, 2917, 2854, 1732, 1448, 1048 cm−1. Anal. Calcd. For C18H28O2: C 78.21, H 10.21; Found C 78.42, H 9.97.
4.14 (3β,5α,14β)-3-Hydroxyestran-17-one cyclic-(1,2-ethanediyl acetal) (7)
A solution of compound 6 (0.41 g, 1.49 mmol), ethylene glycol (2.2 g, 35 mmol) and p-TsOH (60 mg, 0.32 mmol) in benzene (35 mL) was refluxed using a Dean-Stark trap for 12 h. The mixture was cooled to room temperature, diluted with ether (50 mL), washed with saturated NaHCO3 (2 × 20 mL) and brine (2 × 20 mL). The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure to give a crude product which was purified by column chromatography (silica gel; EtOAc/hexanes, 1:1) to yield compound 7 (406 mg, 85%) as an oil: [α] 20D = +73.9 (c = 0.065, CHCl3); 1H NMR δ 3.86 (m, 4H), 3.55 (m, 1H), 0.92 (s, 3H); 13C NMR δ 16.1, 19.7, 25.3, 27.8, 28.9, 30.3, 32.1, 33.6, 35.3, 39.4, 39.7, 40.8, 43.0, 45.1, 46.4, 46.8, 63.7, 65.0, 69.9, 120.4; IR νmax 3368, 2920, 2856, 1447, 1367, 1031 cm−1. Anal. Calcd. For C20H32O3: C 74.96, H 10.06; Found C 75.12, H 9.86.
4.1.5 (5α,14β )-estran-3,17-dione cyclic-17-(1,2-ethanediyl acetal) (8)
To a solution of compound 7 (406 mg, 1.27 mmol) in CH2Cl2 (25 mL) was added NaOAc (0.46 g, 5.6 mmol) and PCC (0.72 g, 3.35 mmol) at 0° C. The mixture was stirred at 0° C for 2 h and allowed to stand overnight. Then the reaction mixture was diluted with Et2O (100 mL) and the mixture was filtered through a pad of silica gel. Removal of solvent gave a residue which was purified by column chromatography (silica gel; hexanes/EtOAc, 5:1) to give compound 8 (302 mg, 75%) as white crystals: mp: 85–86 °C (hexanes/EtOAc); [α] 20D = +103.9 (c = 0.28, CHCl3); 1HNMR δ 3.90 (m, 4H), 2.29 (m, 4H), 0.94 (s, 3H); 13CNMR δ 16.3, 19.8, 25.7, 29.0, 30.0, 30.1, 32.2, 34.2, 39.4, 39.7, 41.1, 43.4, 45.2, 46.1, 46.9, 48.5, 63.9, 65.2, 120.4, 211.6; IR νmax 2935, 2880, 1714, 1470, 1448 cm−1. Anal. Calcd. For C20H30O3: C 75.43, H 9.50; Found C 75.63, H 9.38.
4.1.6 (5α,14β)-2-(Phenylmethylene)estran-3,17-dione cyclic-17-(1,2-ethanediyl acetal) (9)
A mixture of compound 8 (1.60 g, 5.03 mmol), benzaldehyde (1.75 g, 16.5 mmol) and KOH (400 mg, 7.14 mmol) in EtOH (30 mL) and water (5 mL) was stirred at room temperature in the dark for 24 h. Then the reaction mixture was poured into ice-water (50 mL), and the product was extracted with CH2Cl2 (2 × 25 mL), the organic phase was washed with water (15 mL),brine (15 mL) and dried over Na2SO4. The solvent was removed under reduced pressure to give a residue which was purified by column chromatography (silica gel; hexanes/EtOAc, 6:1) to give compound 9 (1.51 g, 74%) as light yellow crystals: mp: 153–155 °C (EtOAc); [α] 20D = +43.0 (c = 0.33, CHCl3); 1H NMR δ 7.47 (d, J = 2.1 Hz,1H), 7.40 (m, 5H), 3.89 (m, 4H), 3.36 (dd, J = 16.2, 2.7 Hz, 1H), 2.63 (dd, J = 16.8, 4.2 Hz, 1H), 0.91 (s, 3H); 13C NMR δ 16.4, 19.9, 25.7, 29.0, 29.9, 32.3, 33.2, 33.7, 39.0, 39.1, 40.8, 44.4, 45.3, 47.0, 47.4, 64.0, 65.3, 120.5, 128.3 (2 × C), 128.5, 130.3 (2 × C), 135.4, 135.6, 135.8, 201.5; IR νmax 2918, 2860, 1682, 1594, 1572, 1491, 1085, 735 cm−1. Anal. Calcd. For C27H34O3: C 79.76, H 8.34; Found C 79.66, H 8.35.
