

Abstract
Poly(ADP-ribose) polymerases (PARPs) are members of a family of enzymes that utilize NAD+ as substrate to form large ADP-ribose polymers (PAR) in the nucleus. PAR has a very short half life due to its rapid degradation by poly(ADP-ribose) glycohydrolase (PARG). PARP-1 mediates acute neuronal cell death induced by a variety of insults including cerebral ischemia, MPTP-induced Parkinsonism, and CNS trauma. While PARP-1 is localized to the nucleus, PARG resides in both the nucleus and cytoplasm. Surprisingly, there appears to be only one gene encoding PARG activity, which has been characterized in vitro to generate different splice variants, in contrast to the growing family of PARPs. Little is known regarding the spatial and functional relationships of PARG and PARP-1. Here we evaluate PARG expression in the brain and its cellular and subcellular distribution in relation to PARP-1. Anti-PARG (α–PARG) antibodies raised in rabbits using a purified 30 kDa C-terminal fragment of murine PARG recognize a single band at 111 kDa in the brain. Western blot analysis also shows that PARG and PARP-1 are evenly distributed throughout the brain. Immunohistochemical studies using α-PARG antibodies reveal punctate cytosolic staining, whereas anti-PARP-1 (α–PARP-1) antibodies demonstrate nuclear staining. PARG is enriched in the mitochondrial fraction together with manganese superoxide dismutase (MnSOD) and cytochrome C (Cyt C) following whole brain subcellular fractionation and Western blot analysis. Confocal microscopy confirms the co-localization of PARG and Cyt C. Finally, PARG translocation to the nucleus is triggered by NMDA-induced PARP-1 activation. Therefore, the subcellular segregation of PARG in the mitochondria and PARP-1 in the nucleus suggests that PARG translocation is necessary for their functional interaction. This translocation is PARP-1 dependent, further demonstrating a functional interaction of PARP-1 and PARG in the brain.
Author Keywords: NMDA, mitochondria, nucleus, immunostaining, subcellular fractionation
INTRODUCTION
Poly(ADP-ribose) polymerase-1 (PARP-1; EC 2.4.2.30) is a member of a growing family of enzymes that utilize nicotinamide adenine dinucleotide (NAD+) as substrate to transfer ADP-ribose moieties onto glutamic acid residues of proteins to form ADP-ribose polymers (PAR) of various lengths and complexity (Ame et al., 2004). Accounting for 80–90% of cellular PARP activity, PARP-1 is activated by DNA strand breaks due to a variety of genotoxic stressors, including oxygen radicals, ionizing radiation, or alkylating agents (Lautier et al., 1993, de Murcia et al., 1994, Shall and de Murcia, 2000). Many nuclear proteins are poly(ADP-ribosyl)ated by PARP-1, including histones, topoisomerase, p53, and PARP-1 itself in an automodification reaction (D’Amours et al., 1999). The actions of PARP-1 are significant to a number of cellular events, including transcriptional activation (Hassa and Hottiger, 1999, Ju et al., 2004), chromatin relaxation (de Murcia et al., 1986, Kim et al., 2004), mitosis (Kanai et al., 2003, Chang et al., 2004), and DNA maintenance (D’Amours et al., 1999, Dantzer et al., 2000). Because PARP-1 activation in response to DNA damage facilitates DNA repair and cellular recovery (Lautier et al., 1993, de Murcia et al., 1994, Shall and de Murcia, 2000, Rouleau et al., 2004), poly(ADP-ribosyl)ation has the important role of maintaining genomic integrity.
In the nervous system, PARP-1 activation plays a critical role in acute neuronal cell death elicited by a variety of insults, including cerebral ischemia (Zhang et al., 1994, Eliasson et al., 1997, Endres et al., 1997), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism (Cosi and Marien, 1999, Mandir et al., 1999), and traumatic brain injury (Whalen et al., 1999, Whalen et al., 2000, LaPlaca et al., 2001). PARP-1-mediated neuronal cell death plays a primary role in glutamate excitotoxicity through N-methyl-D-aspartate (NMDA) glutamate-receptor activation, as mice lacking PARP-1 are highly resistant to excitotoxicity induced by NMDA, but are equally susceptible to alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) excitotoxicity as wild-type mice (Mandir et al., 2000). Recent observations reveal that PARP-1-mediated cell death is linked to the translocation of the cell death effector, apoptosis inducing factor (AIF), from the mitochondria to the nucleus (Yu et al., 2002, Du et al., 2003, Hong et al., 2004) through the actions of PAR (Andrabi et al., 2006, Yu et al., 2006). Further, AIF translocation occurs during excitotoxic neuronal injury in vivo following NMDA receptor stimulation, suggesting AIF can substitute as caspase executioner in PARP-1-dependent cell death (Wang et al., 2004). Therefore, PARP-1 mediates cell death in the nervous system at least in part through AIF, with other apoptotic or necrotic mechanisms occurring downstream of AIF translocation.
Following PARP-1 activation, the appearance of PAR is transient due to its rapid degradation by poly(ADP-ribose) glycohydrolase (PARG) into free ADP-ribose residues (Jonsson et al., 1988a, Brochu et al., 1994a, Davidovic et al., 2001). While there exists a family of PARP homologs capable of synthesizing PAR, to date only one PARG has been shown to catabolize PAR in vivo in mammals. Oka, et al., suggest that there may be an additional PARG gene (Oka et al., 2006). However the specific PARG activity was quite low and no knock-down or over expression studies were performed to confirm the hypothesized function of this gene. Isolation and characterization of the PARG cDNA from several species demonstrated only one mRNA transcript which encodes a 110–111 kDa protein (Lin et al., 1997, Shimokawa et al., 1999). However, recent studies revealed the existence of multiple splice variants of PARG, with full-length PARG encoding a protein of 111 kDa and two shorter forms of 102 and 99 kDa (Meyer-Ficca et al., 2004). PARG has been purified to homogeneity from different tissues of different species revealing important differences in molecular weight (ranging from 50 to 110 kDa) and catalytic activity (Tavassoli et al., 1983, Hatakeyama et al., 1986, Tanuma and Endo, 1990, Maruta et al., 1991, Uchida et al., 1993, Abe and Tanuma, 1996). Since there has not been any molecular evidence of shorter forms of PARG, it is likely that the previous reports describing shorter forms of purified PARG were probably descriptions of degradation fragments. Indeed, PARG degradation fragments (two C-terminal fragments of 85 and 74 kDa) are generated by caspase-3 during apoptosis (Affar et al., 2001), suggesting the possible generation of proteolytic PARG fragments in vivo or during tissue preparation.
The emerging role of PARG is to facilitate cell survival (Koh et al., 2005). Previous reports demonstrating a role for PARG in facilitating cell death by the prevention or re-activation of automodified PARP-1 (Ying and Swanson, 2000, Ying et al., 2001) proved to be inconclusive, since the PARG inhibitors utilized in these studies were later demonstrated to be non-specific and non-selective (Falsig et al., 2004). Characterization of the complete absence of functional PARG protein in mice via disruption of the Parg gene demonstrated that PARG is required for the proper cellular response to DNA damage, since PARG null trophoblast stem (TS) cells derived from these mice were hypersensitive to sublethal doses of DNA damaging agents (Koh et al., 2004). Further, PARG was shown to be essential for normal embryonic development and normal homeostatic cellular functions, since PARG null embryos did not develop past embryonic day 3.5 (E3.5) and PARG null TS cells did not remain viable in the absence of stress, respectively (Koh et al., 2004). Although other studies regarding the disruption of the Parg gene report the survival of PARG knockout animals, these mice are actually hypomorphs expressing functional PARG protein (Cortes et al., 2004). Thus, the viability of these mice confirms the critical role of PARG to the organism. Together with other reports demonstrating a role for PARG in development (Hanai et al., 2004), normal circadian function (Panda et al., 2002), and the response to DNA damage (Cortes et al., 2004), PARG appears to have a protective role and its activity therefore leads to viability.
In vitro studies demonstrate a predominantly cytoplasmic localization of PARG (Meyer-Ficca et al., 2004), while most PARPs have a nuclear localization. However, very little is known about the anatomical or subcellular distribution of PARG in the brain, and nothing is yet known regarding its spatial and functional relationship to PARP-1. The message for PARG is present in the brain (Shimokawa et al., 1999) and active PARG is present in cultured neurons and astrocytes (Sevigny et al., 2003). The purification of PARG from nuclear and post-nuclear extracts suggests that PARG could be localized both in the cytoplasmic and the nuclear compartments. Overexpressed PARG (Winstall et al., 1999) and the 102 and 99 kDa PARG splice variants display a cytoplasmic localization in vitro, while the 111 kDa PARG splice variant displays a nuclear localization (Meyer-Ficca et al., 2004). Other observations demonstrated that green fluorescent protein-PARG fusion protein (GFP-PARG) overexpressed in NIH3T3 cells is exclusively expressed in the nucleus during interphase and that GFP-PARG shuttles between nucleus and cytoplasm during the cell cycle (Ohashi et al., 2003). Therefore, the subcellular localization of PARG and its redistribution throughout the cell is likely to play an important role in the regulation of regional poly(ADP-ribose) metabolism. The subcellular localization of PARG could then possibly depend on many factors, including the splice variant expressed, the tissues or cells studied, and/or the phase of the cell cycle. We report here for the first time the cellular immunolocalization of endogenous PARG in the brain. Using immunocytochemical techniques and subcellular fractionation, we show that PARG is co-localized with PARP-1 throughout the brain, but in different subcellular compartments. In the brain, PARP-1 is primarily enriched in the nuclear fraction and PARG is primarily enriched in the mitochondrial fraction, suggesting that their subcellular segregation would necessitate PARG redistribution for their functional interaction. We also show that NMDA-induced PARP-1 activation leads to PARG redistribution to the nucleus, and that PARG translocation is a PARP-1-dependent phenomenon, confirming their functional interaction in the brain.
EXPERIMENTAL PROCEDURES
Rats and Mice
All experiments were approved and conformed to the guidelines set by the Institutional Animal Care Committee. To avoid differences caused from strain effect or divergent genetic lines, PARP-1 KO mice used in this study were on a pure 129 Sv/Ev background (Wang et al., 1997) with the colony maintained by outbreeding with purebred 129 Sv/Ev wild-type (WT) controls (Taconic, Germantown, NY). Thus, the PARP-1 KO mice are of the same strain as controls, and inbreeding effects are minimized.
