

SUMMARY
Glutamatergic and dopaminergic inputs converge on medium spiny neurons in nucleus accumbens and regulate the excitability of these projections to target areas including the cholinergic basal forebrain. NMDA receptors situated on these projections are locally modulated by D1- and D2-like receptors. We previously reported that the D1-like positive modulation of NMDA receptor activity is expressed trans-synaptically in the control of basal forebrain cholinergic projections to prefrontal cortex. The present experiments tested the hypothesis that D2-like receptors in accumbens negatively modulate cortical ACh release. Perfusion of NMDA (150 μM) into the shell region of the accumbens produced a sustained increase (150–200%) in ACh release in prefrontal cortex. This increase was completely blocked by co-perfusion with the D2-like agonist quinpirole (100 μM). Perfusion of quinpirole also reduced basal ACh release (~50%) in prefrontal cortex. The contribution of D2 receptors to the quinpirole effect was assessed in two additional studies. The first study revealed that co-perfusion of the D2 antagonist haloperidol (100 μM) blocked the quinpirole-induced attenuation of NMDA mediated ACh release. The second experiment demonstrated that intra-accumbens perfusion of quinelorane (100 μM), a more selective D2 agonist than quinpirole, also attenuated the NMDA mediated ACh release. Collectively, these studies demonstrate that D2 receptors in accumbens negatively modulate basal and NMDA mediated increases in ACh release in prefrontal cortex. This negative modulation may contribute to the integration of normal attentional processing and goal directed behavior and to the therapeutic effects of antipsychotic medication on cognition in psychopathologies such as schizophrenia.
Keywords: ACh, D2 receptors, NMDA receptors, accumbens, prefrontal cortex, basal forebrain, schizophrenia
INTRODUCTION
There is abundant evidence indicating that striatal NMDA receptor function is modulated by dopaminergic inputs. Anatomically, dopamine (DA) and NMDA receptors converge onto dendritic spines of medium spiny projection neurons (MSNs) in the dorsal and ventral striatum (Ariano et al, 1997; Pennartz et al, 1994; Sesack and Pickel, 1990). By virtue of their more proximal position, relative to the soma, DA receptors are in position to modulate the neuronal effects of the more distal NMDA receptors.
Interactions between DA and NMDA receptors have been extensively studied in the context of the positive and negative modulation of NMDA receptor function by D1 and D2 receptors, respectively (Flores-Hernandez et al, 2002; Cepeda et al, 1998; Snyder et al, 1998). The intra-cellular transduction mechanisms that link D1 receptor activation with the high-affinity phosphorylated state of the NR1 subunit and D2 receptor activation with the lower affinity de-phosphorylated state have been well characterized (Snyder et al, 1998). This bidirectional dopaminergic modulation of NMDA receptor function is also expressed at the level of MSN excitability. The MSNs of the nucleus accumbens (NAC) exhibit bipolar fluctuations in membrane potential resulting in a more depolarized (i.e. more excitable) ‘UP’ state and a more hyperpolarized (i.e. less excitable) ‘DOWN’ state (Grace, 2000). Activation of D1 receptors has been shown to bias accumbens MSNs in the ‘UP’ state whereas D2 receptor activity biases these neurons toward the ‘DOWN’ state (Charara and Grace, 2003; Goto and O’Donnell, 2001a,b).
Finally, DA-NMDA receptor interactions in NAC have long been discussed in the mediation of motivated behavior. Mesolimbic DA inputs selectively gate the cortico-limbic afferents to NAC MSNs (mediated, in part, by NMDA receptors) and this gating results in the biasing toward certain behavioral responses at the expense of others (Goto and Grace, 2005; Grace, 2000; O’Donnell, 1999; Pennartz et al, 1994). Interactions between DA and NMDA receptors in NAC have recently been demonstrated in the linkage of incentive-laden stimuli and appropriate goal-directed behavior (Deadwyler et al, 2004; Nicola et al, 2004; Yun et al, 2004a,b). Our theoretical perspective is that such coordination between relevant stimuli and appropriate behavioral outcome requires an integration of motivational factors and selective biases in the allocation of attentional resources. We speculate that the DA-NMDA-based regulation of NAC projections to the basal forebrain and the subsequent control of ACh release in prefrontal cortex (PFC) provides a neuronal platform for the integration of motivation and attention in the selection of appropriate goal-directed behavior (Sarter et al, 2005). The positive and negative modulation of NAC NMDA receptor function by D1 and D2 receptors, respectively, might provide a mechanism that biases highly salient cortical projections to NAC for subsequent modulation of the basal forebrain cortical cholinergic system (BFCS). In addition, this bi-directional D1-D2 modulation of NMDA receptors may allow varying levels of extracellular dopamine (based on changing firing rates or DA from synaptic vs extrasynaptic release sites) to differentially modulate cortical ACh release. In testing these hypotheses, we have recently reported that perfusions of NMDA into the shell region of the NAC produce a marked stimulation of ACh release in PFC (Zmarowski et al, 2005). Moreover, D1 receptor activation positively modulates this ability of NMDA receptors to stimulate cortical ACh release. In the present study, we tested the hypothesis that D2 receptors in NAC negatively modulate the ability of NMDA to evoke cortical ACh release. We first determined the effects of intra-NAC perfusions of the D2/D3 agonist quinpirole on cortical ACh as a function of the degree of NMDA receptor activation. Thus, the effects of quinpirole on both basal and NMDA-evoked cortical ACh release were characterized. Finally, we assessed the contributions of D2 receptors to the actions of quinpirole by determining the ability of the D2 antagonist haloperidol to attenuate the quinpirole effect on basal ACh release as well as the ability of another agonist (quinelorane) that has a higher affinity for D2 than D3 (Eaton et al, 1993; Foreman et al, 1989) to mimic the effects of quinpirole.
MATERIALS AND METHODS
Subjects
Two strains of male adult rats (body weights of 350–450 gm) were used in these experiments. Fisher 344/Brown Norway F1 hybrid rats (Harlan Laboratory, Indianapolis, IN) served as subjects for the initial experiment while subsequent experiments were performed using Wistar rats (Charles River Laboratory: Wilmington, MA). The change in strain was necessitated by problems with consistent availability of the hybrid strain. Although the number of subjects did not allow for a statistical comparison no apparent differences were observed in the results obtained by these two strains. Subjects were housed individually and were maintained in a temperature (21–23° C) - and humidity (40–60%)-controlled environment on a 12 hour light (lights on at 6:30 am): dark cycle with food and water available ad libitum. Animals were handled for one week prior to surgery, during which time they were habituated (4–6 hr/day) to their microdialysis testing environment of clear plastic concentric dialysis bowls [35 cm high × 38 cm deep: Carnegie Medicin (CMA), Stockholm, Sweden] lined with corn-cob bedding (Harlan Teklad, Madison WI, USA). Animal care and experimentation were performed in accordance with protocols approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee and consistent with the NIH Guide for the Care and Use of Laboratory Animals.
