

Abstract
Smart biomaterials composed of pH responsive polymers, poly((meth)acrylic acid), were synthesized using a precipitation polymerization technique. The microparticles were grafted with poly(ethylene glycol) (PEG) chains that are capable of complexing with the hydroxyl groups of the polyacid and interpenetrating into the mucus gel layer upon entry into the small intestine. Upon introduction of an alkaline solution, these materials imbibe a significant amount of water and create a highly viscous suspension. These materials have the necessary physicochemical properties to serve as mucoadhesive controlled release drug carriers for the oral delivery of drugs.
Keywords: Poly(acrylic acid), poly(methacrylic acid), poly(ethylene glycol), hydrogels, pH responsive
1. Introduction
Tethered polymer networks have received considerable attention for their use in modifying surface adhesion and friction [1-3], drug delivery systems [4-8], and bioadhesives [1,2,9,10]. Biomimetic systems, biomaterials that mimic a biological environment to elicit a desired cellular response [11], have also utilized tethered macromolecules to effectively enhance cellular adhesion [12-14] or prevent protein adsorption [15]. Certain tethered polymer surfaces have been theorized to act as adhesion promoters by interpenetrating into the mucous gel layer, bridging the interface between the hydrogel-based drug delivery system and the absorption site [16].
Hydrogels have been frequently used in biomedical applications because of the many shared characteristics with natural tissue. The gel/gel adhesion between the hydrogel and the natural tissue can be controlled with the use of tethered polymers at the surface of the biomaterial. Since hydrogels are porous penetrable materials much like natural tissue, the tethered along with the bulk structure of the polymer requires a fundamental understanding to evaluate the structure-property relationships and dynamics.
Synthesis of polymer microparticles as the geometry for the hydrogel material is preferred due to their ease of incorporation into traditional dosage forms such as tablets and capsule formulations. Drug-loaded microparticles can be used on their own as drug delivery systems due to their relatively high loading capacity and ease of administration. Thermally initiated free-radical precipitation polymerizations are typically utilized as the preparation technique for obtaining microparticles possessing the necessary compressibility characteristics needed for successful tablet compression [17].
The incorporation of a (meth)acrylic monomer possessing a carboxylic acid moiety imparts pH responsive behavior into the hydrogel network. The anionic behavior of the polymer allows for ionic repulsion to occur along the polymer backbone, causing the hydrogel networks to expand and collapse depending on whether the pH of the environment is above or below the pKa of the polymer. The addition of a polymer tether capable of participating in interpolymer complexation may affect the swelling behavior, mechanical properties, and solute transport characteristics dependent upon the state of the network, complexed or uncomplexed.
Interpolymer complexation forms between electron deficient moieties, such as the carboxylic acid groups found along the backbone of polyacids, and moieties containing regions of high electron density, such as the ether groups comprising poly(ethylene glycol) (PEG). The complexation behavior is a result of the non-covalent association between the polyacid and the tether as a result of hydrogen-bonding between the carboxyl group and the ether oxygen. This interpolymer complexation is a thermodynamically favorable event and is reversible in nature. The stability of the complex formed is strongly dependent on the structure of the polymers and their relative amounts incorporated into the network.
For complexation to occur, the pH of the environment must be substantially lower than the pKa of the polyacid network to allow for sufficient protonation of the carboxylic acid group. Upon approach to the pKa, deprotonation occurs causing the network to decomplex. Upon decomplexation, the dynamics of the network are significantly altered, allowing for increased swelling, a decrease in the mechanical intergrity of the network, and increased solute diffusion. Due to these interesting characteristics, these polymer networks can be utilized in biosensing applications, molecular recognition, and drug delivery systems.
In our research, we studied the synthesis and characterization of microparticles composed of polymers possessing the ability to form interpolymer complexes stabilized by hydrogen bonding. These tethers are also present to act as adhesion promoters between the hydrogel and other gel networks such as the mucous gel layer covering the epithelium of the gastrointestinal tract. The conditions necessary for a successful polymerization are elucidated. The effects of the concentration and size of the polymer tether on the dynamics of the polymer networks are evaluated. Also, the behavior of two different polyacids in conjunction with the polymer tether is studied.