4.1.7 (3β,5α,14β)-3-Hydroxy-2-(Phenylmethylene)estran-17-one cyclic-(1,2-ethanediyl acetal) (10)
To a solution of compound 9 (1.51 g, 3.72 mmol) in THF (40 mL) and MeOH (25 mL) was added NaBH4 (70 mg, 1.85 mmol) at room temperature. The mixture was stirred for 30 min. Most of solvent was removed under reduced pressure and EtOAc (35 mL) was added to the residue. The organic phase was washed with water (20 mL), brine (20 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 3:1) to give compound 10 (1.50 g, 99%) as a thick oil: [α] 20 D = +67.2 (c = 0.49, CHCl3); 1H NMR δ 7.34 (m, 5H), 6.57 (s, 1H), 4.12 (m, 1H), 3.88 (m, 4H), 3.10 (dd, J = 13.5, 2.4 Hz, 1H), 2.43 (brs, 1H), 0.91 (s, 3H); 13C NMR δ 16.4, 19.9, 25.3, 29.0, 30.3, 31.2, 32.3, 33.2, 39.3, 40.3, 41.4, 44.7, 45.3, 47.0, 48.5, 63.9, 65.2, 72.4, 118.3, 120.6, 125.9, 128.0 (2 × C), 128.8 (2 × C), 137.9, 144.1; IR νmax 3435, 2917, 2859, 1445, 1087 cm−1; Anal. Calcd. For C27H36O3: C 79.37, H 8.88; Found C 79.54, H 8.96.
4.1.8 (3β,5α,14β)-3-(Acetyloxy)-2-(phenylmethylene)estran-17-one cyclic-(1,2-ethanediyl acetal) (11)
A mixture of compound 10 (1.50 g, 3.67 mmol), acetic anhydride (4.5 mL, 480 mmol), pyridine (4.5 mL) and 4-dimethylaminopyridine (10 mg, 0.082 mmol) was stirred at room temperature for 5 min. The reaction mixture was poured into ice-water (50 mL) and was extracted with EtOAc (2 × 30 mL). The combined organic extract was washed with water (2 × 20 mL), 10% NaHCO3 (10 mL), brine (20 mL) and dried over Na2SO4. After solvent removed under reduced pressure, the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 4:1) to give compound 11 (1.48 g, 90%) as white crystals: mp 141–143 °C (hexanes/EtOAc); [α] 20D = +105.0 (c = 0.20, CHCl3); 1H NMR δ 7.33 (m, 5H), 6.36 (s, 1H), 5.32 (m, 1H), 3.88 (m, 4H), 3.24 (dd, J = 13.5, 3.3 Hz, 1H), 2.16 (s, 3H), 0.91 (s, 3H); 13C NMR δ 16.4, 19.9, 21.2, 25.3, 29.1, 30.3, 31.5, 32.3, 33.1, 39.5, 40.3, 41.0, 41.2, 45.3, 47.0, 48.3, 64.0, 65.3, 73.8, 118.9, 120.6, 126.2, 128.1 (2 × C), 128.8 (2 × C), 137.5, 139.3, 170.1; IR νmax 2931, 2860, 1738, 1599, 1241, 1046 cm−1. Anal. Calcd. For C29H38O4: C 77.30, H 8.50; Found 77.50, H 8.61.