Surgical Procedures
The day before surgery, animals were deprived of food in preparation for surgery. Animals were anesthetized with inhalant isofluorane (2%, 0.6 L/min, O2) and two microdialysis guide cannula were stereotaxically implanted. The first cannula was targeted for the medial prefrontal cortex (PFC; in mm from bregma: AP +4.2, LM +0.6, DV −0.6 with the tip of the guide cannula tip pointing 20° rostral) and the second cannula was targeted for the ipsilateral shell of the nucleus accumbens (NAC; in mm from bregma: AP +1.3, LM +1.0, DV −5.8). Coordinates were determined from the atlas of Paxinos and Watson (1986). Guide cannulae were fixed to the skull by three stainless steel screws (Small Parts Inc., Miami Lakes, FL.) and dental cement. Following the conclusion of the surgery, animals received a prophylactic dose of the antibiotic amoxicillin (100 mg/kg, s.c.). Animals recovered from anesthesia before being returned to their home cage.
General Microdialysis Procedures
Microdialysis sessions began the fourth day post surgery and were conducted using a repeated perfusion paradigm. Animals received a different drug on each microdialysis test day, with a day of recovery in between each session. This paradigm allows each animal to receive multiple concentrations of a drug (e.g. dose-response studies) or combinations of drugs (e.g. receptor agonist/antagonist studies). Importantly, this testing paradigm also allows each subject to act as its own control. We have extensively validated this protocol for a number of neurotransmitters (e.g. ACh, GABA, Glu, DA) in several brain regions (for review see Bruno et al, 1999). Relevant to the present experiments, our previous studies demonstrated that basal cortical ACh efflux does not change significantly across multiple dialysis sessions. In addition, neither the magnitude of drug or behaviorally-induced changes interacts with the sequence of multiple dialysis sessions (Nelson et al, 2005; Arnold et al, 2003; Moore et al, 1995).
On the fourth day following surgery, animals were placed in their testing bowls and allowed to acclimate for 30 min. Following the acclimation period, animals’ stylets were removed and probes (Sci Pro, Inc., 0.2mm o.d., 3.0 mm membrane tip for mPFC, 2.0 mm membrane tip for NAC) were inserted into the appropriate guide cannula. Probes were perfused (1.25 μL/min) with artificial cerebral spinal fluid (aCSF) (containing in mM: NaCl 166.5, NaHCO3 27.5, KCL 2.4, CaCl2 1.2, Na2SO4 0.5, KH2PO4 0.5, glucose 1.0, pH 7.1) continuously during the microdialysis session. Due to the sensitivity of our high performance liquid chromatography (HPLC) detection system, no acetylcholinesterase inhibitor was utilized during these experiments. A 3 hr “wash-out” period was observed prior to baseline collections. This equilibration period results in a cortical ACh efflux that maximally reflects neuronal depolarization-induced synaptic release (Moore et al, 1995). In the present studies, one of three distinct experimental sequences were conducted using the same general format; four baseline collections, six drug collections, and three post drug recovery collections.
HPLC Analysis
Upon the completion of each microdialysis session, dialysates were stored at −80º C until analyzed. ACh levels in dialysates collected from the mPFC were quantified using HPLC with electrochemical detection. Once thawed, 15 μL of each dialysate was injected by autosampler (ESA Inc., Chelmsford, MA, USA). Using a sodium phosphate mobile phase transport solution (in mM: Na2HPO4, 50.0, 0.5% microbicide reagent Proclin, pH= 8.5; flow rote of 0.3 mL/min), ACh and choline were separated by a UniJet microbore column (BASi, West Lafayette, IN.; 1 × 530 mm). The use of a pre-column immobilized enzyme reactor (IMER; BASi) allowed for a greater separation between ACh and choline peaks. ACh was hydrolyzed by a post-column enzyme reactor (BASi) and converted to H2O2 (Potter et al, 1983) that was then oxidized and quantified using a peroxidase-wired ceramic glassy carbon electrode (Huang et al, 1995), with an applied potential of −200 mV. The detection limit for ACh under these conditions was approximately 2.0 fmol/15μL injection.
Histology
Following the last microdialysis session, animals were given an overdose of sodium pentobarbital and transcardially perfused with 0.9% heparinized saline followed by 10% formalin. The brains were extracted and stored in 10% formalin for 24 hr, transferred to a 30% sucrose solution and then refrigerated for at least 3 days. Once cryoprotected, brains were sectioned using a cryostat. Sections (50 μm) were mounted on slides, stained using Cresyl Violet, and examined under a light microscope. Only subjects whose probe placements were located within the mPFC and ipsilateral NAC shell were included in the final data analysis.
Data Analysis
An initial statistical analysis was performed on absolute basal ACh efflux (fmol/15 μL). Baseline efflux was determined by averaging the values from the first four 15 minute collections taken after the three hr “wash out” session. In order to determine whether baselines differed among the treatment CONDITIONs, a one-way analysis of variance (ANOVA) was conducted with treatment CONDITION as the within subject factor. A similar ANOVA, on basal values, was conducted across session number (1–4). Following a determination that there were no significant CONDITION or session differences across absolute baselines, a two-way ANOVA was conducted with the data expressed as percent change from baseline. This overall ANOVA examined drug effects with treatment CONDITION and TIME as within-subject measures. Following an overall significant effect of treatment CONDITION more targeted two way ANOVAs were conducted in order to determine the source of the main effects. A minimum number of t-tests were conducted as post-hoc analyses in order to highlight drug effects as a function of time. Significance was defined as α < 0.05, and the Huynh-Feldt correction was utilized to reduced Type 1 errors associated with repeated measures ANOVAs (Vasey and Thayer, 1987). All statistical tests were performed using SPSS for windows (version 14.0; Chicago, IL).