2. Experimental
2.1 Synthesis
Methacrylic acid (MAA, inhibited with 250 ppm hydroquinone), anhydrous potassium carbonate (K2CO3), and ethyl acetate (EtAc) were obtained from Fisher Scientific and used as received. Acrylic acid (AA, inhibited with 200 ppm hydroquinone), poly(ethylene glycol) methyl ether methacrylate (PEGMMA, Mn ~ 300, 1100) and PEGMMA solution (Mn ~ 2080, 50 wt % in H2O) was obtained from Sigma Aldrich (Milwaukee, WI). The PEGMMA 50 wt % in H2O solution was freeze dried to obtain anhydrous PEGMMA Mn ~ 2080. Allyl pentaerythritol (APE), pentaerythritol triacrylate (PETA), and di(4-tert-butylcyclohexyl) peroxydicarbonate (BCHPC) were kindly supplied by Perstorp Polyols (Toledo, OH), Sartomer (Exton, PA), and Degussa Initiators (Elyria, OH), respectively. All other chemicals were of reagent grade and used as received.
In a typical thermally-initiated free radical precipitation polymerization, MAA, K2CO3, PEGMMA, and deionized distilled water (ddH2O) were combined and mixed to form a homogeneous mixture, allowing the escape of the neutralization by-product carbon dioxide17. The crosslinking agent, APE or PETA, was dissolved in ethyl acetate. The monomer mixture and crosslinking agent were added to a four-necked round bottom flask equipped with an overheard stirrer, nitrogen purge, and condenser containing the polymerization solvent EtAc. Following a 20 minute purge with nitrogen, the initiator BCHPC dissolved in the polymerization solvent was added to the vessel and further purged for an additional 10 minutes.
The molecular structures of the monomer, crosslinking agent, and thermal initiatior are shown in Figure 1. The vessel was placed in a thermostatic bath at 50°C ± 0.5°C where precipitation was evident in a matter of minutes. The reaction was allowed to proceed for 16 hours to ensure a high percentage of monomer conversion. Following the polymerization, the particle slurry was centrifuged and washed with fresh ethyl acetate and dried using a rotary evaporator at elevated temperature and reduced pressure (90°C/40 mmHg).
Figure 1.
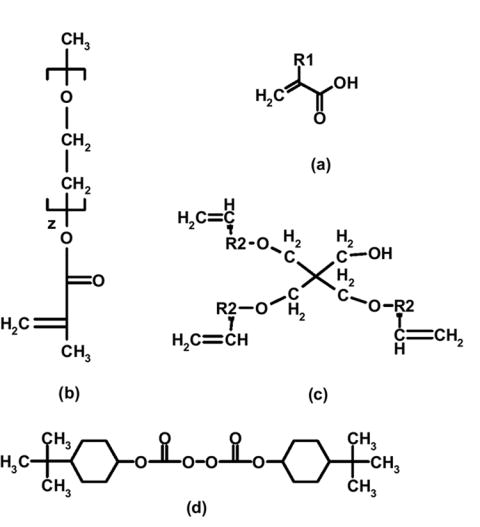
Molecular structures of (a) the carboxylic acid-containing monomer (R1 = H for AA or R1 = CH3 for MAA), (b) a poly(ethylene glycol) tether (z ~ 5, 23, and 45), (c) the tri-functional pentaerythritol (R2 = CH for APE or R2 = COH for PETA), and (d) the thermal initiator di(4-tert-butylcyclohexyl) peroxydicarbonate (BCHPC).
2.2 Characterization
Discs containing 1 mg of sample and 150 mg of KBr were prepared on a Carver laboratory press using a 15,000 lb compression force. Infrared spectra of the microparticles were obtained in the wavenumber range of 400-4000 cm-1 on a Fourier transform infrared spectrophotometer (FT-IR, Thermo Mattson Infinity, Thermo Electron Corp., Waltham, MA) in transmission mode equipped with a KBr beamsplitter and DTGS detector. Each spectrum is an average of 64 scans at a resolution of 1 cm-1.