4.1.9 (3β,5α,14β)-3-(Acetyloxy)estran-2,17-dione cyclic-17-(1,2-ethanediyl acetal) (12)
A solution of compound 11 (1.48 g, 3.29 mmol) in MeOH (60 mL) and EtOAc (30 mL) was treated with ozone at −78 °C until a blue color persisted (ca 30 min). Oxygen was passed through the solution for 20 min until the blue color disappeared, and Me2S (10 mL) was added to the solution, the reaction mixture was stirred overnight from −78 °C to room temperature. Solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 3:1) to give compound 12 (0.98 g, 80%) as white crystals: mp 188–189 °C (hexanes-EtOAc); [α] 20D = +125.0 (c = 0.45, CHCl3); 1H NMR δ 5.20 (m, 1H), 3.88 (m, 4H), 2.70 (d, J = 13.2 Hz, 1H), 2.13 (s, 3H), 0.91 (s, 3H); 13C NMR δ 16.1, 19.6, 20.3, 25.0, 28.6, 29.8, 32.0, 32.1, 38.5, 38.7, 39.9, 40.6, 43.3, 45.0, 46.5, 48.0, 63.7, 65.0, 75.5, 120.1, 169.5, 203.8; IR νmax 2933, 2863, 1748, 1728, 1238, 1092 cm−1. Anal. Calcd. For C22H32O5: C 70.18, H 8.57; Found C 69.93, H 8.80.
4.1.10 (5α,14β)-estran-2,17-dione cyclic-17-(1,2-ethanediyl acetal) (13), (2β,5α,14β)- 2-hydroxyestran-17-one cyclic-(1,2-ethanediyl acetal) (14a) and (2α,5α,14β)-2-hydroxyestran-17-one cyclic-(1,2-ethanediyl acetal) (14b)
Iodine (2.06 g, 8.12 mmol) in dried THF (30 mL) was added to samarium filings (1.50 g, 10 mmol) by syringe under Ar. The mixture was stirred at room temperature for 30 min to give a black blue solution. Then compound 12 (0.92 g, 2.45 mmol) in dried THF (15 mL) and MeOH (5 mL) was added to the SmI2-THF solution and the reaction mixture was stirred for 15 min. The reaction mixture was poured into 10% Na2CO3 (70 mL) and was extracted with EtOAc (3 × 30 mL), the combined organic extract was washed with water (20 mL), brine (20 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; CH2Cl2/EtOAc, 10:1) to give compound 13 (0.32 g, 41%), compound 14a (0.16 g, 20%) and compound 14b (0.20 g, 26%).
Compound 13 was obtained as white crystals: mp 143–145 °C (hexanes/EtOAc); [α] 20D = +91.4 (c = 0.45, CHCl3); 1H NMR δ 3.89 (m, 4H), 2.59 (dd, J = 15.6, 2.7 Hz, 1H), 2.35 (m, 2H), 0.91 (s, 3H); 13C NMR δ 16.2, 19.8, 24.9, 28.8, 30.0, 32.2, 32.6, 33.3, 38.8, 41.0, 41.1, 41.2, 45.0, 45.1, 46.7, 47.7, 63.9, 65.1, 120.4, 211.7; IR νmax 2948, 2858, 1714, 1455, 1088 cm−1. Anal. Calcd. For C20H30O3: C 75.43, H 9.50; Found C 75.29, H 9.70.
Compound 14a was obtained as white crystals: mp 136–138 ºC (hexanes/EtOAc); [α] 20D = +105.5 (c = 0.12, CHCl3); 1H NMR δ 4.13 (m, 1H), 3.89 (m, 4H), 0.91 (s, 3H); 13C NMR δ 16.4, 19.9, 25.1, 27.6, 29.1, 30.5, 32.3, 32.5, 33.8, 36.3, 39.7, 40.1, 40.8, 42.6, 45.2, 47.2, 64.0, 65.2, 66.6, 120.8; IR νmax 3436, 2918, 2860, 1305, 1095 cm−1. Anal. Calcd. For C20H32O3: C 74.96, H 10.06; Found C 74.94, H 9.94.