Specific Experimental Procedures
Experiment 1: Effects of intra-NAC perfusion of quinpirole on basal and NMDA-stimulated cortical ACh release
The first group of rats (Fisher 344/Brown Norway, n = 8) was used to study the ability of the D2/D3 agonist quinpirole to modulate cortical ACh release under basal levels of NMDA receptor activity in NAC. Animals received intra-NAC perfusions of the D2/D3 agonist quinpirole (1, 10, and 100 μM) or its vehicle (aCSF) in a pseudo randomized order every other day. Upon completion of the 3 hr “wash out” period, 4 baseline collections were taken. Each collection interval lasted 15 min. During the aCSF control session, aCSF was perfused during all 4 baselines, all 6 ‘drug’, and 3 additional ‘post-drug’ collections. During each quinpirole session, baseline collections were followed by a switch from aCSF to quinpirole (1, 10, or 100 μM quinpirole). A 15 minute “wash out” period was imposed to account for the dead volume of the tubing and microdialysis swivel. Following the wash out period, quinpirole was perfused during all six drug intervals and was then replaced with aCSF. Following a second wash out, 3 additional collections were taken. Following the last collection, probes were removed, the stylets were reinserted, and the animals were returned to their home cage.
A second group of rats (Fisher 344/Brown Norway, n = 8) was tested in order to determine the ability of quinpirole to modulate cortical ACh release under conditions in which the NAC NMDA receptor activity was increased following local perfusions of NMDA. A perfusion concentration of 150 μM was selected based on our previous observation that this concentration of NMDA elicited a sustained increase in cortical ACh release and this evoked release was modulated by co-perfusion with the D1 antagonist SCH 23390 (Zmarowski et al., 2005). The basic design of the repeated perfusion experiments was identical to that described above except for the following changes in procedure. Each animal received three different, pseudo randomized treatments [aCSF, NMDA (150 μM), or NMDA (150 μM) + quinpirole (100 μM)] every other day. During the NMDA session, aCSF was perfused across 4 baseline collections and 2 additional collection intervals. Following these 6 collections, the syringe delivering aCSF to the NAC was switched to one containing NMDA (150 μM). After the 15 minute “wash out” collection, NMDA was perfused across the remaining 4 drug collections and was then replaced with aCSF. Following a second “wash out” period, 3 additional collections were taken to determine if efflux would return to baseline values. During the NMDA + quinpirole session, aCSF was replaced by quinpirole (100 μM) during collections 5 and 6 to ensure that quinpirole concentrations were maximal at the onset of NMDA perfusion. This perfusate was then replaced with one containing NMDA (150μM) + quinpirole (100μM) and a second 15 min wash out was imposed. This drug combination was perfused across 4 additional collection intervals and was then replaced with aCSF followed by 3 additional collections were taken.
Experiment 2: The contribution of D2 receptors to the modulatory effects of quinpirole
An additional group of animals was used to assess the contribution of D2 receptors in NAC to quinpirole’s modulation of basal cortical ACh release. Two strategies were employed to highlight the aspect of quinpirole’s actions that was dependent upon D2 receptor activation. First, we determined whether the D2 antagonist haloperidol would attenuate the effects of quinpirole. Second, we determined whether the effects of quinpirole could be mimicked by an agonist that exhibits a higher selectivity for D2 receptors than does quinpirole (Eaton et al., 1993). A combined group of Fisher 344 Brown Norway (n = 3) + Wistar (n = 5) rats received one of four pseudo randomized intra-NAC perfusions [quinpirole (100μM), the more selective D2 agonist quinelorane (100μM), the D2 antagonist haloperidol (100μM) + quinpirole (100μM)], or aCSF] every other day. The structure of individual dialysis sessions with respect to number of baselines, duration of collection intervals, and wash out periods during drug changes was the same as described in Experiment 1. During the aCSF, quinpirole, or quinelorane sessions aCSF was perfused during collections 5 and 6 in order to provide control conditions against which the effects of haloperidol perfusion alone could be assessed prior to switching to the perfusion of haloperidol + quinpirole.
RESULTS
Guide Cannulae Placements
All subjects included in these studies had cannula placements located in the mPFC and shell region of the ipsilateral NAC. Figure 1 depicts a representative photomicrograph of probe placements within the NAC shell and mPFC. These placements were very similar to those previously reported on the D1 positive modulation of NMDA-mediated cortical ACh release (Zmarowski et al, 2005).
Figure 1.
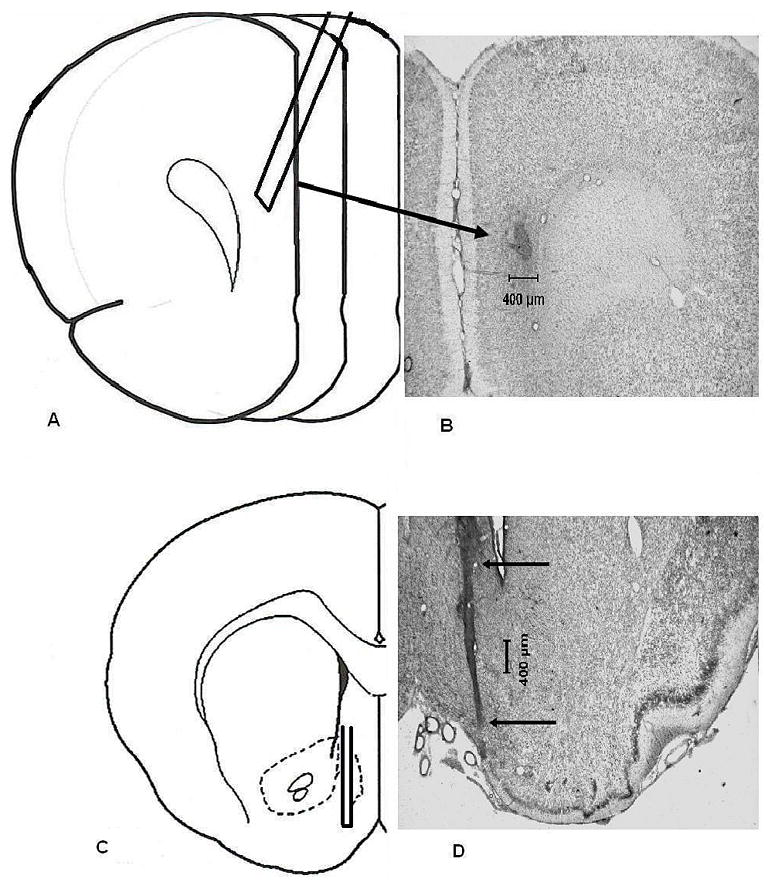
Representative histology for mPFC and NAC shell probe placements. (Panel A) a schematic depiction of a mPFC placement. Guides were implanted so that when probes were inserted, the membrane tip (3.0 mm) was located at AP +4.2, LM +0.6, DV −0.6 from dura pointing 20° rostral. (Panel B) a representative photomicrograph from a subject with the mPFC placement that is illustrated in panel A. Due to the angle at which the guide cannula and probe were positioned, the arrow points to the site of termination of the dialysis probe. (Panel C) a schematic depiction of a NAC shell placement. Guides were implanted so that when probes were inserted, the membrane tip (2.0 mm ) was located at AP + 1.3, LM +1.0, DV −5.8 from dura mater. (Panel D) a representative photomicrograph from a subject with the NAC shell placement illustrated in panel C. The arrows indicate the tract of the guide cannula. All coordinates were based upon Paxinos and Watson (1998).