The thermal properties of the microparticles were characterized using a differential scanning calorimeter (DSC, MDSC 2920, TA Instruments, New Castle, DE). Approximately 10-15 mg samples were analyzed at a sample rate of 10°C/min over the range of 80°C to 160°C for poly(acrylic acid) and 80°C to 300°C for poly(methacrylic acid) using a heat/cool/heat method to erase the thermal history.
The equilibrium weight swelling ratio of the polymeric hydrogel microparticles was determined by carefully weighing 50 mg of dried particles and combining them with 35 mL of NaHCO3 solution (1.5 g / 100 mL). The suspension was agitated for 60 min and then centrifuged for 60 min at 2000 rpm, carefully discarding the supernatant. The pellet was resuspended in an additional 35 mL of NaHCO3 solution and agitated for 60 min. The suspension was centrifuged at 2000 rpm for 60 min, carefully removing the supernatant, and the weight of the gelled mass was determined. This procedure was also carried out in a 0.1 N HCl solution.
Approximately 4.0 g of the prepared polymer microparticles previously dried at 30°C and 28 inHg were added to 350 mL of an agitated aqueous solution containing the appropriate amount of NaCl to achieve the specified ionic strength. After a 15 minute hydration period, the pH of the microparticle slurry was adjusted accordingly using 1 N NaOH, and a sufficient volume of ddH2O was added to make the final volume of liquid 400 mL.
One mL of the polymer gel was placed on the lower of the two cylindrical (D = 19 mm) agarose sample mounts (0.5 g agarose in 50 mL of ddH2O) in a tensile tester (Instron 4301, 10 N load cell, Canton, MA) at 22°C. The upper sample mount was brought into contact with a 42 mN impingement force achieved through resting the upper sample mount on the gel sample and lower sample mount, and the gel was allowed to relax for five minutes. The upper sample mount was raised at 6 mm/min until failure of the gel bond occurred, and the detachment force was measured as a function of displacement. The area under the curve of force versus length up until the peak force is defined as the work of cohesion, and the area post peak force is defined as the work of cohesion.
3. Results and Discussion
3.1 Precipitation Polymerization for Synthesis of Tethered Microparticles
The main requirement for a precipitation polymerization is the presence of an inert diluent which dissolves the monomer but precipitates the polymer as it is formed. In the formation of the tethered microparticles, the monomer, crosslinking agent, tether, and initiator are dissolved in ethyl acetate which is a non-solvent for the formed polymer [18]. Polymer precipitates out and forms agglomerated microparticles. These types of polymerizations are different than those of dispersion polymerizations in that there is no polymeric dispersant present to stabilize the polymer particles as they are formed. This stabilizer forms a dissolved protective layer around the polymer particles as they grow, and its absence presents an unstable dispersion which results in flocculation of the growing polymeric particles. The resultant microparticle is composed of primary polymeric particles which have agglomerated together.
The processes involved with precipitation polymerizations depend on the solubility characteristics of the polymer, especially those processes related to the formation and growth of polymer particles. The free energy change ΔGm must be negative for the monomers and the diluent to mix. More specifically the heat of mixing ΔHm must be smaller than the entropy factor TΔSm since the entropy of mixing will always be positive as can be seen in equation (1).
| (1) |
The total cohesion energy, the total energy needed to separate the molecules of each liquid to a distance infinitely far from one antother, is given by equation (2).
| (2) |
The solubility parameter δ is defined as the square root of the cohesive energy density since the interaction between the unlike molecules can be taken as the geometric mean of the cohesive energy densities of the two components.
| (3) |
The heat of mixing can be obtained by subtracting the contributions of a mixture from the cohesive energies of the separate unmixed components as shown in equation (4) where Vm is the average molar volume of the mixture. Because the interactions between like molecules are counted twice, a factor of 2 is added.
| (4) |
From these equations, the heat of mixing can be expressed in the following form and is minimized when the difference between the solubility parameters of the liquids are as small as possible.
| (5) |
Flory-Huggins solution theory shows that the free energy of mixing (6) is highly dependent on the molecular weight of the polymer.
| (6) |
χ is the Flory polymer-solvent interaction parameter which is related to the solubility parameters of the components by equation, where β is a correction to the Flory combinatorial entropy with a value between 0.2-0.4.
| (7) |
Further evaluation of precipitation and phase separation can be described as an equilibrium process by the Flory-Huggins theory and one can derive the chemical potential for each component of the precipitation polymerization. These derivations yield that the solubility of a polymer in a liquid diluent falls off very rapidly as the molecular weight is increased and as the difference in solubility parameters for each component increases [18].