Preparation of rabbit polyclonal α–PARG antibodies
Polyclonal α–PARG antibodies were produced in rabbit using a purified His-tagged 30 kDa C-terminal fragment of murine PARG (amino acids 718–978) as antigen. Murine PARG cDNA was kindly provided by Dr. E.L. Jacobson (University of Arizona). The 30 kDa C-terminal fragment was subcloned by PCR at the EcoR1 and Sal1 sites (EcoR1: 5′-CCG CCG GAA TTC ATG ACA CGC TTA CAC GTC; Sal1: 5′-GGA ACA AGA GTC GAC TTC AGG TGC CTG CCT TC) into the pET28a expression vector (Novagen, Madison WI), overexpressed, and purified according to the company’s protocol. Anti-PARG antibodies were affinity purified using the purified 30 kDa His-tagged C-terminal PARG antigen. PARG antiserum was incubated overnight with the purified PARG antigen affinity matrix, and after several washes with PBS, the bound antibody was eluted with 4 M MgCl2. The purified antibody was dialyzed overnight against 1000 volumes of dialysis buffer (PBS, 20% sucrose, 1 mM EDTA, and 0.02% NaN3), then dialyzed overnight against 1000 volumes of dialysis buffer without EDTA. The antibody was then stored at 4°C until used.
Preparation of whole tissue extracts
Various tissues from CD male rats (150–250 g; Charles River, Wilmington, MA) were rapidly dissected and homogenized for 30 s with a Polytron homogenizer Diax 900 (Heidolph, Germany) in buffer containing 10 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 6 M urea, and 1X protease inhibitor cocktail (Roche, Indianapolis, IN). Protein concentrations were determined by the Bradford assay (Pierce, Rockford, IL). Protein samples were then resuspended in loading buffer (2% SDS, 62.5 mM Tris-HCl pH 6.8, 6 M urea, 10% glycerol, 0.003% bromophenol blue, and 5% 2-mercaptoethanol), incubated 15 min at 65°C, resolved on a SDS-PAGE gel, and subjected to Western blot analysis.
Subfractionation protocol
Tissues were rapidly dissected, minced, and homogenized in subfractionation buffer (50 mM Tris-HCl pH 7.5, 25 mM KCl, 5 mM MgCl2, 0.25 M sucrose, and 1X protease inhibitor cocktail) using 10 up-down strokes at 1700 rpm with a motor-driven Teflon pestle. The homogenate was filtered through four layers of cheesecloth and centrifuged 10 min at 600 x g. The pellet was then washed four times with subfractionation buffer containing 0.1% NP-40 in order to eliminate mitochondrial contamination. This pellet (P1) constituted the nuclear fraction. The supernatant (S1) was centrifuged 30 min at 10,000 x g. The pellet (P2), which constituted the mitochondrial fraction, was washed once with homogenization buffer. The supernatant (S2) constituted the post-mitochondrial fraction. Samples were resuspended in loading buffer without urea and incubated 5 min at 95°C. Samples were subjected to SDS-PAGE and Western blot analysis as before. For salting out experiments, before the washing step, the mitochondrial pellet (P2) was aliquoted in separate tubes and centrifuged. The individual pellets were resuspended in 200 μl of subfractionation buffer containing increasing concentrations of NaCl. The mitochondrial suspensions were centrifuged 30 min at 10,000 x g. The proteins contained in the supernatants and pellets were subjected to SDS-PAGE/Western blot.
Overexpression of PARG in HEK-293 cells
Human embryonic kidney cells (HEK-293; ATCC #CRL-1573), plated the night before at 100,000 cells/well of a 24-well plate, were transfected with 1 μg of plasmid using Lipofectamine 2000 (Invitrogen, Carlsbad, Ca) according to the manufacturer’s protocol. Twenty-four hours later, cells were washed with cold PBS, harvested in loading buffer without urea, and incubated 5 min at 95°C. Samples were subjected to SDS-PAGE/Western blot. Plasmid pCMV2α-PARG expresses myc-tagged full-length murine PARG and pCMVtag-hPARG102, a gift from Dr. Guy Poirier, expresses flag-tagged exon 1 deleted PARG. For adenoviral infection, a stock of adenovirus expressing flag-tagged full-length human PARG was received from Dr. G.G. Poirier at an approximate titer of 3.9 × 1010 pfu/ml. HEK-293 cells were plated as above, and on the day of viral infection, cells were washed once with PBS and adenovirus was added to each well at 75 multiplicity-of-infection in antibiotic-free DMEM containing 2% FBS. After 48 hr of infection, the cells were collected as above.
Immunohistochemistry
For PARP-1 immunohistochemistry, CD rat brains were perfusion-fixed with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB), pH 7.4, as described previously (Mandir et al., 2000). After post-fixation in the same fixative solution and cryoprotection in 20% sucrose in PB, brains were frozen and serially sectioned in 40 μm increments. The sections were then permeabilized in TBS buffer (50 mM Tris-HCl pH 7.5, 1.5% NaCl) containing 0.4% Triton X-100 for 30 min at 4°C then stained as free-floating sections. For PARG immunohistochemistry, CD rat brains were rapidly dissected, frozen on powdered dry ice, serially sectioned (20 μm thick), then mounted and stained on glass slides. Fresh frozen sections were fixed with ice-cold methanol for 10 min at −20°C and rehydrated in TBS for 30 min at 4°C. The methanol fixation was required for PARG immunostaining due to the difficulty to obtain high quality, low background staining with the PARG antibody following PFA fixation. After blocking with blocking buffer (TBS, 4% normal goat serum (NGS), 0.2% Triton X-100, and 0.02% NaN3) for 30 min at 4°C, PFA fixed sections and methanol fixed sections were incubated overnight at 4°C respectively with α–PARP-1 antibodies (1 μg/ml) and α–PARG antibodies (10 μg/ml) diluted in blocking buffer containing 2% NGS. Sections were washed 3 × for 10 min with TBS+1% NGS, incubated 45 min with 1/500 biotinylated-conjugated goat α–rabbit IgG (111-066-006, Jackson ImmunoResearch) or 1/500 α–mouse IgG (115-066-006, Jackson ImmunoResearch), diluted in TBS+1.5% NGS, and then incubated 45 min with horseradish-peroxidase-conjugated avidin/biotin complex (Vector ELITE; Vector Laboratories) diluted in TBS. Staining was developed with DAB (Gibco). For immunofluorescence and confocal microscopy, rat neuronal cultures were fixed with 4% PFA and methanol for PARP-1 and PARG (and others antibodies), respectively. Cells were permeabilized or rehydrated, blocked and incubated with primary antibodies α–PARP-1 (1 μg/ml), α–PARG (10 μg/ml), α–Cyt C (1/100) (6H2.B4, Pharmingen), α–GFAP (G3893, Sigma), and α–MAP2 (M4403, Sigma) as described for the brain sections. After washing 3 × 10 min with TBS+1% NGS, neurons were incubated with fluorescein isothiocyanate (FITC) or Texas red conjugated α–mouse or α–rabbit secondary antibodies (Jackson ImmunoResearch). Nuclei were stained with 1 μM Toto-3 (Molecular Probes) for 10 min after the secondary antibody incubation and rinsed twice with TBS. Confocal microscopy was performed on a LSM510 laser scanning microscope (Zeiss) equipped with an argon and two HeNe lasers. Images were obtained by scanning stained cells under 40X oil objectives.
Primary neuronal culture
Primary cortical cell cultures were prepared from gestational day 14 fetal rats or day 16 fetal mice as described previously (Dawson et al., 1993, Dawson et al., 1996). The cortex was dissected under a microscope, incubated for 20 min in 0.0027% trypsin and saline solution (5% PBS; 40 mM sucrose, 30 mM glucose, 10 mM HEPES, pH 7.4). Rat cortex was transferred to modified Eagle’s medium (MEM), 10% horse serum, 10% fetal bovine serum, and 2 mM glutamine, and cells were dissociated by trituration. Mouse cortex was dissected and the cells were dissociated by trituration in MEM, 20% horse serum, 25 mM glucose, and 2 mM L-glutamine after a 30 min digestion in 0.027% trypsin and saline solution (Life Technologies, Gaithersburg, MD). Cells were plated in 24 well plates (Nunc, Roskilde, Denmark) coated with polyornithine at a density of 3–4 × 105 cells per well. Four days after they were plated, the cells were treated with 10 μg/ml of 5-fluoro-2′-deoxyuridine (Sigma) for 3 days to inhibit proliferation of non-neuronal cells. Rat cultures were maintained in MEM, 5% horse serum, and 2 mM glutamine in 8% CO2, humidified, 37°C atmosphere. Murine cultures were maintained in MEM, 10% horse serum, 25 mM glucose, and 2 mM L-glutamine in a 5% O2, 8% CO2, humidified, 37°C incubator. The medium was changed twice a week. Mature neurons (10–14 days in culture) were used for all experiments. In mature cultures, neurons represent 70–90% of the total number of cells (Dawson et al., 1993, Dawson et al., 1996). Untreated rat cortical neurons were used for immunofluorescence and confocal analysis. For PAR Western blot, and PARG translocation experiments, WT and PARP-1 KO cortical cultures were washed with control salt solution (CSS) (120 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 25 mM Tris-HCl, 15 mM glucose, pH 7.4) and treated 5 min with either 100–500 μM NMDA (in CSS) or with CSS alone. The CSS was then replaced by culture media and the cells were harvested at different times after the treatment. For PAR Western blot, after the treatment, cells were washed once with ice cold PBS and harvested in 100 μl PAGE-loading buffer/well, and processed for SDS-PAGE and Western blot analysis. For NAD+ assays, cells were washed with ice cold PBS and NAD+ was extracted and quantified according to the cycling method described by Jacobson and Jacobson (Jacobson and Jacobson, 1997). For PARG immunostaining, cells were washed once with ice cold PBS, fixed with methanol and stained as described for fresh frozen brains. For subcellular fractionation experiments, the neurons were washed with ice-cold PBS, resuspended in hypotonic homogenation buffer (10 mM KCl, 1.5 mM MgCl2, 1 mM Na-EDTA, 1 mM Na-EGTA, 1 mM dithiothreitol, 0.1 mM PMSF and 10 mM Tris-HCl, pH 7.4) containing protease inhibitor cocktail (Roche) and left on ice for 30 min. NP-40 was added just before the homogenation (0.1% final concentration) and the neurons were homogenized by 40 full strokes in a 2 ml Dounce homogenizer with the Teflon pestle. The homogenation process was monitored under light microscopy and > 90% cell were homogenized. The nuclei fraction was fractionated at 720 × g for 5 min and the supernatant was used as post-nuclear fraction. The nuclei fraction was washed twice with the isotonic homogenation buffer (0.25 M sucrose in hypotonic buffer). Samples were then processed for SDS-PAGE/Western blot.