Experiment 1a: The effects of intra-NAC perfusions of a D2-like agonist on basal cortical ACh efflux
Basal levels (mean ± S.E.M., fmol/15μL) of cortical ACh efflux for rats in the aCSF and quinpirole (1, 10, and 100 μM) treatment conditions (collapsed across sessions) were 5.2 ± 1.0, 9.6 ± 1.6, 5.9 ± 0.6, and 7.8 ± 1.0, respectively. ACh levels were equivalent across treatment conditions (F3,15 = 2.69, p = 0.10). Basal ACh levels for sessions one, two, three, and four (collapsed across conditions) were 9.0 ± 1.7, 6.9 ± 0.9, 7.9 ± 1.0, and 4.7 ± 0.7, respectively. ACh levels were equivalent across all dialysis sessions (F3,15= 2.162, p = 0.17). Given that there were no significant differences in basal levels of ACh efflux among the various treatment conditions or dialysis sessions, subsequent levels of cortical ACh efflux were expressed as percent change from the within session baseline.
Figure 2 illustrates the effects of intra-NAC perfusion of the D2-like agonist quinpirole on cortical ACh release under basal, non-activated conditions. Perfusions of quinpirole led to concentration-dependent decreases, relative to aCSF controls, in cortical ACh efflux (GROUP, F3,15 = 4.809, p = 0.02). These effects varied across collection interval (TIME, F12,60 = 3.176, p = 0.01), however, no significant CONDITION X TIME interaction was observed (F36,180 = 1.288, p = 0.16). A series of 2-way ANOVAs was conducted to determine the source of the overall differences among quinpirole concentrations. Quinpirole (100 μM) significantly decreased cortical ACh levels below the aCSF vehicle (CONDITION, F1,5 =18.038, p = 0.01; TIME, F12,60 = 3.067, p = 0.002; CONDITION X TIME, F12,60 = 2.809, p = 0.005). This concentration of quinpirole significantly lowered cortical ACh levels below aCSF within the first 30 minutes of perfusion (collection 6, t5 = 3.987, P = 0.010) and levels remained lowered throughout the perfusion period (collection 10, t5 = 3.463, p = 0.02). Perfusion of the two smaller concentrations of quinpirole (1 and 10μM) did not significantly decrease cortical ACh efflux as compared to the aCSF vehicle (all p’s > 0.05).
Figure 2.
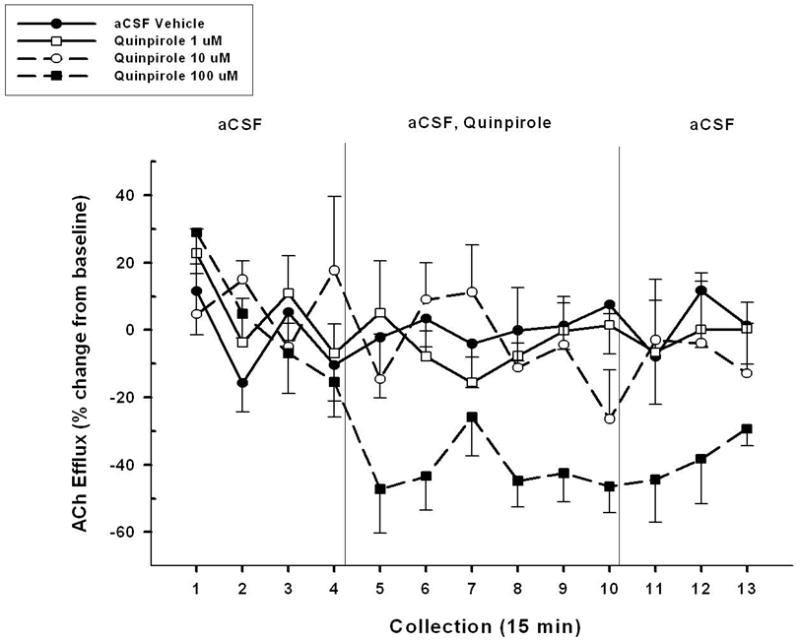
Mean (± S.E.M.) ACh efflux in the mPFC of animals (n=6) receiving, in a pseudo-randomized order, perfusions of vehicle (aCSF) and three concentrations of the D2-like agonist quinpirole (1, 10, and 100 μM) into the NAC shell during four separate dialysis sessions. Following baseline collections (collections 1–4), vehicle or one of three concentrations of quinpirole was perfused for 90 min (collections 5–10). Upon conclusion of this perfusion period, aCSF was perfused for 45 min until the end of the dialysis period (collections 11–13). The largest concentration of quinpirole (100 μM) significantly reduced cortical ACh levels below those seen following aCSF vehicle or the two lower concentrations of drug. Perfusions of 1 and 10 μM had no effect on cortical ACh efflux.
A comparison between 100 μM quinpirole and each of the two lower concentrations (1 and 10 μM) revealed differences in ACh efflux. Specifically, the effect of 100 μM quinpirole on cortical ACh release significantly differed from the effect of 1 μM (CONDITION, F1,5 = 9.557, p = 0.03; TIME, F12,60 = 4.035, p<0.001) and 10 μM (CONDITION, F1,5 = 9.981, p = 0.02; TIME, F12,60 = 4.750, p = 0.004).
Experiment 1b: Effects of intra-NAC perfusions of a D2-like receptor agonist on NMDA-evoked cortical ACh release
Basal ACh levels (mean ± S.E.M., fmol/15 μL) for rats in the aCSF, NMDA (150 μM), and NMDA (150 μM) + quinpirole (100 μM) treatment conditions (collapsed across sessions) were 6.4 ± 1.3, 6.3 ± 1.1, and 10.2 ± 1.6, respectively. Basal levels of cortical ACh were equivalent across all three treatment conditions (F2,14 = 2.35, p = 0.13). Basal ACh levels (mean ± S.E.M., fmol/15 μL) for session one, session two, and session three (collapsed across conditions) were 7.4 ± 1.0, 7.9 ± 1.2, and 7.6 ± 2.1, respectively. These levels of cortical ACh were equivalent across all three dialysis sessions (F2,14 = 0.02, p = 0.98). Given that there were no significant differences among baseline levels of ACh efflux for treatment condition or session, subsequent values are expressed as percent change from the within session baseline.