The precipitation polymerization of tethered microparticles synthesized in this work occurs through the following stages: (a) the polymer formed is insoluble in the diluent and polymer particles precipitate from the initially homogeneous reaction solution; (b) opalescence is detected almost immediately after initiation of polymerization due to the formation of polymer particles at very low conversions; (c) high molecular weight polymers are obtained due to restricted radical termination in the formed glassy particle19.
Due to the biomedical application of the particles, absence of a dispersant was desired to eliminate the need for removal. The resulting size distribution is rather polydisperse with 95% of the particles being smaller than 30 μm (data not shown). The addition of the low molecular weight PEG used in these studies did not act as a polymerizable stabilizer as can be noted from the presence of the agglomeration and the absence of individual primary particles.
For AA precipitation polymerizations, it is necessary to preneutralize a small portion of the acrylic acid monomers to form potassium acrylate. Potassium acrylate imparts the proper solubility characteristics onto the formed polymer to precipate out of solution. Without the addition of potassium acrylate to the monomer solution, polymerization and marginal precipitation occurs, but the large agglomerates formed are undesirable. The polymerization proceeds as a solution polymerization without production of any microparticles. For PMAA, the preneutralization is not necessary due to the solubility characteristics of the formed polymer, but can be utilized to increase the microclimate pH of the particles formed. However, a large concentration of potassium acrylates and methacrylates is insoluble in ethyl acetate, resulting in a heterogeneous monomer/diluent mixture.
For the P(AA-g-PEG) polymerizations, the PEG does affect the solubility of the formed microparticles. With PEG being soluble in ethyl acetate, its incorporation results in a higher propensity for salvation of the growing polymer chains. This effect can be kinetically controlled by decreasing the reaction temperature after initiation to decrease the extent of agglomeration that occurs. For P(MAA-g-PEG) polymerizations, this effect is negligible due to large extent of the insolubility of PMAA in ethyl acetate.
The choice of initiator relies on the fact that a lower reaction temperature is needed for increasing concentrations of PEG. The peroxydicarbonate provides this element, however, lauroyl peroxide can also be utilized as the initiator for systems composed of a high AA:EG ratio or for all MAA systems. The choice of crosslinking agent is also dependent on the monomer employed. Due to the reactivity ratios of acrylic and methacrylic acid, an allyl or acrylate functionalized crosslinking agent, respectively, must be used to achieve a crosslinked network.
3.2 FT-IR Spectroscopy of Poly(ethylene glycol)-tethered Biomaterials
Figure 3 shows the IR spectra for crosslinked poly(acrylic acid) (PAA) microparticles containing the PEGMMA tether at various concentrations and potassium acrylate (P(AA-g-PEG)) and Table 1 lists the vibrational assignments for the various peaks present in the spectra. All spectra exhibit the characteristic C=O stretching, with crosslinked PAA stretching occurring at 1710 cm-1 and moving to a slightly higher wavenumber with increasing PEG tether concentration (1740 cm-1 for AA:EG 50:50). This indicates a higher prevalence of obtaining cyclic hydrogen-bonded COOH groups in dimeric form with a decreasing concentration of the PEG tether [20]. As the concentration of PEG increases in the network, the concentration of free (non-hydrogen-bonded) COOH groups increases due to the disruption or prevention of dimeric formation with the addition of the PEG macromolecule into the structure. The asymmetric vibration of the methyl group present at the end of the PEG tether is exhibited on the FT-IR spectrum at approximately 2890 cm-1. The characteristic C-O-C stretching of the PEG tether is clearly evident at approximately 1110 cm-1.
Figure 3.

FT-IR spectrum of (a) crosslinked PAA (0.75 mol % APE), (b) P(AA-g-PEG), PEG-1000, AA:EG 98:2, (c) 90:10, (d) 83:17, (e) 60:40, (f) 50:50.