Compound 14b was obtained as white crystals: mp 141–143 °C (hexanes/EtOAc); [α] 20D = +81.2 (c = 0.07, CHCl3); 1H NMR δ 3.89 (m, 4H), 3.55 (m, 1H), 0.91 (s, 3H); 13C NMR δ 16.4, 20.0, 25.3, 29.2, 30.5, 32.0, 32.4, 33.3, 35.4, 39.2, 39.5, 40.0, 41.9, 45.3, 45.8, 47.1, 64.0, 65.3, 71.2, 120.8; IR νmax 3368, 2920, 2857, 1305, 1097, 1031 cm−1. Anal. Calcd. For C20H32O3: C 74.96, H 10.06; Found C 74.92, H 10.18.
14.1.11 (2β,5α,14β)-2-hydroxyestran-17-one cyclic-(1,2-ethanediyl acetal) (Compound 14a)
To a solution of compound 13 (406 mg, 1.28 mmol) in dried THF (35 mL) was added 1M K-Selectride (6.5 mL, 6.5 mmol) in THF at −78 °C under N2 and the mixture was stirred for 1 h at this temperature. Then the reaction was quenched by adding 10% NaOH (8 mL) and 30% hydrogen peroxide (8 mL), and the reaction was continued for other 30 min. The mixture was extracted with EtOAc (3 × 30 mL). The combined organic phase was washed with water (10 mL), brine (2×10 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; CH2Cl2/EtOAc, 10:1) to give compound 14a (346 mg, 85%) whose properties were identical to those for this product when isolated directly from the SmI2 reduction.
14.1.12 (2β,5α,14β)-2-(Acetyloxy)estran-17-one cyclic-(1,2-ethanediyl acetal) (15)
A mixture of compound 14 (476 mg, 1.49 mmol), acetic anhydride (2 mL), dry pyridine (2 mL) and 4-dimethylaminopyridine (10 mg) was stirred at room temperature for 5 min. The reaction mixture was poured into ice-water (15 mL) and was extracted with EtOAc (2 × 15 mL). The organic phase was washed successively with saturated NaHCO3 (5 mL), water (5 mL), 10% aqueous HCl (5 mL), water (5 mL), brine (5 mL) and dried over Na2SO4. The solvent was removed under reduced pressure to give oily compound 15 (496 mg, 92%) that was not purified and was directly converted to compound 16.
14.1.13 (2β,5α,14β)-2-(Acetyloxy)estran-17-one (16)
The mixture of compound 15 (496 mg, 1.37 mmol), p-TsOH (50 mg, 0.26 mmol) and water (36 mg, 2 mmol) in acetone (15 mL) was stirred at room temperature overnight. After the solvent was removed under reduced pressure, EtOAc (35 mL) was added to the residue. and the organic phase was washed with saturated NaHCO3 (10 mL), water (10 mL), brine (10 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by re-crystallization (hexanes/EtOAc) to give compound 16 (430 mg, 99%) as white crystals: mp 144–146 °C (hexanes/EtOAc); [α] 20D = +92.3 (c = 0.12, CHCl3); 1H NMR δ 5.10 (s, 1H), 2.44 (dd, J = 18.6, 7.2 Hz, 1H), 2.05 (s, 3H), 1.08 (s, 3H); 13C NMR δ 18.2, 19.4, 21.1, 24.3, 27.7, 28.0, 29.4, 30.0, 33.1, 33.4, 35.6, 39.2, 40.2, 41.3, 41.9, 47.4, 48.1, 69.6, 170.1, 222.3; IR νmax 2910, 2850, 1727, 1250, 1020 cm−1. Anal. Calcd. For C20H30O3: C 75.43, H 9.50; Found C 75.27, H 9.39.
14.1.14 (2β,5α,14β)-2-(Acetyloxy)estran-17-one oxime (17)
The mixture of compound 16 (460 mg, 1.47 mmol), hydroxylamine hydrochloride (1.00 g, 14.4 mmol) and NaOAc (1.15 g, 14.0 mmol) in MeOH (35 mL) was refluxed for 1.5 h. The solvent was removed partially under reduced pressure. Water (20 ml) was added to the residue. Then the mixture was extracted with EtOAc (2 × 20 mL), the combined organic extract was washed with water (20 mL), brine (20 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by re-crystallization (hexanes/EtOAc) to give compound 17 (476 mg, 97%) as white crystals: mp 186–188 °C (hexanes/EtOAc); [α] 20D = +76.4 (c = 0.89, CHCl3); 1H NMR δ 9.44 (s, 1H), 5.10 (s, 1H), 2.06 (s, 3H), 1.18 (s, 3H); 13C NMR δ 20.7, 21.5, 21.8, 25.2, 25.3, 28.2, 29.6, 31.0, 32.0, 33.5, 33.8, 39.1, 40.3, 41.7, 42.2, 43.9, 50.3, 70.1, 173.0, 176.4; IR νmax 3305, 2918, 2857, 1737, 1369, 1242, 937 cm−1. Anal. Calcd. For C20H31NO3: C 72.04, H 9.37, N 4.20; Found C 72.18, H 9.12, N 4.10.