Figure 3 depicts the effects of aCSF vehicle, NMDA (150 μM), and NMDA (150 μM) + quinpirole (100 μM) on cortical ACh efflux (n= 8). Overall, significant differences among treatment conditions and collection time intervals were observed (CONDITION, F2,14 = 40.982, p < 0.001; TIME, F12,84 = 3.123, p =0.005; CONDITION X TIME, F24,168 = 6.777, p < 0.001). Given the overall differences among conditions, a series of 2-way ANOVAs was conducted to determine significant differences between pairs of treatment conditions. The administration of NMDA (150 μM) significantly increased cortical ACh levels above the aCSF vehicle (CONDITION, F1,7 = 23.2, p = 0.002; TIME, F12,84 = 7.4, p< 0.001; CONDITION X TIME, F12,84 = 6.98, p < 0.001). The stimulatory effects of NMDA administration were apparent within the first 15 minutes of perfusion (collection 7, t7 = 8.04, p< 0.001) and ACh levels remained elevated throughout the ensuing three, 15 minutes collection periods (collection 10, t7 = 5.28, p = 0.001). Cortical ACh levels returned to basal levels within the first 15 minutes following the removal of NMDA (collection 11, t7 = 1.79, p = 0.12).
Figure 3.
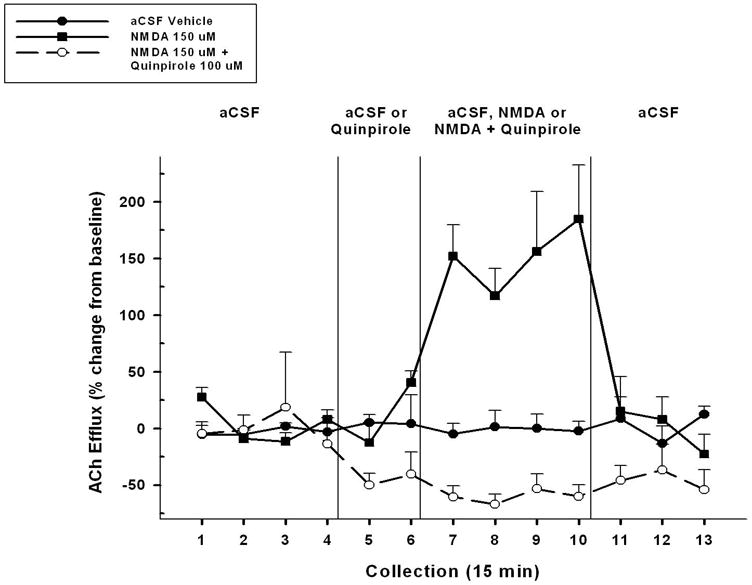
Mean (± S.E.M.) ACh efflux in the mPFC of animals (n=8) receiving, in a pseudo-randomized order, perfusions of vehicle (aCSF), NMDA alone (150 μM), and co-administration of a D2-like agonist quinpirole (100 μM) + NMDA into the NAC shell during three separate dialysis sessions. Following baseline collections (collections 1–4), vehicle or the agonist was perfused for 30 min (collections 5–6). This was followed by administration of aCSF, NMDA, or quinpirole + NMDA for 60 min (collections 7–10). Upon conclusion of the 60 min perfusion period, aCSF was perfused alone for 45 min until the end of the dialysis period (collections 11–13). Perfusion of NMDA significantly increased ACh efflux above that seen in following aCSF vehicle. Co-administration of quinpirole + NMDA completely blocked this NMDA effect as well as maintained a significant decrease in ACh release below that seen in the aCSF vehicle control condition.
Administration of quinpirole (100 μM) blocked the increase in cortical ACh evoked by the administration of NMDA. This is evidenced by the significant difference between NMDA (150 μM) and the combined administration of NMDA (150 μM) + quinpirole (100 μM) (CONDITION, F1,7 = 61.34, p < 0.001; TIME, F12,84 = 3.89, p = 0.002; CONDITION X TIME, F12,84 = 8.92, p < 0.001). The elimination of the NMDA effect by quinpirole was apparent within the first 15 minutes of dual perfusion of both drugs (collection 7, t7 = 6.23, p < 0.001) and was maintained throughout all four collection periods (collection 10, t7 = 3.73, p = 0.01). Cortical ACh levels between the two conditions did not differ within the first 15 minutes following the removal of drugs during the NMDA and NMDA + quinpirole sessions (collection 11, t7 = .189, p = 0.86).
Finally, the results obtained from Experiment One, that quinpirole administration reduced basal ACh efflux, were replicated in collections 5 and 6 prior to the addition of NMDA to quinpirole.
Experiment 2: The contribution of D2 receptors to the quinpirole effects on basal cortical ACh release
The contribution of D2 receptor activation to the inhibitory effects of quinpirole on basal cortical ACh release was assessed in two ways. First, we determined the ability of the D2 antagonist haloperidol to block the effects of quinpirole. Second, we determined whether a different agonist, quinelorane, with a greater affinity for D2 than D3 receptors (Eaton et al, 1994; Foreman et al, 1989) would mimic the effects of quinpirole on basal cortical ACh release. The results of these two studies are illustrated in Figure 4.
Figure 4.