Table 1.
Vibrational assignments for crosslinked PAA and the effect of PEG tether addition on the spectra of P(AA-g-PEG) microparticles
| Wavenumber (cm-1) | Intensity1 | Assignment2 | |
|---|---|---|---|
| x-PAA | PAA-g-PEG | ||
| 3440 | v | υ (O-H) free groups | |
| ~3200 | m | υ (O-H) associated w/water | |
| 2970 | w | υa (C-H) CH2 | |
| 2890 | m-s | υa (CH3) | |
| ~2620 | w | υ (O-H) bonded groups | |
| 1710 | shifts to slightly higher | s | υ (C=O) |
| 1560 | s | υ carboxylate ion | |
| 1460 | 1460 | w | δ (CH2) out of plane |
| 1260 | s | υ (C=O) or δ (O-H) | |
| 1190 | s | related to 1260 | |
| 1110 | s | υ (C-O-C) | |
| 820 | w | γ (CH2) | |
Intensity: v, variable, w, weak, m, medium, s, strong.
Vibrtations: υ, stretching, a, asymmetric, δ, bending, γ, rocking.
3.3. Differential Scanning Calroimetry
The glass transtition temperature, Tg, of the dry crosslinked PAA is approximately 131°C, which is significantly higher than that reported for linear PAA (Figure 4). As reported previously [17], this can be attributed to the presence of the pentaerythritol crosslinking agent and potassium acrylate. Upon incorporation of small amounts of the PEG tether (AA:EG 98:2 and 90:10), the ΔCp is decreased with no change or appearance of a second Tg observed. It is not until the ratio of AA:EG is increased to 83:17 that a second Tg is observed. This is due to a heterogeneous network consisting of crosslinked PAA rich domains and domains containing both crosslinked PAA and the PEG tether. Upon further increase of the PEG concentration, a single lower Tg is observed, indicating the return to a more homogenous network.
Figure 4.
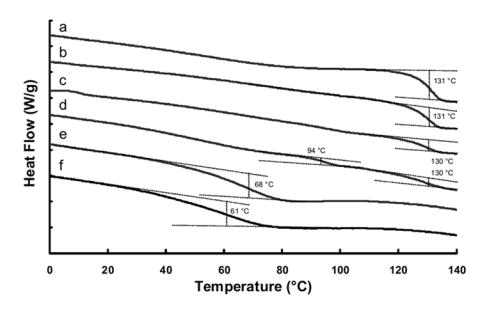
DSC thermogram of (a) crosslinked PAA (0.75 mol % APE), (b) PAA PAA-g-PEG, PEG-1000, AA:EG 98:2, (c) 90:10, (d) 83:17, (e) 60:40, (f) 50:50.
3. 4. Swelling Behavior of PEG-tethered Hydrogel Networks
To evaluate the swelling properties of the PEG-tethered hydrogel networks, the amount of water uptake of the microparticles, q, was calculated according to equation (8), where Ws and Wd are the weight of the swollen polymer and the dry polymer, respectively.
| (8) |
The polymer volume fraction after equilibrium swelling, υ2,s, was calculated according to equation (9), where ρd and ρd are the density of the polymer (1.41 g/cm3) and water (0.995 g/cm3), respectively.
| (9) |
The molecular weight between crosslinks, , was calculated according to the Flory-Rehner equation, (10), where is the number average molecular weight of the uncrosslinked polymer (20,000 for PMAA, 50,000 for PAA), is the specific volume of the polymer (0.71 cm3/g), V1 is the molar volume of the swelling medium (18.1 cm3/mol), and χ is the Flory polymer-solvent interaction parameter in water which is calculated as a weighted average for the values PMAA, χ = 0.5987, PAA, χ = 0.495, and PEG, χ = 0.55.
| (10) |
After determining the number of links between two crosslinks, n, was calculated according to equation (11), where Mr is the average molecular weight of the repeat unit (MAA, 86.09; AA, 72.06; EG, 44).
| (11) |
The value of the root mean squared end-to-end distance of the polymer chain in the freely jointed state was calculated using equation (12) where ℓ is the carbon-carbon bond length (1.54 Å).