SDS-PAGE and Western blot analysis
The different protein extracts were size-separated through denaturing polyacrylamide gel electrophoresis (SDS-PAGE). For most blots, equal amounts of protein for each sample (50 μg of proteins) were loaded onto 10% polyacrylamide gels and run overnight at 65 V using a Hoeffer model SE 600 electrophoresis unit. For the overexpression of flag-tagged PARG isoforms, only 20 μg protein was loaded for each sample onto a 7.5% SDS-PAGE gel and separated using a Bio-Rad Mini-PROTEAN 3 electrophoresis system. The proteins were then electrotransferred to a nitrocellulose membrane (Bio-Rad) in tris-glycine-methanol buffer for 3 hours at 0.6 A. The membrane was blocked for 1 hr at room temperature in PBS-T pH 7.5 (PBS 0.1% Tween-20) containing 5% nonfat dry milk. The membrane was then incubated overnight at 4 C with different primary antibodies in blocking solution: rabbit purified α–PARG 1 μg/ml, rabbit α–MnSOD antiserum diluted 1/20,000, rabbit α–Cyt C 1/5,000 (sc7159, Santa Cruz), mouse α–β-tubulin 1/5,000 (T-4026; Sigma), mouse α–PARP-1 1/2,000 (7D3-6 and C2-10, Pharmingen), mouse α–GAPDH 1/5,000 (Santa Cruz), rabbit α–β-actin 1/5,000 (Sigma), α–histone 1/200 (US Biological), and mouse HRP-linked α–Flag 1/10,000 (Sigma). The membrane was washed with blocking buffer 3 × 10 min and incubated for 1 hr at room temperature with the secondary goat anti-rabbit IgG peroxidase-conjugated antibody 1/10,000 (31463, Pierce), or goat anti-mouse IgG peroxidase-conjugated antibody 1/1,000 (A-4416, Sigma). The membranes were washed 3 × 10 min with blocking buffer and 2 × 10 min with PBS-T, and then processed for chemiluminescence analysis using the SuperSignal West Pico detection kit (Pierce).
PARG immunoprecipitation and one-dimensional PARG activity zymogram
HEK-293, mouse brain, and testis were homogenized using 15 strokes in a 2 ml Dounce homogenizer in buffer containing 10 mM Tris/HCl pH 7.5; 1 mM EDTA; 10 mM NaF; 0.3 M sucrose, 1 mM β-mercaptoethanol, 1 × protease inhibitors cocktail and 0.1% NP-40 and incubated on ice for 30 min. The homogenates were then centrifuged 30 min at 20,000 x g and the protein concentration of the supernatants was determined by the Bradford assay. HEK-293 cells lysate (1 mg/ml) and tissue lysates (2.5 mg/ml) were immunoprecipitated for 3 hr at 4 C with either 1/100 diluted α–PARG antiserum or non immune serum using protein G sepharose (GammaBind Plus, 17-0886-01, Amersham). The immunoprecipitated complexes were washed twice with PBS and resuspended in loading buffer. The samples were analyzed on an one-dimensional PARG activity zymogram. The activity zymogram consisting of a SDS-PAGE separation of PARG samples using a Mini-PROTEAN 3 system with a gel containing 32P-automodified PARP-1 prepared according to Brochu et al. (Brochu et al., 1994b) using purified PARP-1 (Trevigen, Gaithersburg, MD). After renaturation, the gel was dried, and the PARG activity was determined by the disappearance of the 32P-automodified PARP-1 in the gel.
RESULTS
Characterization and Specificity of α–PARG antibody
Anti-PARG polyclonal antibodies were raised in rabbits using a purified recombinant 30 kDa C-terminal His-tagged fragment of mouse PARG (amino acid 718–978). The α–PARG antibodies were affinity purified and display a high affinity and specificity for PARG (Figure 1). Specificity of the antibody is evident in Western blots that display a prominent 110–111 kDa molecular weight band corresponding to full length PARG in whole rat brain homogenates (Figure 1A). Preabsorption with excess PARG antigen completely eliminates the PARG immunoreactive band (Figure 1A).
Figure 1.
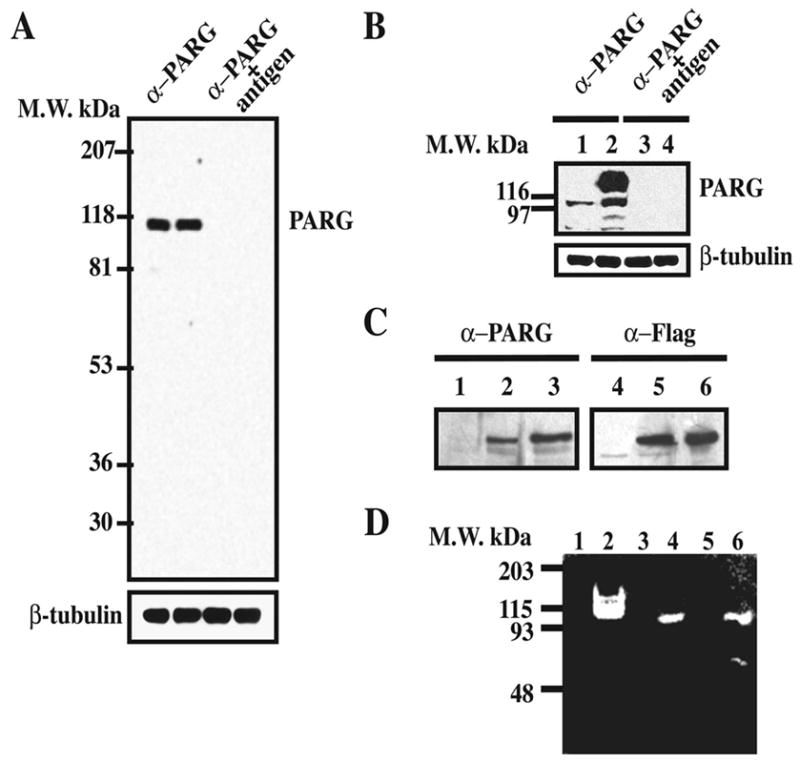
Characterization of α–PARG antibodies. Polyclonal α–PARG antibodies were produced in rabbits as described in the methods. A) Proteins from whole rat brain extract (50 μg/well) were separated by SDS-PAGE, transferred to nitrocellulose membranes and blotted with 1 μg/ml of affinity purified α–PARG antibodies (left) or antigen preadsorbed α–PARG antibodies (right). The membrane was then stripped and reprobed with an α–β-tubulin antibody (1:5,000). B) HEK-293 cells were transfected with pCMV2α (lanes 1 and 3) and pCMV2α-PARG (2 and 4) and proteins from transfected cells were separated, transferred and blotted as in A). C) HEK-293 cells were either untreated (lanes 1 and 4), transfected with pCMVtag-hPARG102 (lanes 2 and 5), which overexpresses flag-tagged 102 kDa PARG, or infected with adenovirus expressing flag-tagged 111 kDa PARG (lanes 3 and 6). Proteins (20 μg/well) were separated by 7.5% SDS-PAGE and transferred to nitrocellulose as in A). D) Zymogram of PARG activity in HEK-293 cells (lanes 1–2), mouse brain (lanes 3–4) and testis (lanes 5–6). Proteins extracts from cells and tissues were immunoprecipitated with non-immune serum (lanes 1,3,5) or α–PARG anti-serum 1:100 (lanes 2,4,6). Immunoprecipitated proteins were loaded on the 32P-automodified PARP-1 containing gel and the PARG activity, revealed by the absence of radioactive signal, was observed after gel renaturation. These results are representative of at least three independent experiments that gave similar results.
To further test the specificity of the α–PARG antibody, HEK-293 cells were transfected with pCMV2-PARG to overexpress a myc-tagged mouse full length PARG (Figure 1B). Vector transfected cells (lane 1) exhibit a 110–111 kDa immunoreactive band for PARG and a minor band at 80 kDa that probably represents a proteolytic breakdown product or a processed form of PARG. Forced overexpression of myc-tagged PARG (lane 2) yields an intense immunoreactive band of PARG that has a retarded migration pattern on SDS-PAGE consistent with the myc tag (Figure 1B). Pre-adsorption with PARG antigen completely eliminates the PARG immunoreactive bands in both vector and PARG transfected cells (Figure 1B lane 3 and 4).
To determine whether the antibody is capable of detecting splice variants of PARG, HEK-293 cells were either transfected with pCMVtag-hPARG102, which overexpresses flag-tagged 102 kDa PARG, or infected with adenovirus expressing flag-tagged 111 kDa PARG (Figure 1C). Transfected cells (lane 2) exhibit an immunoreactive band representing exon 1 deleted PARG, which is correlated with a comparable band as detected by α–flag antibody (lane 5). As a positive control, the overexpression of flag-tagged human 111 kDa PARG following adenoviral infection yields immunoreactive bands representing full-length PARG after α–PARG (lane 3) or α–flag (lane 6) detection.