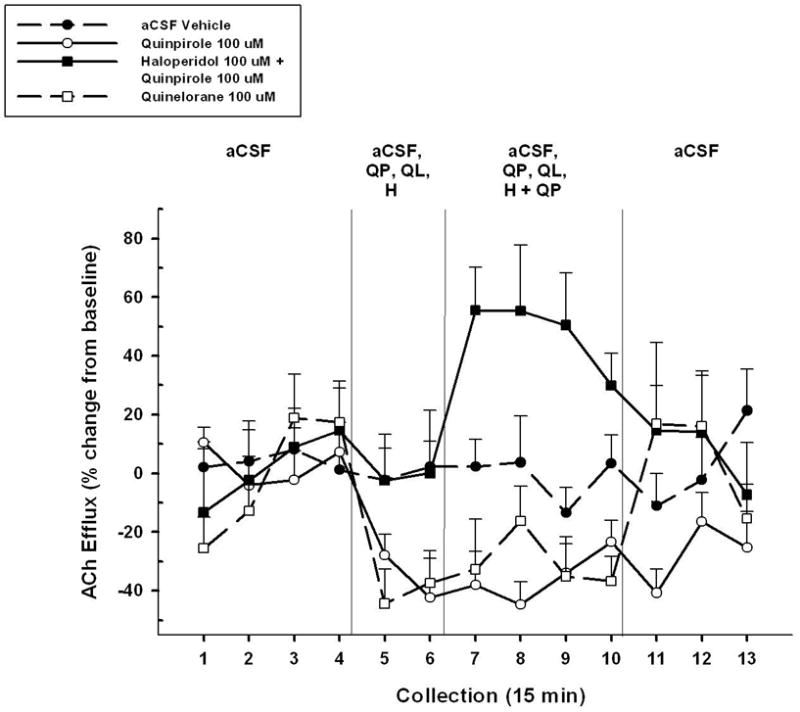
Mean (± S.E.M.) ACh efflux in the mPFC of animals (n=7) receiving, in pseudo-randomized order, perfusions of vehicle (aCSF), a D2/D3 agonist quinpirole alone (QP, 100 μM), a more selective D2 agonist quinelorane (QL, 100 μM), and co-administration of quinpirole + a D2-like antagonist haloperidol (H, 100 μM) into the NAC shell during four separate dialysis sessions. Following baseline collections (collections 1–4), vehicle, agonists, or antagonist was perfused for 30 min (collections 5–6). This was followed by administration of vehicle, quinpirole, quinelorane, or quinpirole + haloperidol for 60 min (collections 7–10). Upon conclusion of the 60 min perfusion period, aCSF was perfused alone for 45 min until the end of the dialysis period (collections 11–13). Quinpirole perfusion decreased ACh efflux below aCSF vehicle levels as previously observed. Similar to quinpirole, quinelorane also decreased ACh efflux below aCSF levels. Co-administration of haloperidol + quinpirole blocked the observed effect of quinpirole on cortical ACh release and actually increased ACh levels above the aCSF vehicle condition. Perfusion of haloperidol alone (collections 5 and 6) did not affect basal ACh levels.
Basal ACh levels (mean ± S.E.M., fmol/15μl), collapsed across microdialysis session, for aCSF vehicle, quinpirole (100 μM), haloperidol (100 μM) + quinpirole (100 μM), and quinelorane (100 μM) were 5.86 ± 0.56, 6.37 ± 1.00, 7.60 ± 2.14 and 8.07 ± 2.49, respectively. Basal levels of cortical ACh were equivalent across treatment conditions (F3,18 = 0.276, p= 0.77). Basal ACh levels (mean ± S.E.M., fmol/15μl) for session one, session two, session three, and session four (collapsed across treatment conditions) were 5.55 ± 0.60, 5.67 ± 1.52, 7.94 ± 1.67, and 8.73 ± 2.43, respectively. Basal levels of cortical ACh were equivalent across the dialysis sessions (F3,18 = 0.714 , p= 0.55). Given that there were no significant differences across baseline levels of ACh efflux for treatment condition or session, subsequent ACh values were expressed as percent change from baseline.
The first analysis of these data compared the effects of aCSF (control), quinpirole, and quinpirole + haloperidol on ACh release in PFC. An overall ANOVA revealed significant differences among the CONDITIONS (F2,72 = 14.53, p < 0.001). A series of two-way ANOVAs were conducted to identify the sources of this overall difference. Perfusion of qiunpirole significantly reduced basal ACh levels seen under the aCSF control condition (F1,72 = 20.297, p = 0.004), replicating the results seen in Experiment 1a. This decrease was evident during the first collection period (#5, t6 = 2.83, p = 0.03) and persisted until the drug was removed (#11, t6 = 2.203, p = 0.07).
Figure 4 also illustrates that co-perfusion of the D2 antagonist haloperidol completely blocked the inhibitory effects of quinpirole (CONDITION, F1,72 = 17.758, p = 0.01; CONDITION x TIME, F12, 72 = 3.121, p = 0.003). The blockade of quinpirole’s effect by haloperidol was seen during the first collection interval (#7, t6 = −4.776, p = 0.03) and persisted until the removal of the antagonist (#11, t6 = −0.763, p = 0.07). Unexpectedly, the combined perfusion of quinpirole and haloperidol actually resulted in cortical ACh release that was significantly higher than the aCSF control baseline (CONDITION, F1,72 = 11.248, p = 0.005; CONDITION x TIME, F12,72 = 3.847, p = 0.004). The ability of combined drug perfusion to stimulate cortical ACh release above baseline was evident during the first double-drug collection interval (#7, t6 = −3.932, p = 0.01) and lost by the final drug collection interval (#10, t6 = −1.774, p = 0.13). Importantly, the perfusion of haloperidol alone (during collections 5 and 6) did not affect basal levels of cortical ACh efflux.
A final set of analyses on the data summarized in Figure 4 revealed that perfusion of the more selective D2 agonist quinelorane reduced basal ACh release relative to that seen following aCSF control perfusions (CONDITION, F1,72 = 5.906, p = 0.05; CONDITION x TIME, F12,72 = 2.349, p = 0.01). As with quinpirole, the inhibitory effects of quinelorane were apparent after the first drug perfusion (#5) and persisted throughout the final drug perfusion (#10, both p’s < 0.05). Importantly, the overall effect of quinelorane was not significantly different from that seen following quinpirole perfusion (p < 0.05).
DISCUSSION
These experiments tested the hypothesis that D2 receptors modulate NMDA receptors in the nucleus accumbens (NAC) and that this modulation is expressed trans-synaptically at the level of the basal forebrain cortical cholinergic system (BFCS). Three major findings are reported. First, activation of D2/D3 receptors in NAC with quinpirole attenuated BFCS activity as evidenced by a decrease in basal ACh release in prefrontal cortex (PFC). Second, quinpirole blocked NMDA-evoked cortical ACh release, suggesting that D2/D3 receptors negatively modulate NMDA receptor activity in NAC. Finally, the modulatory effects of quinpirole on basal ACh release reflect the drug’s activation of D2 receptors as the effect was eliminated following co-administration of the D2 antagonist haloperidol and mimicked by intra-NAC perfusion of quinelorane. The discussion that follows addresses the potential mechanisms underlying this interaction between D2 and NMDA receptors and the cognitive implications of a D2-NMDA modulation of the BFCS.