| (12) |
The root mean squared end-to-end distance of the polymer chain in the unperturbed state was calculated according to equation (13) where Cn is the Flory characteristic ratio or rigidity factor of the polymer (PMAA, Cn = 14.6, PAA, Cn = 14.6, and PEG, Cn = 3.8).
| (13) |
Lastly, the mesh size of the hydrogel network, ξ, was determined according to equation (14).
| (14) |
The results for the swelling dynamics of PAA microparticles are summarized in Table 2. Loosely crosslinked PAA (0.43 mol % APE) imbibes a significant amount of water in the sodium carbonate buffer (pH~9) as compared to 0.1 N HCl. As the amount of crosslinking agent increases, the degree of swelling in the carbonate buffer decreases. The results for the swelling dynamics of PMAA microparticles (Table 3) show a similar trend. At similar crosslinking levels, PMAA microparticles imbibe a significantly less amount of water due to the presence of the methyl group along the backbone of polyacid. Swelling for both networks occurs due to deprotonation of the carboxyl group resulting in ionic repulsion of the neighboring chains. This causes the network to expand and imbibe more water. In the 0.1 N HCl buffer, the carboxylic acid groups remain protonated and water uptake is minimal. This expansion is also evident through the significant increase in the mesh size for the polymers in the carbonate buffer as compared to the 0.1 N HCl solution. As crosslinking is increased, the mesh size decreases which corresponds to the lower solution uptake.
Table 2.
Equilibrium swelling ratio of crosslinked PAA microparticles
| 0.1 N HCl | NaHCO3 Buffer | |||
|---|---|---|---|---|
| Theoretical
Crosslinking Ratio (Xtheo) |
Weight
swelling ratio (q) |
Mesh Size
(ξ, Å) |
Weight swelling
ratio (q) |
Mesh Size
(ξ, Å) |
| 0.0043 | 17.0 ± 0.3 | 420 | 91.4 ± 0.8 | 780 |
| 0.0075 | 15.0 ± 0.2 | 400 | 64.6 ± 1.7 | 700 |
| 0.0148 | 14.4 ± 0.2 | 390 | 47.6 ± 0.2 | 630 |
| 0.0309 | 16.5 ± 0.7 | 420 | 34.7 ± 2.2 | 561 |
Table 3.
Equilibrium swelling ratio of crosslinked PMAA microparticles
| 0.1 N HCl | NaHCO3 Buffer | |||
|---|---|---|---|---|
| Theoretical
Crosslinking Ratio (Xtheo) |
Weight
swelling ratio (q) |
Mesh Size
(ξ, Å) |
Weight
swelling ratio (q) |
Mesh Size
(ξ, Å) |
| 0.0043 | 11.6 ± 0.6 | 270 | 40.5 ± 0.4 | 350 |
| 0.0079 | 11.9 ± 0.5 | 270 | 34.8 ± 0.8 | 340 |
| 0.0148 | 11.8 ± 0.6 | 270 | 26.6 ± 0.6 | 310 |
| 0.0309 | 11.2 ± 0.7 | 260 | 20.8 ± 1.1 | 290 |
Upon incorporation of PEG, the swelling dynamics of the PMAA microparticles changes significantly (Table 4). The swelling capacity of the tethered hydrogels is significantly lower as compared to crosslinked PMAA microparticles containing no tether. The degree of swelling is similar with a slightly smaller mesh size. The ratio of the mesh size in carbonate buffer versus 0.1 N HCl is significantly greater for tethered hydrogels. This allows for a dramatic increase in the amount of water uptake in higher pH solutions compared with lower pH ones providing a stimuli responsive material.
Table 4.