To determine whether the antibody is capable of immunoprecipitating and further confirm the specificity of the antibody, we assessed the ability of the α–PARG antibody to immunoprecipitate PARG activity from HEK-293 cells, brain and testis (Figure 1D, lane 2, 4 and 6). PARG activity was assessed using a one-dimensional zymogram assay (Brochu et al., 1994b). The α–PARG antibody is capable of immunoprecipitating PARG activity, and yields a catalytically active PARG band that migrates at 110 kDa from HEK-293 cells and from brain; and two catalytically active PARG bands, a major 110 kDa band and a minor 65 kDa band from testis (Figure 1D) corresponding to a proteolytic breakdown product. Pre-immune serum immunoprecipitates obtained from HEK-293 cells, brain and testis (lane 1, 3 and 5) are devoid of PARG activity. In some experiments, a 65 kDa catalytically active fragment of PARG was also observed in PARG immunoprecipitates obtained from brain extract, and HEK cell extract with a concomitant decrease of the PARG full length activity (data not shown). Furthermore, Western blot analysis performed with our antibody demonstrated a complete loss of the PARG immunoreactivity in PARG KO trophoblasts (Koh et al., 2004). Taken together, these results indicate that our α–PARG antibody is specific and selective for PARG and can recognize full length active PARG as well as proteolytic breakdown products of PARG.
Expression profile of PARG and PARP-1 in peripheral tissues and in the brain
PARG expression profile was evaluated by Western blot analysis from various tissues and compared to the distribution of PARP-1 (Figure 2A). The distribution of PARP-1 was monitored with the monoclonal antibody 7D3-6 (Ranjit et al., 1995, Yu et al., 2002). Full length PARG is expressed in most tissues examined. The highest level of expression appears to be brain, heart and stomach, whereas the testis and kidney express moderate levels, and the spleen and pancreas express low levels of PARG. In liver, we were only able to detect a 85 kDa form of PARG (also detected in kidney, spleen and stomach) (Figure 2A). PARG was not detected in lung, which may be due to very low expression levels of PARG or to proteolytic processing as full length PARP-1 was also not detected in lung (Figure 2A). High PARG mRNA levels were previously detected by Northern blot in rat testis, brain, kidney and lung, while moderate levels and undetectable levels were observed in stomach and heart, and liver respectively (Shimokawa et al., 1999). The distribution of full length PARP-1 is also ubiquitous, but PARP-1 expression levels are distributed in a different pattern from those of PARG including the testis, spleen, liver, and pancreas, which express relatively much higher levels of PARP-1 than PARG (Figure 2A). The PARP-1 expression profile obtained by Western blot analysis was similar to PARP-1 tissue distribution evaluated by either activity assay or Northern blot analysis, as previously reported (Ogura et al., 1990, Menegazzi et al., 1991). Western blot analysis of different brain regions reveals that full length PARG is ubiquitously expressed in the brain at relatively uniform levels, except for slightly decreased levels in midbrain (Figure 2B). PARP-1 expression is very similar to PARG with relatively uniform levels throughout the brain, except for decreased levels in the midbrain (Figure 2B).
Figure 2.
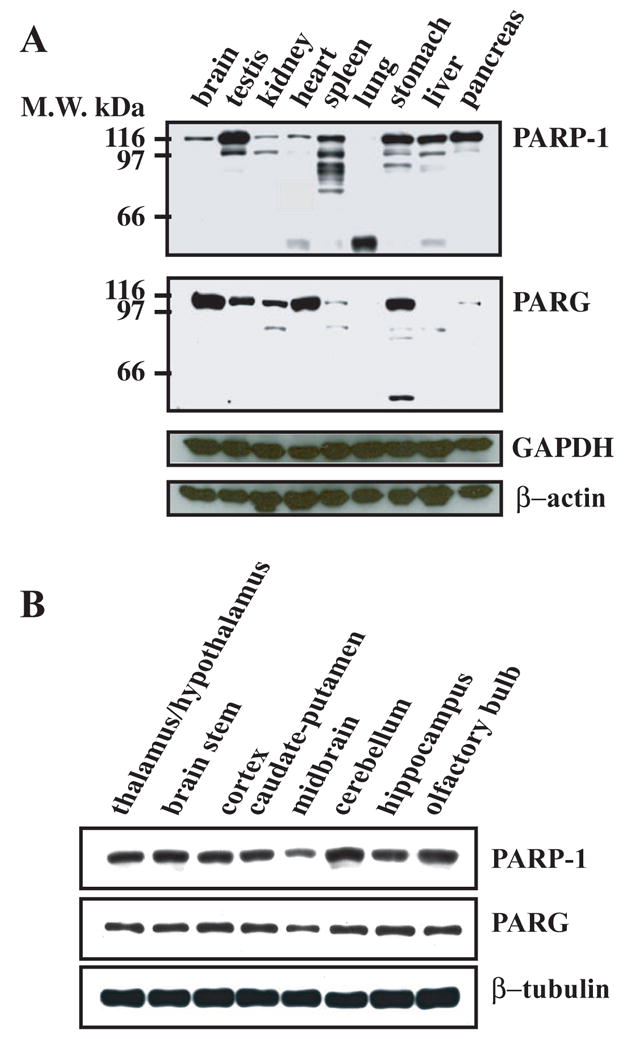
PARP-1 and PARG expression profile in rat tissues. CD1 rats were rapidly dissected and the different tissues and the different parts of the brain were homogenized in urea buffer. Proteins from the different samples (50 μg/well) were separated by SDS-PAGE, transferred to nitrocellulose membrane and blotted with affinity purified α–PARG antibodies (1 μg/ml), α–PARP-1 monoclonal antibodies 7D3-6 (0.5 μg/ml), monoclonal α–GAPDH antibodies (Santa Cruz), and polyclonal α–β-actin antibodies (Sigma). Western blots were developed by chemiluminescence using SuperSignal Pico reagent (Pierce). These results were replicated three times with similar results
Immunohistochemical Localization of PARG and PARP-1 in Rat Brain
Immunohistochemical analysis of the distribution of PARP-1 and PARG in rat brain reveals that PARP-1 and PARG are uniformly distributed in all brain structures examined, including olfactory bulb, cortex, basal ganglia, thalamus, hypothalamus, hippocampus, cerebellum, midbrain, and brain stem (data not shown). In frontoparietal cortex, PARP-1 exhibits a nuclear staining pattern (Figure 3A). Specificity of PARP-1 immunostaining is demonstrated by the absence of staining with mouse IgG (Figure 3A) and the complete absence of PARP-1 immunostaining in PARP-1 knockout mice (data not shown). In striking contrast, PARG immunoreactivity is primarily cytoplasmic and perinuclear in the frontoparietal cortex (Figure 3A). Specificity of the α–PARG immunoreactivity is demonstrated by the relatively complete absence of immunostaining following preadsorption with excess PARG antigen (Figure 3A). In the striatum, we observe a similar staining pattern for PARP-1 and PARG with PARP-1 being primarily concentrated in the nucleus and PARG being primarily concentrated within the cytoplasm and perinuclear regions (Figure 3B). The cerebellum displays high densities of PARG in the cytoplasmic and perinuclear region of Purkinje cell bodies, as well as high densities within basket cells within the molecular layer of the cerebellum and moderate levels of staining within the granule cells (Figure 3C). In addition, there are cells within the white matter tracks that intensely stain with PARG that may represent oligodendrocytes (Figure 3C). In all the cell types, the distribution of PARG is primarily concentrated within the cytoplasm and perinuclear region (Figure 3C). In contrast, PARP-1 immunostaining is primarily nuclear with high levels of PARP-1 densities in Purkinje cells and moderate levels in the molecular and granule cell layers of the cerebellum (Figure 3C). In the hippocampus, both PARP-1 and PARG are highly concentrated with the pyramidal cells of all hippocampal subfields (Figure 3D). Shown are representative examples of PARP-1 and PARG staining within the CA1 subfield of the hippocampus (Figure 3D). PARP-1 exhibits a nuclear staining pattern, whereas PARG exhibits primarily a cytoplasmic perinuclear-staining pattern (Figure 3D). Both PARP-1 and PARG are also highly concentrated within white matter structures and shown are representative examples of PARP-1 and PARG immunostaining in the corpus callosum (Figure 3E). PARP-1 has primarily a nuclear staining pattern and PARG has a cytoplasmic and perinuclear staining pattern within cells of the corpus collosum (Figure 3E).
Figure 3.
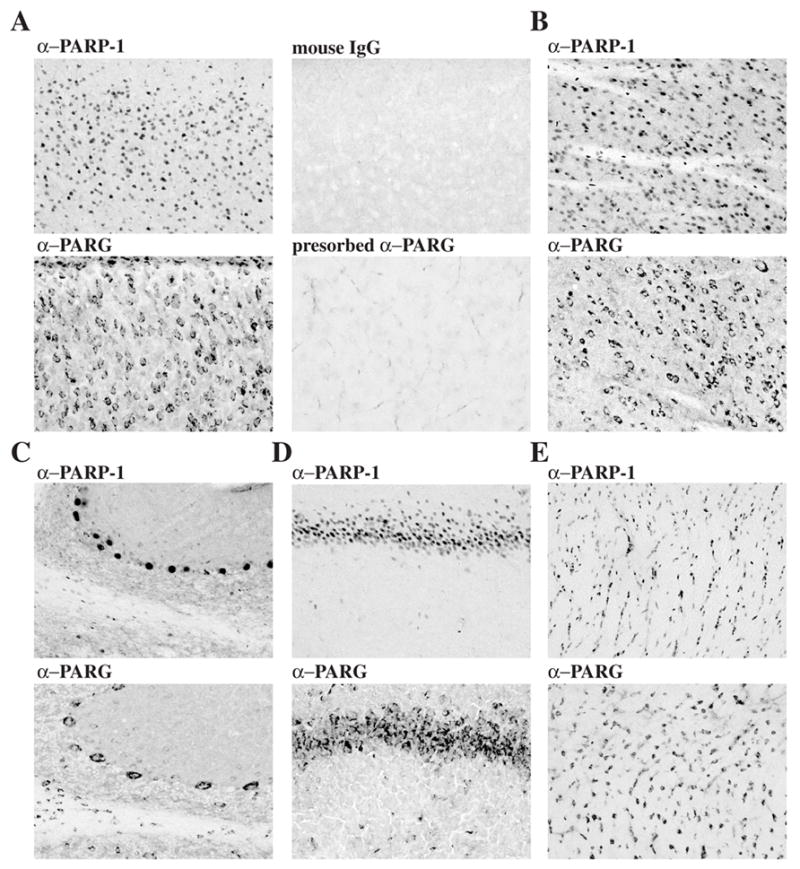
PARP-1 and PARG immunohistochemical localization in rat brain. PFA perfused rat brain sections (40 μm) and frozen rat brain sections (20 μm) were stained with mouse α–PARP-1 and mouse IgG (1 μg/ml) or with rabbit α–PARG and antigen-preadsorbed α–PARG (10 μg/ml) respectively. Sections were then incubated with biotinylated anti-rabbit antibodies and with the ABC solutions. PARG immunostaining was developed with DAB. A) cortex; B) striatum; C) cerebellum; D) CA1 region of hippocampus; E) corpus collosum.