Mechanisms contributing to the D2-NMDA receptor interactions
The present microdialysis experiments do not permit an assessment of the relative locations of the D2 and NMDA receptors within the shell of the NAC that are affecting cortical ACh release. These two receptor subtypes have been shown to be co-localized on medium spiny projection neurons (Sesack and Pickel, 1990) but they may also be segregated on different NAC afferents, interneurons, and MSN projection pathways (Alcantara et al, 2003; Guzman et al, 2003). It is well-established that D2 receptors can negatively modulate NMDA receptor function locally within the NAC and that this modulation is mediated by multiple mechanisms. First, D2 agonists can decrease the affinity of co-localized NMDA receptors to glutamate. D2 receptor activation biases the de-phosphorylated and reduced affinity state of the NR1 subunit of the NMDA receptor (Snyder et al,1998). Second, D2 receptors located presynaptically on glutamatergic terminals within NAC might decrease the release of glutamate (West et al, 2003; Wang and Pickel, 2002; Hsu et al, 1995) or DA (Roberts et al, 2006; Shilliam and Heidbreder 2003) within NAC. Such decreases in release could diminish the ability of AMPA (Hernandez-Echeagaray et al, 2004) or D1 receptor activation (Zmarowski et al., 2005) to provide the necessary excitation and removal of Mg+2 blockade of NMDA receptors.
The observation that intra-NAC perfusion with quinpirole reduced cortical ACh release to the same extent under basal and NMDA-evoked conditions was unexpected. These experiments were guided by two hypotheses. First, we predicted that the established D2-mediated negative modulation of NMDA receptor function would be expressed trans-synaptically at the level of cortical ACh release. Our previous study (Zmarowski et al, 2005) demonstrated this to be the case with the D1-mediated positive modulation of NMDA receptor function. Co-perfusion of the D1 antagonist SCH 23390 markedly attenuated the NMDA-induced cortical ACh release, suggesting that D1 receptor activation (presumably as a result of NMDA-induced DA release) contributes to the trans-synaptic effect of NMDA on the BFCS.
Second, we predicted that the ability of the D2/D3 agonist quinpirole to dampen NMDA receptor function would be inversely related to the level of NMDA receptor activity. We further speculated that NMDA receptor activity in NAC could be varied by testing animals under basal conditions, in which tonic levels of NMDA in NAC contribute to cortical ACh release (Neigh et al, 2004) and following perfusion of NMDA. Perfusion of quinpirole (100 μM) significantly reduced basal cortical ACh release (Figure 3). We expected that this concentration of quinpirole would attenuate NMDA-evoked release but not that the stimulatory effects of NMDA would be fully blocked. These data suggest a more complex underlying mechanism than simply a D2-mediated bias toward a low affinity NR1 binding site (Snyder et al, 1998) which might have predicted, as was the case with D1 and NMDA receptors (Zmarowski et al, 2005), that high levels of NMDA receptor activity would over-ride a DA modulation. Additional experiments are clearly needed to resolve these mechanisms.
The potency of the intra-NAC quinpirole effect and its ability to totally block NMDA-evoked ACh release raises the issue of whether D2/D3 receptor prevents activation of the cortical cholinergic transmission following other stimuli. Systemic administration of amphetamine stimulates ACh release in cortex (Arnold et al, 2000; Day and Fibiger, 1992). While the mechanisms underlying this effect are not well understood (Arnold et al, 2000), the magnitude of the release is comparable to that seen following intra-NAC infusions of NMDA (~150-200%). We have recently reported that intra-NAC perfusion of quinpirole does not attenuate the ability of systemically-administered amphetamine, to stimulate cortical ACh release (Brooks et al, 2005, SFN). Thus, quinpirole’s blockade of NMDA-evoked ACh release does not reflect a generalized dampening of BFCS excitability.
Specifying the relative contributions of D2 vs D3 receptors to the reported effects of quinpirole on basal and stimulated cortical ACh release is complicated by the fact that the drug exhibits mixed affinities for each DA receptor subtype (Gackenheimer et al, 1995; Levant et al, 1993). The results of Experiment 2 clearly suggest that quinpirole’s ability to negatively modulate NMDA receptors is due to the drug’s ability to stimulate D2 receptors. Co-perfusion of the D2 antagonist haloperidol totally blocked the inhibitory effects of the drug. Moreover, intra-NAC perfusion of quinelorane, a drug with greater affinity for D2 receptors than quinpirole [Eaton et al. (1994) and Foreman et al. (1989)], mimicked the inhibitory effects of quinpirole. Unexpectedly, perfusion of haloperidol not only completely blocked the effects of quinpirole but the co-perfusion actually resulted in a significant increase in basal cortical ACh release. The mechanism underlying this reversal of quinpirole’s actions are not known. It is tempting to speculate that the addition of haloperidol to quinpirole reveals a residual D3 receptor activation and that this subtype is responsible for the activation of the BFCS. We are currently testing this hypothesis with local administration of selective D3 agonists.
Implications of D2 modulation of NMDA function for cognitive behavior
DA receptors gate glutamatergic NAC afferents (Goto and Grace, 2005; Brady and O’Donnell, 2004; Charara and Grace, 2003) and this integration serves to select appropriate ensembles of projection neurons (Deadwyler et al, 2004; O’Donnell, 2003). We proposed that the DA- and NMDA-mediated gating of information processing within this distributed neural system underlies the role of these receptors in the integration of attentional processing and goal-directed behavior.
Attention demanding signals are amplified by activation of the BFCS. The activity of the BFCS is modulated by PFC (Zaborszky et al, 1997) and this top-down regulation is involved in the knowledge-based augmentation of detection and in the filtering of irrelevant information (Sarter et al, 2005, 2001). We have recently reported that cortical ACh release is enhanced, during execution of a sustained attention task, under conditions in which performance demands are augmented (Kozak et al, 2006). We propose that the ability to enhance cortical cholinergic transmission and to recover performance under these challenging conditions involves a recruitment of a PFC-NAC-BF circuit. Currently, we are determining whether intra-NAC infusions of quinpirole attenuate task-induced increases in cortical ACh release and whether such infusions impair performance under conditions in which the sustained attention task demands have been increased.