14.1.15 (1R,4aR,4bS,6S,8aS,10aS)-6-(Acetyloxy)tetradecahydro-2-methylene-1-phenanthrenepropanenitrile (18)
To a solution of compound 17 (476 mg, 1.43 mmol) and trimethyl orthoformate (2.8 mL, 25.6 mmol) in dried THF (35 mL) was added dropwise trifluoroacetic acid (0.26 mL, 35 mmol) at 60 °C for 10 min, and the reaction was continued for another 30 min. Then saturated Na2CO3 (4.5 mL) was added, the solvent was partially removed under reduced pressure and EtOAc (50 mL) was added. The organic phase was washed with saturated NaHCO3 (10 mL), water (10 mL), brine (10 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; hexanes/EtOAc, 4:1) to give compound 18 (356 mg, 79%) as white crystals: mp 103–105 °C (EtOAc); [α] 20D = +21.5 (c = 0.13, CHCl3); 1H NMR δ 5.10 (s, 1H), 4.74 (t, J = 2.1 Hz, 1H), 4.72 (d, J = 9.0 Hz, 1H), 2.03 (s, 3H); 13C NMR δ 15.3, 21.5, 22.0, 28.2, 29.7, 29.9, 30.2, 31.7, 33.6, 33.7, 40.2, 42.1, 42.4, 46.7, 47.8, 70.0, 109.7, 112.0, 149.2, 170.6; IR νmax 2920, 2856, 2244, 1733, 1647, 1246 cm−1. Anal. Calcd. For C20H29NO2: C 76.15, H 9.27, N 4.44; Found C 76.13, H 9.32, N 4.19.
14.1.16 (1R,4aR,4bS,6S,8aS,10aR)-6-(Acetyloxy)tetradecahydro-2-oxo-1-phenanthrenepropanenitrile (19)
A solution of compound 18 (356 mg, 1.13 mmol) in MeOH (60 mL) and EtOAc (35 mL) was treated with ozone at −78 °C until a blue color persisted (ca 30 min). Oxygen was passed through the solution for 20 min until the blue color disappeared, and Me2S (4.5 mL) was added at −78 °C and the resultant mixture was stirred overnight while warming to room temperature. Solvent was removed under reduced pressure and the residue was purified by column chromatography (silica gel; hexanes/EtOAc/CH2Cl2, 4:1:1) to give compound 19 (206 mg, 58%) as white crystals: mp 157–159 °C (hexanes/EtOAc); [α] 20D = +24.4 (c = 0.13, CHCl3); 1H NMR 5.11 (s, 1H), 2.46 (m, 1H), 2.04 (s, 3H); 13C δ NMR δ 15.1, 21.2, 22.6, 27.7, 29.2, 29.4, 30.4, 33.0, 33.6, 37.2, 39.1, 41.5, 41.7, 45.8, 54.5, 69.5, 118.8, 170.3, 213.1; IR νmax 2922, 2850, 2244, 1727, 1699, 1379, 1241 cm−1. Anal. Calcd. For C19H27NO3: C 71.89, H 8.57, N 4.41; Found C 71.64, H 8.73, N 4.49.