Subcellular localization of PARG and PARP-1
The immunohistochemical studies suggest that PARP-1 and PARG have different subcellular localizations with PARP-1 being concentrated in the nucleus and PARG being concentrated primarily within the cytoplasm and perinuclear structures. Subcellular fractionation studies were conducted to identify the subcellular localization of PARG (Figure 4). Whole brain homogenates were separated into nuclear, mitochondrial and post-mitochondrial fractions (Figure 4). In rat brain, PARG is primarily contained within the mitochondrial fraction, little immunoreactive PARG detected in the nuclear and post-mitochondrial fractions (Figure 4). Overexposed films clearly revealed the presence of small amounts of PARG in both the nuclear and the post-mitochondrial fractions. The integrity of the subcellular fractionation is confirmed by the enrichment of PARP-1 in the nuclear fraction and the enrichment of mitochondrial manganese superoxide dismutase (MnSOD) and cytochrome C (Cyt C) in the mitochondrial fraction (Figure 4). To determine whether PARG is also enriched within the mitochondrial fraction in non-neuronal tissues, the subcellular distribution of PARG in heart, kidney, and testis was examined (Figure 4). In heart, kidney and testis, full length PARG is primarily enriched within the mitochondrial and post-mitochondrial fractions, with less immunoreactivity within the nuclear fraction (Figure 4). The 85 kDa PARG proteolytic fragment observed in some whole tissue extracts (Figure 2A) was observed in the mitochondrial and post-mitochondrial fractions, with variable immunoreactivity within the nuclear fraction of heart, kidney and testis. In heart, kidney and testis, a 90 kDa immunoreactive band was present in the postmitochondrial fraction. The integrity of the nuclear and mitochondrial fractions from heart, kidney and testis are confirmed by the enrichment of PARP-1 in the nuclear fraction and MnSOD in the mitochondrial fraction (Figure 4).
Figure 4.
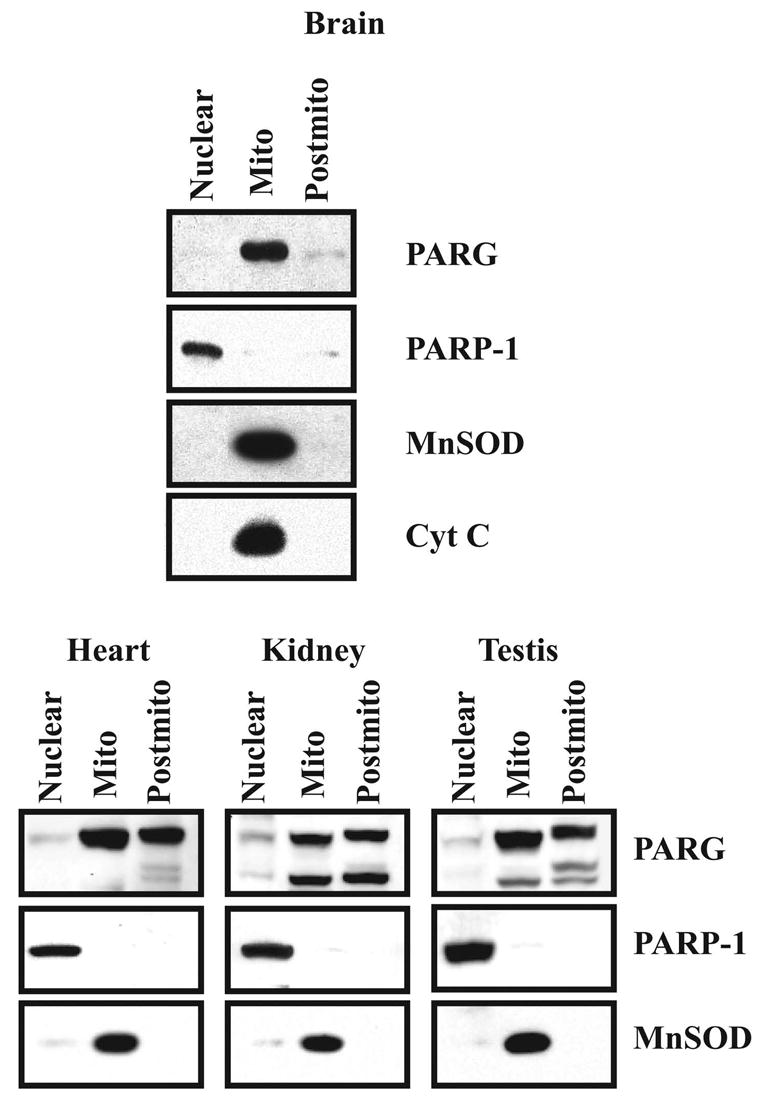
Subcellular localization of PARG. 20 μg of protein of each subcellular fraction from the brain, heart, kidney and testis were separated on SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blotted with α–PARP-1, α–PARG, α–MnSOD and α–Cyt C. Western blots were developed by chemiluminescence using SuperSignal. These results were replicated three times with similar results.
The subcellular localization of PARG to both the mitochondrial and post-mitochondrial fractions of heart, kidney and testis suggests that PARG is not an intramitochondrial protein and that it may be tethered to the outer membrane of the mitochondria. To test whether PARG is an integral mitochondrial membrane protein versus whether it is tethered to the outer membrane of the mitochondria, we conducted de-salting experiments on purified mitochondria and examined the distribution of PARG compared to the distribution of the integral mitochondrial proteins Cyt C and MnSOD (Figure 5). In rat brain mitochondria, increasing concentrations of NaCl shift the distribution of PARG from the mitochondrial pellet to the supernatant without effecting the distribution of Cyt C or MnSOD (Figure 5). The fact that low physiological NaCl concentrations (100 mM NaCl) can washout PARG from the mitochondrial fraction without affecting Cyt C and MnSOD strongly suggests that PARG is associated with the outer surface of the mitochondria and that it can probably be released under physiological conditions.
Figure 5.
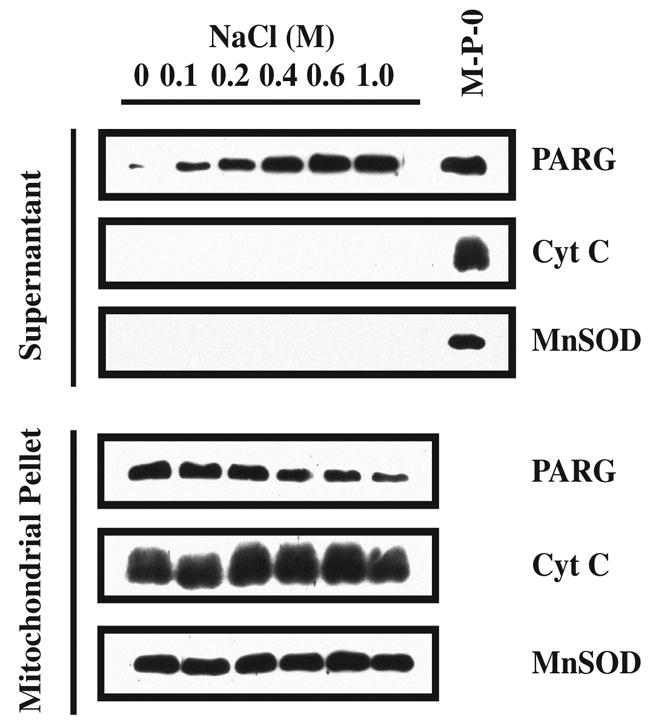
Mitochondrial PARG salting out. The mitochondrial pellet (P2) was aliquoted in separate tubes and centrifuged. The individual pellets were then resuspended in 200 μl of subfractionation buffer containing increasing concentrations of NaCl. The mitochondrial suspensions were centrifuged 30 min at 10,000 x g. The proteins contained in the supernatants and pellets were resolved on SDS/PAGE gel, subjected to Western blot analysis and blotted with α–PARG, α–MnSOD and α–Cyt C. Western blots were developed by chemiluminescence using SuperSignal. These results were replicated two times with similar results. M-P-0: mitochondrial pellet at 0 M NaCl.
To confirm that PARG is a mitochondrial-associated protein, we conducted immunohistochemical co-localization studies using confocal microscopy of primary cortical neuronal cultures (Figure 6). To identify the nucleus, we used the DNA binding dye, TOTO-3. To identify the mitochondria, we used Cyt C immunohistochemistry. As previously reported, PARP-1 is exclusively localized to the nucleus, as it dramatically co-localizes with TOTO-3 in rat cortical neurons. PARG immunostaining is primarily concentrated within the cytoplasm and perinuclear regions with slight staining in the nucleus. As such, the majority of PARG immunoreactivity fails to co-localize with TOTO-3 staining. In contrast, PARG immunoreactivity is strikingly co-localized with the mitochondrial protein Cyt C (Figure 6A). We also find that PARG also co-localizes with the astrocytic marker GFAP and that it co-localizes with the neuronal marker MAP-2 confirming the immunohistochemical localization of PARG to glia and neurons in the brain (Figure 6B).
Figure 6.