Equilibrium swelling of crosslinked PEG-tethered PMAA microparticles (PEG-1000)
| 0.1 N HCl | NaHCO3 Buffer | |||
|---|---|---|---|---|
| MAA:EG | Weight
swelling ratio (q) |
Mesh Size
(ξ, Å) |
Weight
swelling ratio (q) |
Mesh Size
(ξ, Å) |
| 83:17 | 6.1 ± 0.2 | 61 | 41.6 ± 0.9 | 300 |
| 69:31 | 5.8 ± 0.3 | 54 | 42.0 ± 0.5 | 290 |
| 50:50 | 5.3 ± 0.4 | 45 | 43.4 ± 1.2 | 270 |
3.5. Gel/Gel Adhesion of Tethered Hydrogels
Crosslinked PAA hydrogel microparticles, upon neutralization, produce a highly viscous suspension at low concentrations (1 wt % polymer). This viscous gel suspension possesses tack which is capable of adhering to other hydrogel materials. This characteristic is exhibited in Figures 5-7 for crosslinked PAA microparticles containing a PEG tether. The gel adhesion remains relatively unchanged upon incorporation of low amounts of the PEG tether (AA:EG 83:17). However, as the concentration is increased to a ratio of AA:EG 60:40, a significant decrease in the adhesive characteristics is obtained. The viscosity of the neutralized hydrated gel is the key factor that attributes to the ability of the gel to act as a bioadhesive. With higher concentrations of PEG, the amount of ionizable gropus present in the network are decreased which leads to a significant reduction in the viscosity of the gel. The lowered viscosity in turn causes the adhesive characteristics to be lower than hydrogels containing lowered amounts of PEG.
Figure 5.
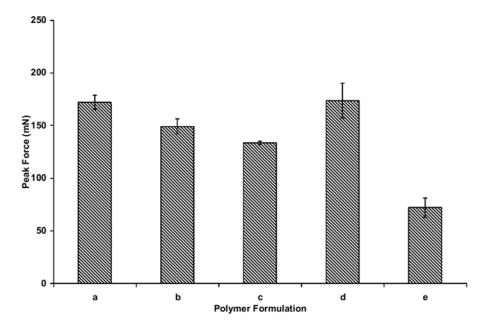
Evaluation of the peak force obtained from neutralized hydrated gels containing (a) crosslinked PAA (0.75 mol % APE), (b) PAA-g-PEG, PEG-1000, AA:EG 98:2, (c) 90:10, (d) 83:17, (e) 60:40, (f) 50:50.
Figure 7.

Evaluation of the work of cohesion of neutralized hydrated gels containing (a) crosslinked PAA (0.75 mol % APE), (b) PAA-g-PEG, PEG-1000, AA:EG 98:2, (c) 90:10, (d) 83:17, (e) 60:40, (f) 50:50.
4. Conclusions
The fundamental understanding of the behavior of tethered gel networks has implications in tissue engineering, adhesion, and drug delivery. In this research, ionizable hydrogel networks containing a PEG tether were successfully synthesized using a thermally initiated free radical precipitation polymerization. The effects of the PEG tether on the structure of the microparticles were evaluated. The addition of the PEG was shown to cause disruption of the dimer formation of PAA. Differential scanning calorimetry exhibited a concentration dependent decrease in the Tg of xerogels. At low concentrations, the network exhibits a heterogeneous behavior which is evident through a lowered ΔCp and the presence of two distinct Tg’s.
The gel adhesion of a neutralized gel was shown to be dependent on the amount of PEG incorporated into the network, which in turn affected the viscosity of the gel. These materials possess interesting swelling properties which can beneficial in the development of pH responsive drug delivery systems.
Figure 2.
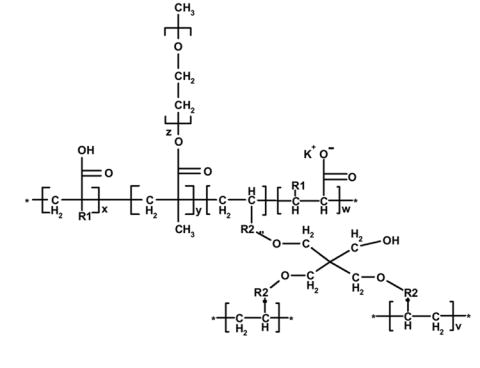
Molecular structure of the crosslinked PEG-tethered polyacid hydrogel prepared through a thermally initiated free radical precipitation polymerization.
Figure 6.