14.1.17 (2β,5α,14β)-2-(Acetyloxy)-13-hydroxy-18-norestran-17-one (20) and (2β,5α,14β)-2-(Acetyloxy)gonan-17-one (21)
Iodine (240 mg, 0.95 mmol) in dried THF (30 mL) was added to samarium filings (225 mg, 1.50 mmol) by syringe under Ar. The mixture was stirred at room temperature for 30 min to give a black blue solution. Then a solution of compound 19 (93 mg, 0.30 mmol) and 2-methyl-2-propanol (38 mg, 0.51 mmol) in THF (10 mL) was added to the freshly made SmI2-THF solution under Ar at 0° C. The reaction mixture was stirred at 0–10 °C while irradiated with a 500 W tungsten lamp for 8 h and then the reaction mixture was poured into 5% aqueous HCl (10 mL), and extracted with EtOAc (2 × 15 mL). The combined organic extract was washed with water (10 mL), 10% NaHCO3 (10 mL), brine (10 mL), and dried over Na2SO4. Solvent removal gave crude compound 20, (46 mg) which was used without purification or characterization.
Under Ar, a solution of crude compound 20 in dried THF (5 mL) and MeOH (0.5 mL) was added to a freshly made SmI2-THF (0.1 M, 15 mL) solution by syringe. The reaction mixture was stirred at room temperature for 10 min, quenched by adding 5% aqueous HCL (10 mL) and extracted with EtOAc (2 × 10 mL). The combined organic extract was washed with water (10 mL), 10% NaHCO3 (10 mL), brine (10 mL) and dried over Na2SO4. After solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; CH2Cl2/EtOAc, 20:1) to give compound 21 (15.6 mg, 17% from compound 19) as white crystals: mp 118–120 °C (EtOAc); 1H NMR δ 5.11 (s, 1H), 2.38 (dd, J = 18.9, 8.1Hz, 1H), 2.05 (s, 3H). From the 13C NMR data this product was determined to be a 9:1 mixture of epimers at C14. The 13C NMR resonances for the major isomer were δ 20.7, 21.5, 22.9, 28.0, 28.3, 29.7, 30.2, 33.5, 33.7, 37.1, 40.6, 41.5, 41.7, 41.8, 42.3, 49.2, 70.0, 170.6, 221.4; IR νmax 2918, 2855, 1738, 1245 cm−1.
14.1.18 (2β,5α,13β,14β)-2-Hydroxygonan-17-one (1)
The mixture of compound 21 (15.6 mg, 0.05 mmol), NaOH (2 mg, 0.05 mmol) and water (18 mg, 1 mmol) in MeOH (12 mL) was refluxed for 2 h. The reaction mixture was chilled, and most of the MeOH was removed under reduced pressure to give a residue to which was added EtOAc (20 mL). The organic phase was washed with 10% aqueous HCl (5 mL), water (5 mL), brine (5 mL) and dried over Na2SO4. After the solvent was removed under reduced pressure, the residue was purified by column chromatography (silica gel; hexanes/CH2Cl2/EtOAc, 3:1:1) to give compound 1 (11 mg, 82%) as white crystalline needles: mp 123–124 °C (EtOAc/hexanes); [α] 20D = +140 (c = 0.30, CHCl3); 1H NMR δ 4.16 (m, 1H), 2.38 (dd, J = 18.9, 8.1Hz, 1H); 13C NMR δ 20.7, 23.0, 27.6, 28.0, 30.3, 32.5, 33.7, 36.3, 37.2, 40.6, 40.8, 41.7, 41.8, 42.6, 49.2, 66.7, 221.5; IR νmax 3565, 2923, 2851, 1720 cm−1. Anal. Calcd. For C17H26O2: C 77.82, H 9.99; Found: C 77.68, H 10.02.
Scheme 4.