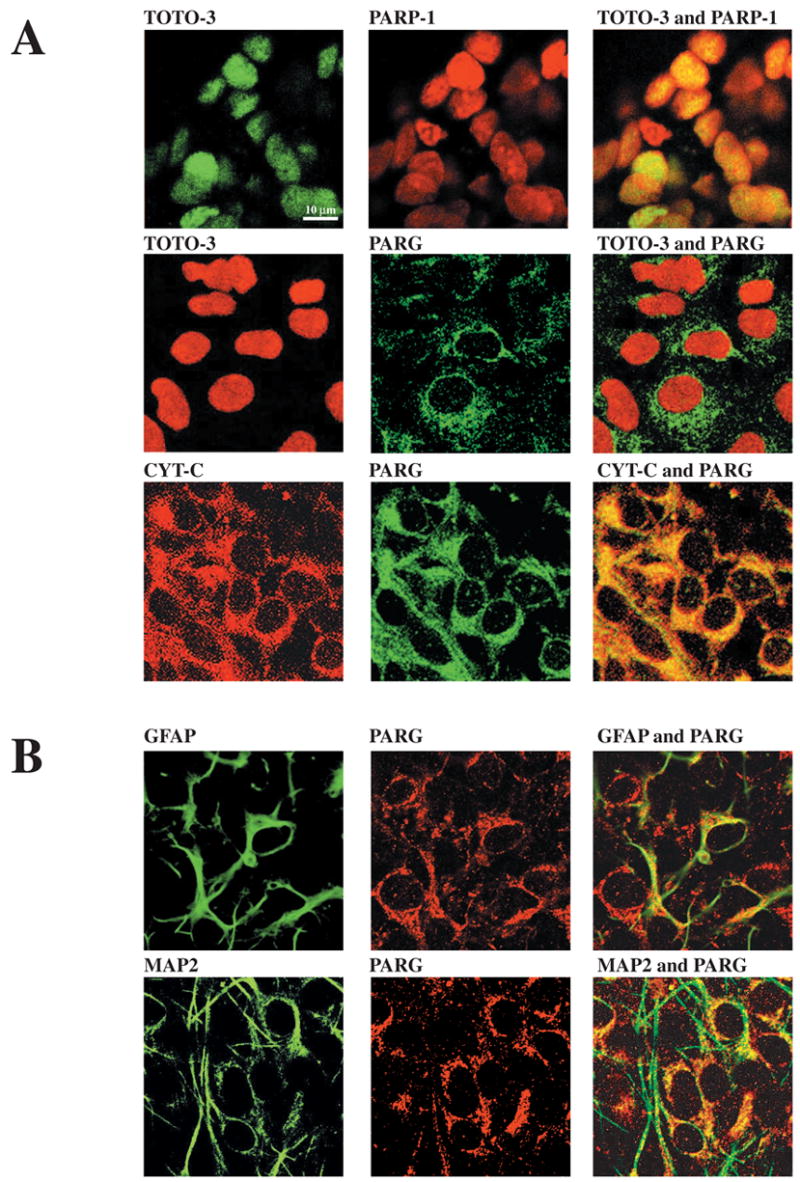
Mutually exclusive cellular localization of PARP-1 and PARG. Confocal laser scanning microscopy analysis of PARP-1 and PARG in rat neuronal cortical cultures. Upper panels: Mixed neuronal cultures were stained with mouse α–PARP-1, α–Cyt C, rabbit α–PARG and with the DNA binding dye TOTO-3. Lower panels: Mixed neuronal cortical cultures were stained with rabbit α–PARG and mouse α–GFAP (glial marker) or α–MAP2 (neuronal marker). Confocal laser scanning microscopy shows that PARP-1 and PARG display a mutually exclusive cellular localization and that PARG is expressed in both neurons and astrocytes.
The subcellular segregation of PARP-1 and PARG suggest that PARG would need to relocalized to the nucleus in order to allow its functional interaction with PARP-1. It is well known that PARP-1 activation only leads to a transient increase in nuclear PAR accumulation due to the activity of PARG, further suggesting that PARG would need to relocalize to the nucleus in order to cleave the PAR after PARP-1 activation. In order to test this hypothesis, WT mouse cortical neurons were treated with NMDA under excitotoxic conditions that potently lead to PARP-1 activation (Mandir et al., 2000, Yu et al., 2002), and PAR formation and PARG translocation to the nucleus was monitored and compared to PARP-1 knockout (KO) cortical neurons (Figure 7). Western blot analysis with α–PAR antibody 96-10 (Affar et al., 1998, Yu et al., 2002) shows that treatment of WT cortical neurons with 500 μM NMDA (5 min) induces a transient increase of PAR that reaches maximal levels 1 hour after NMDA treatment and returns to basal levels after 4–6 hours (Figure 7A), clearly demonstrating the presence of a PARG activity after PARP-1 activation. As expected, NMDA also induces an important reduction of cellular NAD+ levels (Figure 7B) as a consequence of PARP-1 activation. No PAR accumulation and no reduction in cellular NAD+ levels are observed in PARP-1 KO cortical neurons treated with NMDA, clearly showing that PAR accumulation and NAD+ consumption induced by NMDA is PARP-1 dependent. PAR levels are gradually reduced between 2 and 6 hours after NMDA treatment, with almost no PAR remaining at 6 hours, suggesting that there is an increase of PARG activity in the nucleus over time after NMDA treatment. Although it is possible that the small amount of PARG observed in the nucleus could be involved in polymer degradation after NMDA treatment, we reasoned that recruitment of cytosolic PARG might explain the increase PARG activity in the nucleus (6 hours after NMDA treatment). Therefore there should be an enrichment of PARG immunostaining in the nucleus after NMDA treatment. PARG immunostaining performed in WT and PARP-1 KO cortical neurons (Figure 7C) reveals cytosolic and perinuclear localization of PARG in untreated WT and PARP-1 KO and in NMDA-treated PARP-1 KO cortical neurons. In contrast, there is a strong perinuclear and nuclear localization of PARG in NMDA treated WT cortical neurons. Furthermore, subcellular fractionation experiments (Figure 7D) followed by PARG Western blot analysis confirmed the enrichment of full length PARG in the nuclear fraction in WT cortical neurons treated with NMDA, and this PARG translocation was NMDA dose-dependent (Figure 7E). These data suggest that full length PARG translocates from the cytosolic compartment to the nucleus following PARP-1 activation and that this PARG relocalization is PARP-1 dependent.
Figure 7.
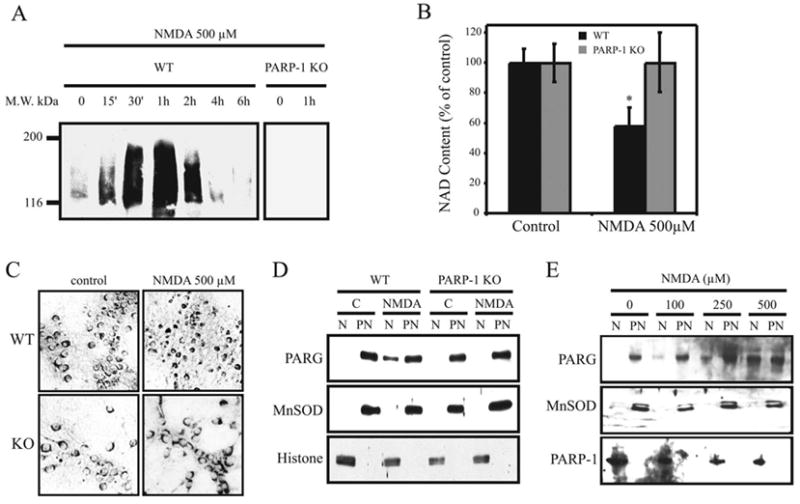
NMDA induced PARG translocation is PARP-1 dependent. WT and PARP-1 KO cortical cultures were treated 5 min with the indicated doses of NMDA and harvested at different time points for PAR Western blot analysis (A), or at 6 hours for cellular NAD level evaluation (B), PARG immunostaining (C), and subcellular fractionation experiments followed by PARG Western blotting (D & E). α–MnSOD antibodies and α–histone antibodies were respectively used as markers for post-nuclear and nuclear fractions, except in (E) where the nuclear fraction was indicated using α–PARP-1 (C2-10). These results are representative of at least two independent experiments that gave similar results.
DISCUSSION
The major finding of this study lies in the differential subcellular localization of PARP-1 and PARG and the PARP-1 dependent nuclear translocation of PARG after NMDA-induced PARP-1 activation. Our results also constitute the first report of immunolocalization of endogenous PARG in the brain. As demonstrated by immunohistochemistry and subcellular fractionation of the brain, PARP-1 and PARG mainly occur in different subcellular compartments. PARP-1 is enriched in the nuclear fraction whereas PARG is primarily enriched in the mitochondrial fraction (associated with the outer mitochondrial membrane). Throughout the brain we find high densities of PARP-1 and PARG in all neuronal structures examined. In all structures studied, PARP-1 and PARG mainly reside in different subcellular compartments. We also demonstrate that PARP-1 and PARG reside in different subcellular compartments in non-neuronal tissue including the heart, testis, and kidney, where PARP-1 occurs primarily in the nucleus while PARG occurs mainly in the mitochondrial/cytosolic fraction. In agreement with our observations, Sevigny et al (Sevigny et al., 2003) demonstrated that in neuronal cultures, full length PARG was present only in the post-nuclear fraction where higher PARG activity was observed. Recent observations showed that full length PARG overexpressed in NIH 3T3 cells is localized exclusively in the nucleus during interphase and that it shuttles between nucleus and cytoplasm during the cell cycle (Ohashi et al., 2003). Meyer-Ficca et al (Meyer-Ficca et al., 2004) also showed that full length PARG (the 111 kDa PARG variant) is expressed in the nucleus, while splice variants 102 and 99 kDa are expressed in the cytosol. Thus, the subcellular targeting of PARG could depend on many factors, including the splice variant expressed, the tissues or cells studied, the phase of the cell cycle, and potentially on the presence of PARG interacting proteins that could differentially relocalize PARG under different conditions.
PARGs with different molecular weight have been purified from various cellular compartments of organs, revealing the presence of PARG activity in both nuclear and postnuclear fractions (Tavassoli et al., 1983, Hatakeyama et al., 1986, Tanuma and Endo, 1990, Maruta et al., 1991, Uchida et al., 1993, Abe and Tanuma, 1996), suggesting that there might be several different forms of PARG. We found that in rat brain, the full length PARG is the predominant form of the active enzyme (Figure 1D). Interestingly, Sevigny et al, (2003) showed by Western blot analysis that primary neuronal cultures express mainly a short 63 kDa form of PARG. The differences observed could be associated with the different PARG antibodies used and possibly to their preferential affinity for PARG full length or the shorter forms of PARG. Although, following subcellular fractionations in kidney and testis, we did detect shorter forms of PARG, possibly the 99 and 102 kDa splice variants (Figure 4), we cannot exclude the possibility that our antibody has a lower affinity for the shorter forms of PARG, and that this could preclude the detection of shorter forms of PARG by Western blot and immunohistochemistry. At this moment, the identification of a single PARG gene, and the identification of high molecular weight PARG splicing variants (111, 102 and 99 kDa) would still suggest that the different low molecular weight PARGs identified would probably be degradation fragments of one of the PARG splice variants. However, one cannot exclude the possibility that the different PARGs identified would be the products of unidentified genes.