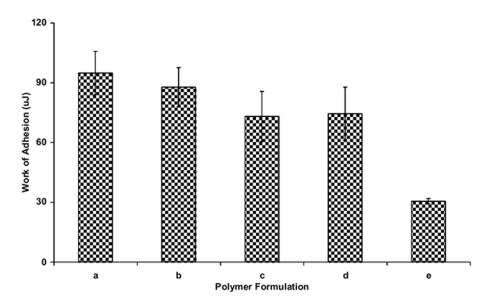
Evaluation of the work of adhesion of neutralized hydrated gels containing (a) crosslinked PAA (0.75 mol % APE), (b) PAA-g-PEG, PEG-1000, AA:EG 98:2, (c) 90:10, (d) 83:17, (e) 60:40, (f) 50:50.
Acknowledgments
This work was supported by an NIH grant EB-000246. We would also like to thank the Welch Foundation. This research was performed while on appointment as a U.S. Department of Homeland Security (DHS) Fellow (to JBT) under the DHS Scholarship and Fellowship Program, a program administered by the Oak Ridge Institute for Science and Education (ORISE) for DHS through an interagency agreement with the U.S Department of Energy (DOE). ORISE is managed by Oak Ridge Associated Universities under DOE contract number DE-AC05-06OR23100.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang YB, Szleifer I, Peppas NA. J Chem Phys. 2001;114:3809. [Google Scholar]
- 2.Huang YB, Szleifer I, Peppas NA. Macromolecules. 2002;35:1373. [Google Scholar]
- 3.Leger L, Raphael E, Hervet H. Polymers In Confined Environments. Vol. 138. Springer-Verlag Berlin; Berlin: 1999. pp. 185–225. [Google Scholar]
- 4.Lowman AM, Cowans BA, Peppas NA. J Polym Sci B-Polym Phys. 2000;38:2823. [Google Scholar]
- 5.Lowman AM, Morishita M, Kajita M, Nagai T, Peppas NA. J Pharm Sci. 1999;88:933. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- 6.Lowman AM, Peppas NA. Macromolecules. 1997;30:4959. [Google Scholar]
- 7.Lowman AM, Peppas NA. J Biomater Sci -Polym Ed. 1999;10:999. doi: 10.1163/156856299x00586. [DOI] [PubMed] [Google Scholar]
- 8.Lowman AM, Peppas NA. Polymer. 2000;41:73. [Google Scholar]
- 9.Bromberg L, Temchenko M, Alakhov V, Hatton TA. Int J Pharm. 2004;282:45. doi: 10.1016/j.ijpharm.2004.05.030. [DOI] [PubMed] [Google Scholar]
- 10.Cleary J, Bromberg L, Magner E. Langmuir. 2004;20:9755. doi: 10.1021/la048993s. [DOI] [PubMed] [Google Scholar]
- 11.Drotleff S, Lungwitz U, Breunig M, Dennis A, Blunk T, Tessmar J, Gopferich A. Eur J Pharm Biopharm. 2004;58:385. doi: 10.1016/j.ejpb.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 12.Mann BK, Schmedlen RH, West JL. Biomaterials. 2001;22:439. doi: 10.1016/s0142-9612(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 13.Mann BK, Tsai AT, Scott-Burden T, West JL. Biomaterials. 1999;20:2281. doi: 10.1016/s0142-9612(99)00158-1. [DOI] [PubMed] [Google Scholar]
- 14.Ruoslahti E, Pierschbacher MD. Science. 1987;238:491. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- 15.Jeon SI, Lee JH, Andrade JD, Degennes PG. J Colloid Interface Sci. 1991;142:149. [Google Scholar]
- 16.Peppas NA, Huang YB. Adv Drug Deliv Rev. 2004;56:1675. doi: 10.1016/j.addr.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Thomas JB, Creecy CM, McGinity JW, Peppas NA. Polym Bull. 2006;57:11. [Google Scholar]
- 18.Barrett KEJ, Thomas HR. In: Dispersion Polymerization in Organic Media. Barrett KEJ, editor. John Wiley & Sons; New York, NY: 1975. pp. 115–200. [Google Scholar]
- 19.Barrett KEJ, Thomas HR. J Polym Sci. 1969;7:2621. [Google Scholar]
- 20.Dong J, Ozaki Y, Nakashima K. Macromolecules. 1997;30:1111. [Google Scholar]