Acknowledgments
This work was supported by NIH grant GM47969. Instrumentation for crystallographic studies was made possible by NSF Grant CHE-0420497. The authors thank Dr. Alex S. Evers, Dr. Steven Mennerick, Dr. Charles F. Zorumski, Ann Benz and Brad D. Manion for the biological evaluations of compound 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Phillips GH. J Steroid Biochem. 1975;6:607. doi: 10.1016/0022-4731(75)90041-2. [DOI] [PubMed] [Google Scholar]
- 2.Covey DF, Evers AS, Mennerick S, Zorumski CF, Purdy RH. Brain Res Rev. 2001;37:91. doi: 10.1016/s0165-0173(01)00126-6. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton NM. Curr Top Med Chem. 2002;2:887. doi: 10.2174/1568026023393570. [DOI] [PubMed] [Google Scholar]
- 4.Belelli D, Lambert JJ. Nat Rev Neurosci. 2005;6:565. doi: 10.1038/nrn1703. [DOI] [PubMed] [Google Scholar]
- 5.Katona BW, Krishnan K, Cai ZY, Manion BD, Benz A, Taylor A, Evers AS, Zorumski CF, Mennerick S, Covey DF. Eur J Med Chem. doi: 10.1016/j.ejmech.2007.02.017. In press. [DOI] [PubMed] [Google Scholar]
- 6.Purdy RH, Morrow AL, Blinn JR, Paul SM. J Med Chem. 1990;33:1572. doi: 10.1021/jm00168a008. [DOI] [PubMed] [Google Scholar]
- 7.Anderson A, Boyd AC, Clark JK, Fielding L, Gemmell DK, Hamilton NM, Maidment MS, May V, McGuire R, McPhail P, Sansbury FH, Sundaram H, Taylor R. J Med Chem. 2000;43:4118. doi: 10.1021/jm000977e. [DOI] [PubMed] [Google Scholar]
- 8.Wittmer LL, Hu Y, Kalkbrenner M, Evers AE, Zorumski CF, Covey DF. Mol Pharmacol. 1996;50:1581. [PubMed] [Google Scholar]
- 9.Han M, Zorumski CF, Covey DF. J Med Chem. 1996;39:4218. doi: 10.1021/jm960304p. [DOI] [PubMed] [Google Scholar]
- 10.Zeng CM, Manion BD, Benz A, Evers AS, Zorumski CF, Mennerick S, Covey DF. J Med Chem. 2005;48:3051. doi: 10.1021/jm049027+. [DOI] [PubMed] [Google Scholar]
- 11.McKinney AR, Ridley, Damon D, Turner P. Aust J Chem. 2003;56:829. [Google Scholar]
- 12.Han M, Hayes BA, Prendergast PT, Gupta S. J Labelled Compd Radiopharm. 2000;43:1149. [Google Scholar]
- 13.Groszek G, Kabat MM, Kurek A, Masnyk M, Wicha J. Bull Pol Acad Sci Chem. 1986;34:313. [Google Scholar]
- 14.Numazawa M, Nagaoka M, Osawa Y. J Org Chem. 1982;47:4024. [Google Scholar]
- 15.Liu Z, Zhang J, Cheng W. Chinese Chem Lett. 1994;5:39. [Google Scholar]
- 16.Leese MP, Newman SP, Purohit A, Reed MJ, Potter BVL. Bioorg & Med Chem Lett. 2004;14:3135. doi: 10.1016/j.bmcl.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Demir AS, Sabol MR, Jeganathan A, Kurt Dolence E, Watt DS, Moldowan JM. Org Prep and Proced Int. 1987;19:197. [Google Scholar]
- 18.Ohloff G, Maurer B, Winter B, Giersch W. Helv Chim Acta. 1983;66:192. [Google Scholar]
- 19.Wang C, Jiang X, Shi J, Lu J, Hu Y, Hu H. J Org Chem. 2003;68:4579. doi: 10.1021/jo034142y. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Wang C, Hu Y, Hu H, Covey DF. J Org Chem. 2000;65:3555. doi: 10.1021/jo991906u. [DOI] [PubMed] [Google Scholar]
- 21.Crystallographic data (excluding structure factors) for the structure in this article have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 634624. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [fax: +44 1223 330633 or email: deposit@ccdc.cam.ac.uk].
- 22.Jiang X, Manion BD, Benz A, Rath NP, Evers AS, Zorumski CF, Mennerick S, Covey DF. J Med Chem. 2003;46:5334. doi: 10.1021/jm030302m. [DOI] [PubMed] [Google Scholar]
- 23.Han M, Covey DF. J Org Chem. 1996;61:7614. doi: 10.1021/jo9611430. [DOI] [PubMed] [Google Scholar]
- 24.Counsell RE. Tetrahedron. 1961;15:202. [Google Scholar]