What might be the functional significance for the differential localization of PARP-1 and PARG in different tissues? In vivo, PARG activity is thought to be in large excess to cellular PARP-1 activity (Menard et al., 1990, Thomassin et al., 1990). Despite this excess, nuclear PAR concentrations are kept within the Km values of the PARG enzyme (Alvarez-Gonzalez and Althaus, 1989), suggesting that PARG and PARP-1 may need to be in different subcellular compartments and/or nuclear PARG would need to be tightly regulated in order to allow adequate PARP-1 mediated poly(ADP-ribosyl)ation of nuclear proteins. It was shown that the half-life of PAR decreases from 7.7 hours to less than 1 min after MNNG-induced DNA-damage (Alvarez-Gonzalez and Althaus, 1989), due to the fact the PARG displays a higher affinity for longer PAR (Hatakeyama et al., 1986). So the rapid degradation of the PAR would prevent its accumulation in the nucleus after DNA damage. In fact, Jonsson et al (Jonsson et al., 1988a, Jonsson et al., 1988b) suggested that PARG activity has to be inhibited by 90% in order for PAR to accumulate in the cell. Considering all these factors, one way to explain the occurrence of a transient PAR profile after DNA damage is that PARG activity would be tightly regulated and part of this regulation could be the recruitment of cytosolic PARG to the nucleus. While low levels of PARG would be sufficient to perform normal nuclear functions, at some point after DNA damage, when the PAR level reaches a certain level, larger amounts of PARG would be needed in order to remove PAR from ADP-ribosylated proteins, and PARG would then be recruited from the cytosolic compartment. In vivo studies suggest that PAR levels need to reach a threshold of 5 μM for the initiation of PAR catabolism (Alvarez-Gonzalez and Althaus, 1989). Consistent with this notion, we failed to observe any PARG enrichment in the nucleus in PARP-1 KO neurons treated with NMDA (where no PAR was detected) (Figure 7). Thus, PAR catabolism in the nucleus could be regulated, at least in part, by a nuclear-cytosolic shuttling of PARG. PARG shuttling to and from the nucleus is probably a very efficient and rapid process that would make it very difficult for the actual detection of PARG translocation. Supporting this notion, Winstall et al. (Winstall et al., 1999) demonstrated that nuclear PAR (induced after DNA damage) was undetectable in cells overexpressing PARG despite the fact that overexpressed PARG was observed exclusively in the cytosol. Sequence analysis of PARG proteins reveals a potential nuclear export signal sequence within the N-terminal portion of PARG at amino acid positions 124–132 that is conserved among mammals as well as a functional nuclear localization signal (NLS) (Lin et al., 1997, Shimokawa et al., 1999). Thus, a balance between the NLS and NES signals on PARG probably controls its nuclear and cytosolic localization. Since poly(ADP-ribosyl)ation of nuclear proteins is an ongoing process within cells, PARG probably constantly shuttles between the nucleus and cytosol depending upon PARP-1 activity. Furthermore, consistent with the notion that there is nuclear-cytoplasmic shuttling of PARG is the observation that leptomycin B, an inhibitor of nuclear export, leads to the enrichment of GFP-PARG in the nucleus (Bonicalzi et al., 2003).
PARG is probably also required for de-ribosylation of proteins modified by other members of the PARP family in different subcellular compartments, preventing the accumulation of free or protein-bound polymer reactions (Burkle, 2001, Smith, 2001). Since the other PARP family member proteins are not exclusively localized to the nucleus, there are probably additional mechanisms, such as chaperones, that participate in the shuttling of PARG to these other subcellular compartments. Therefore, the presence of both NES and NLS in the PARG protein would allow a dynamic shuttling of PARG throughout the cell where PARP activity is found.
In this report, we show that PARG is predominantly localized outside the nucleus, and that its re-localization to the nucleus might be an important mechanism regulating the levels of PAR after NMDA-induced PARP-1 activation (Figure 7). The transient accumulation of PAR, followed by its complete disappearance, suggests a dynamic increase in PARG activity in the nucleus, possibly due to the to the recruitment of cytosolic PARG. As suggested by the results of Winstall et al. (Winstall et al., 1999), PARG shuttling and PAR degradation is a very rapid process, that could occur without a noticeable increase in nuclear PARG. An increase in nuclear PARG might only be seen when the shuttling process is altered by agents such as leptomycin B or when cellular energy is insufficient to allow an efficient shuttling. PARP-1 activation uses large amount of NAD+ and ATP leading to cellular energy depletion. As we show in our study, PARG is observed in the nucleus concomitantly with the disappearance of PAR 6 hours after NMDA-induced PARP-1 activation, when cellular energy levels are low. This suggests that PARG translocation could be at least in part responsible for PAR degradation. The effect of PARP-1 activation on cellular energy supply might alter the PARG shuttling equilibrium, resulting in PARG accumulation in the nucleus.
What could be the signal leading to PARG translocation? PARP-1 activation is required for the translocation of AIF from the mitochondria to nucleus (Yu et al., 2002). This AIF translocation induced by PARP-1 activation is dependent on PARP-1 catalytic activity. It is therefore conceivable that PAR signaling, together with the decrement in NAD+ and/or ATP, could initiate PARG shuttling and cause its accumulation in the nucleus, but other mechanisms may also be responsible. The elucidation of the mechanisms by which PARP-1 activation signals the mitochondria to elicit translocation of PARG would be of paramount importance.
Acknowledgments
We thank Joke Wortel for technical support and Weza Cotman for secretarial support. Supported by grants from the NIH (NS39148), the American Heart Association, and the Mary Lou McIlhaney Scholar Award. M.F.P was supported by NSERC of Canada. G.G.P. is supported by the Medical Research Council of Canada. Under an agreement between the Johns Hopkins University and MGI, T.M.D. and V.L.D. are entitled to a share of sales royalty received by the university from MGI. The terms of this arrangement are being managed by the university in accordance with its conflict-of-interest policies. T.M.D. is the Leonard and Madlyn Abramson Professor of Neurodegenerative Diseases.
Abbreviations
- PARPs
Poly(ADP-ribose) polymerases
- PAR
poly(ADP-ribose) or ADP-ribose polymer
- PARG
poly(ADP-ribose) glycohydrolase
- Cyt C
cytochrome C
- NMDA
N-methyl-D-aspartate
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- NGS
normal goat serum
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe H, Tanuma S. Properties of poly(ADP-ribose) glycohydrolase purified from pig testis nuclei. Arch Biochem Biophys. 1996;336:139–146. doi: 10.1006/abbi.1996.0541. [DOI] [PubMed] [Google Scholar]
- Affar EB, Duriez PJ, Shah RG, Sallmann FR, Bourassa S, Kupper JH, Burkle A, Poirier GG. Immunodot blot method for the detection of poly(ADP-ribose) synthesized in vitro and in vivo. Anal Biochem. 1998;259:280–283. doi: 10.1006/abio.1998.2664. [DOI] [PubMed] [Google Scholar]
- Affar EB, Germain M, Winstall E, Vodenicharov M, Shah RG, Salvesen GS, Poirier GG. Caspase-3-mediated processing of poly(ADP-ribose) glycohydrolase during apoptosis. J Biol Chem. 2001;276:2935–2942. doi: 10.1074/jbc.M007269200. [DOI] [PubMed] [Google Scholar]
- Alvarez-Gonzalez R, Althaus FR. Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res. 1989;218:67–74. doi: 10.1016/0921-8777(89)90012-8. [DOI] [PubMed] [Google Scholar]
- Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonicalzi ME, Vodenicharov M, Coulombe M, Gagne JP, Poirier GG. Alteration of poly(ADP-ribose) glycohydrolase nucleocytoplasmic shuttling characteristics upon cleavage by apoptotic proteases. Biol Cell. 2003;95:635–644. doi: 10.1016/j.biolcel.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Brochu G, Duchaine C, Thibeault L, Lagueux J, Shah GM, Poirier GG. Mode of action of poly(ADP-ribose) glycohydrolase. Biochim Biophys Acta. 1994a;1219:342–350. doi: 10.1016/0167-4781(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Brochu G, Shah GM, Poirier GG. Purification of poly(ADP-ribose) glycohydrolase and detection of its isoforms by a zymogram following one- or two-dimensional electrophoresis. Anal Biochem. 1994b;218:265–272. doi: 10.1006/abio.1994.1177. [DOI] [PubMed] [Google Scholar]
- Burkle A. Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays. 2001;23:795–806. doi: 10.1002/bies.1115. [DOI] [PubMed] [Google Scholar]
- Chang P, Jacobson MK, Mitchison TJ. Poly(ADP-ribose) is required for spindle assembly and structure. Nature. 2004;432:645–649. doi: 10.1038/nature03061. [DOI] [PubMed] [Google Scholar]
- Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, Wang ZQ. Depletion of the 110-Kilodalton Isoform of Poly(ADP-Ribose) Glycohydrolase Increases Sensitivity to Genotoxic and Endotoxic Stress in Mice. Mol Cell Biol. 2004;24:7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosi C, Marien M. Implication of poly (ADP-ribose) polymerase (PARP) in neurodegeneration and brain energy metabolism. Decreases in mouse brain NAD+ and ATP caused by MPTP are prevented by the PARP inhibitor benzamide. Ann N Y Acad Sci. 1999;890:227–239. doi: 10.1111/j.1749-6632.1999.tb07998.x. [DOI] [PubMed] [Google Scholar]
- D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559–7569. doi: 10.1021/bi0003442. [DOI] [PubMed] [Google Scholar]
- Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Kizushi VM, Huang PL, Snyder SH, Dawson TM. Resistance to neurotoxicity in cortical cultures from neuronal nitric oxide synthase-deficient mice. J Neurosci. 1996;16:2479–2487. doi: 10.1523/JNEUROSCI.16-08-02479.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia G, Huletsky A, Lamarre D, Gaudreau A, Pouyet J, Daune M, Poirier GG. Modulation of chromatin superstructure induced by poly(ADP-ribose) synthesis and degradation. J Biol Chem. 1986;261:7011–7017. [PubMed] [Google Scholar]
- de Murcia G, Schreiber V, Molinete M, Saulier B, Poch O, Masson M, Niedergang C, Menissier de Murcia J. Structure and function of poly(ADP-ribose) polymerase. Mol Cell Biochem. 1994;138:15–24. doi: 10.1007/BF00928438. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Falsig J, Christiansen SH, Feuerhahn S, Burkle A, Oei SL, Keil C, Leist M. Poly(ADP-ribose) glycohydrolase as a target for neuroprotective intervention: assessment of currently available pharmacological tools. Eur J Pharmacol. 2004;497:7–16. doi: 10.1016/j.ejphar.2004.06.042. [DOI] [PubMed] [Google Scholar]
- Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, Takahashi H, Miwa M. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. Proc Natl Acad Sci U S A. 2004;101:82–86. doi: 10.1073/pnas.2237114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose) J Biol Chem. 1986;261:14902–14911. [PubMed] [Google Scholar]
- Hong SJ, Dawson TM, Dawson VL. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol Sci. 2004;25:259–264. doi: 10.1016/j.tips.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Jacobson EL, Jacobson MK. Tissue NAD as a biochemical measure of niacin status in humans. Methods Enzymol. 1997;280:221–230. doi: 10.1016/s0076-6879(97)80113-9. [DOI] [PubMed] [Google Scholar]
- Jonsson GG, Jacobson EL, Jacobson MK. Mechanism of alteration of poly(adenosine diphosphate-ribose) metabolism by hyperthermia. Cancer Res. 1988a;48:4233–4239. [PubMed] [Google Scholar]
- Jonsson GG, Menard L, Jacobson EL, Poirier GG, Jacobson MK. Effect of hyperthermia on poly(adenosine diphosphate-ribose) glycohydrolase. Cancer Res. 1988b;48:4240–4243. [PubMed] [Google Scholar]
- Ju BG, Solum D, Song EJ, Lee KJ, Rose DW, Glass CK, Rosenfeld MG. Activating the PARP-1 sensor component of the groucho/TLE1 corepressor complex mediates a CaMKinase IIdelta-dependent neurogenic gene activation pathway. Cell. 2004;119:815–829. doi: 10.1016/j.cell.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Kanai M, Tong WM, Sugihara E, Wang ZQ, Fukasawa K, Miwa M. Involvement of poly(ADP-Ribose) polymerase 1 and poly(ADP-Ribosyl)ation in regulation of centrosome function. Mol Cell Biol. 2003;23:2451–2462. doi: 10.1128/MCB.23.7.2451-2462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gevry N, Lis JT, Kraus WL. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004;119:803–814. doi: 10.1016/j.cell.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Koh D, Dawson V, Dawson T. The Road to Survival Goes through PARG. Cell Cycle. 2005;4 doi: 10.4161/cc.4.3.1559. [DOI] [PubMed] [Google Scholar]
- Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to Degrade Poly(ADP-ribose) Causes Increased Sensitivity to Cytotoxicity and Early Embryonic Lethality. Proc Natl Acad Sci U S A. 2004;101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- Lautier D, Lagueux J, Thibodeau J, Menard L, Poirier GG. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem. 1993;122:171–193. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- Lin W, Ame JC, Aboul-Ela N, Jacobson EL, Jacobson MK. Isolation and characterization of the cDNA encoding bovine poly(ADP-ribose) glycohydrolase. J Biol Chem. 1997;272:11895–11901. doi: 10.1074/jbc.272.18.11895. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, Dawson VL. NMDA but not non-NMDA excitotoxicity is mediated by Poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. doi: 10.1523/JNEUROSCI.20-21-08005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A. 1999;96:5774–5779. doi: 10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruta H, Inageda K, Aoki T, Nishina H, Tanuma S. Characterization of two forms of poly(ADP-ribose) glycohydrolase in guinea pig liver. Biochemistry. 1991;30:5907–5912. doi: 10.1021/bi00238a014. [DOI] [PubMed] [Google Scholar]
- Menard L, Thibault L, Poirier GG. Reconstitution of an in vitro poly(ADP-ribose) turnover system. Biochim Biophys Acta. 1990;1049:45–58. doi: 10.1016/0167-4781(90)90083-e. [DOI] [PubMed] [Google Scholar]
- Menegazzi M, Grassi-Zucconi G, Carcerero De Prati A, Ogura T, Poltronieri P, Nyunoya H, Shiratori-Nyunoya Y, Miwa M, Suzuki H. Differential expression of poly(ADP-ribose) polymerase and DNA polymerase beta in rat tissues. Exp Cell Res. 1991;197:66–74. doi: 10.1016/0014-4827(91)90480-i. [DOI] [PubMed] [Google Scholar]
- Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- Ogura T, Takenouchi N, Yamaguchi M, Matsukage A, Sugimura T, Esumi H. Striking similarity of the distribution patterns of the poly(ADP-ribose) polymerase and DNA polymerase beta among various mouse organs. Biochem Biophys Res Commun. 1990;172:377–384. doi: 10.1016/0006-291x(90)90683-e. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Kanai M, Hanai S, Uchiumi F, Maruta H, Tanuma S, Miwa M. Subcellular localization of poly(ADP-ribose) glycohydrolase in mammalian cells. Biochem Biophys Res Commun. 2003;307:915–921. doi: 10.1016/s0006-291x(03)01272-5. [DOI] [PubMed] [Google Scholar]
- Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281:705–713. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- Panda S, Poirier GG, Kay SA. tej Defines a Role for Poly(ADP-Ribosyl)ation in Establishing Period Length of the Arabidopsis Circadian Oscillator. Dev Cell. 2002;3:51–61. doi: 10.1016/s1534-5807(02)00200-9. [DOI] [PubMed] [Google Scholar]
- Ranjit GB, Cheng MF, Mackay W, Whitacre CM, Berger JS, Berger NA. Poly(adenosine diphosphoribose) polymerase in peripheral blood leukocytes from normal donors and patients with malignancies. Clin Cancer Res. 1995;1:223–234. [PubMed] [Google Scholar]
- Rouleau M, Aubin RA, Poirier GG. Poly(ADP-ribosyl)ated chromatin domains: access granted. J Cell Sci. 2004;117:815–825. doi: 10.1242/jcs.01080. [DOI] [PubMed] [Google Scholar]
- Sevigny MB, Silva JM, Lan WC, Alano CC, Swanson RA. Expression and activity of poly(ADP-ribose) glycohydrolase in cultured astrocytes, neurons, and C6 glioma cells. Brain Res Mol Brain Res. 2003;117:213–220. doi: 10.1016/s0169-328x(03)00325-5. [DOI] [PubMed] [Google Scholar]
- Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- Shimokawa T, Masutani M, Nagasawa S, Nozaki T, Ikota N, Aoki Y, Nakagama H, Sugimura T. Isolation and cloning of rat poly(ADP-ribose) glycohydrolase: presence of a potential nuclear export signal conserved in mammalian orthologs. J Biochem (Tokyo) 1999;126:748–755. doi: 10.1093/oxfordjournals.jbchem.a022512. [DOI] [PubMed] [Google Scholar]
- Smith S. The world according to PARP. Trends Biochem Sci. 2001;26:174–179. doi: 10.1016/s0968-0004(00)01780-1. [DOI] [PubMed] [Google Scholar]
- Tanuma S, Endo H. Purification and characterization of an (ADP-ribose)n glycohydrolase from human erythrocytes. Eur J Biochem. 1990;191:57–63. doi: 10.1111/j.1432-1033.1990.tb19093.x. [DOI] [PubMed] [Google Scholar]
- Tavassoli M, Tavassoli MH, Shall S. Isolation and purification of poly(ADP-ribose) glycohydrolase from pig thymus. Eur J Biochem. 1983;135:449–455. doi: 10.1111/j.1432-1033.1983.tb07672.x. [DOI] [PubMed] [Google Scholar]
- Thomassin H, Jacobson MK, Guay J, Verreault A, Aboul-ela N, Menard L, Poirier GG. An affinity matrix for the purification of poly(ADP-ribose) glycohydrolase. Nucleic Acids Res. 1990;18:4691–4694. doi: 10.1093/nar/18.16.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, Suzuki H, Maruta H, Abe H, Aoki K, Miwa M, Tanuma S. Preferential degradation of protein-bound (ADP-ribose)n by nuclear poly(ADP-ribose) glycohydrolase from human placenta. J Biol Chem. 1993;268:3194–3200. [PubMed] [Google Scholar]
- Wang H, Yu SW, Koh DW, Lew J, Coombs C, Bowers W, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Stingl L, Morrison C, Jantsch M, Los M, Schulze-Osthoff K, Wagner EF. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11:2347–2358. doi: 10.1101/gad.11.18.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham S, Virag L, Hasko G, Stachlewitz R, Szabo C, Kochanek PM. Traumatic brain injury in mice deficient in poly-ADP(ribose) polymerase: a preliminary report. Acta Neurochir Suppl. 2000;76:61–64. doi: 10.1007/978-3-7091-6346-7_12. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham SH, Virag L, Hasko G, Stachlewitz R, Szabo C, Kochanek PM. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1999;19:835–842. doi: 10.1097/00004647-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Winstall E, Affar EB, Shah R, Bourassa S, Scovassi IA, Poirier GG. Preferential perinuclear localization of poly(ADP-ribose) glycohydrolase. Exp Cell Res. 1999;251:372–378. doi: 10.1006/excr.1999.4594. [DOI] [PubMed] [Google Scholar]
- Ying W, Sevigny MB, Chen Y, Swanson RA. Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci U S A. 2001;98:12227–12232. doi: 10.1073/pnas.211202598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Swanson RA. The poly(ADP-ribose) glycohydrolase inhibitor gallotannin blocks oxidative astrocyte death. Neuroreport. 2000;11:1385–1388. doi: 10.1097/00001756-200005150-00007. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]