Finally, altered D2-NMDA receptor interactions in NAC and subsequent dysregulations in cortical cholinergic transmission might contribute to the cognitive deficits seen in psychopathologies such as schizophrenia. In this regard, we have reported that intra-NAC perfusions of NMDA no longer stimulate cortical ACh release in an animal model of schizophrenia (neonatal ventral hippocampal lesions; Alexander et al, 2006). Moreover, NMDA perfusions evoke cortical ACh release in lesioned animals when tested pre-puberty but not when these same animals are tested post-puberty; the developmental time period when others have reported the onset of functional impairments consistent with the information processing deficits seen in schizophrenia (Alexander et al, 2006; Tseng et al, 2006; Goto and O’Donnell, 2003; Lipska and Weinberger, 2002; Lipska et al, 1993). We propose that the emergence of this lesion-induced effect reflects impairments in the ability to recruit the PFC-NAC-BF neuronal circuit.
Collectively, these experiments reveal a negative modulation of the NMDA receptors in the NAC shell by local D2 receptors. This modulation, that has been demonstrated to occur locally within the NAC (Brady and O’Donnell, 2004; Synder et al,1998), can now be extended trans-synaptically at the level of prefrontal ACh release. The stimulation of cortical ACh release by NAC NMDA receptors may mimic the naturally occurring top-down controls of the BFCS by PFC. The D2 modulation of NMDA may serve to filter out low strength excitatory inputs thereby enhancing the signal-to-noise necessary under conditions that require the matching incentive-laden stimuli with activation of particular NAC projection ensembles (Nicola et al., 2004; Yun et al., 2004a,b), and, ultimately, appropriate motor responses.
Acknowledgments
The authors’ research was supported by research grants from the National Institute of Health (MH057436, [JPB, MS]; MH063114 [MS, JPB]; NS37026 [MS, JPB]; and KO2 MH01072 [MS]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcantara AA, Chen V, Herring BE, Mendenhall JM, Berlanga ML. Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Research. 2003;986:22–29. doi: 10.1016/s0006-8993(03)03165-2. [DOI] [PubMed] [Google Scholar]
- Alexander KS, Sarter M, Bruno JP. Neonatal ventral hippocampal lesions produce deficits in attentional processing and the accumbens modulation of cortical ACh release. Neuroscience Meeting Planner; Atlanta, GA. Society for Neuroscience. Online. 2006. [Google Scholar]
- Ariano MA, Larson ER, Noblett KL, Sibley DR, Levine MS. Coexpression of striatal dopamine receptor subtypes and excitatory amino acid subunits. Synapse. 1997;26:400–414. doi: 10.1002/(SICI)1098-2396(199708)26:4<400::AID-SYN8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Arnold HM, Nelson CL, Neigh GN, Sarter M, Bruno JP. Systemic and intra-accumbens administration of amphetamine differentially affects cortical acetylcholine release. Neuroscience. 2000;96:675–685. doi: 10.1016/s0306-4522(99)00590-4. [DOI] [PubMed] [Google Scholar]
- Arnold HM, Nelson CL, Sarter M, Bruno JP. Sensitization of cortical acetylcholine release by repeated administration of nicotine in rats. Psychopharmacology. 2003;165:346–358. doi: 10.1007/s00213-002-1260-6. [DOI] [PubMed] [Google Scholar]
- Brady AM, O'Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. The Journal of Neuroscience. 2004;24:1040–1049. doi: 10.1523/JNEUROSCI.4178-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks JM, Sarter M, Bruno JP. Nucleus accumbens modulation of cortical cholinergic transmission: D2-receptor-mediated inhibition of NMDA-induced ACh release. Neuroscience Meeting Planner; Washington, DC. Society for Neuroscience. Online. 2005. [Google Scholar]
- Bruno JP, Sarter M, Moore AH, Himmelheber AM. In vivo neurochemical correlates of cognitive processes: Methodological and conceptual challenges. Reviews in the Neurosciences. 1999;10:25–48. doi: 10.1515/revneuro.1999.10.1.25. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: Contribution of calcium conductances. Journal of Neurophysiology. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- Charara A, Grace AA. Dopamine receptor subtypes selectively modulate excitatory afferents from the hippocampus and amygdala to rat nucleus accumbens neurons. Neuropsychopharmacology. 2003;28:1414–1421. doi: 10.1038/sj.npp.1300220. [DOI] [PubMed] [Google Scholar]
- Day J, Fibiger HC. Dopaminergic regulation of cortical acetylcholine-release. Synapse. 1992;12:281–286. doi: 10.1002/syn.890120405. [DOI] [PubMed] [Google Scholar]
- Deadyler SA, Hayashizaki S, Cheer J, Hampson RE. Reward, memory, and substance abuse: Functional neuronal circuits in the nucleus accumbens. Neuroscience and Biobehavior Reviews. 2004;27:703–714. doi: 10.1016/j.neubiorev.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Eaton MJ, Lookinlang KJ, Moore KE. Effects of the selective dopaminergic D2 agonist quinelorane on the activity of dopaminergic and noradrenergic neurons projecting to the diencephalons of the rat. The Journal of Pharmacology and Experimental Therapeutics. 1993;268:645–652. [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annual Review of Psychology. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- Flores-Hernandez J, Cepeda C, Hernandez-Echeagaray E, Calvert CR, Jokel ES, Fienberg AA, Greengard P, Levine MS. Dopamine enhancement of NMDA currents in dissociated medium-sized striatal neurons: Role of D1 receptors and DARPP-32. Journal of Neurophysiology. 2002;88:3010–3020. doi: 10.1152/jn.00361.2002. [DOI] [PubMed] [Google Scholar]
- Foreman MM, Fuller RW, Hynes MD, Gidda JS, Nichols CL, Schaus JM, Kornfeld EC, Clemens JA. Preclinical studies on quinelorane, a potent and highly selective D2-dopaminergic agonist. Pharmacology. 1989;1:227–235. [PubMed] [Google Scholar]
- Gackenheimer SL, Schaus JM, Gehlert DR. [3H]-Quinelorane binds to D2 and D3 dopamine receptors in the rat brain. Journal of Pharmacology and Experimental Therapeutics. 1995;274:1558–1565. [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-dirceted behavior. Nature Neuroscience. 2005;8:805–812. doi: 10.1038/nn1471. [DOI] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Altered prefrontal cortex-nucleus accumbens information processing in a developmental animal model of schizophrenia. Annals of the New York Academy of Sciences. 2003;1003:398–401. doi: 10.1196/annals.1300.035. [DOI] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Synchronous activity in the hippocampus and nucleus accumbens in vivo. Journal of Neuroscience. 2001a;24:121–131. doi: 10.1523/JNEUROSCI.21-04-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, O’Donnell P. Network synchrony in the nucleus accumbens in vivo. Journal of Neuroscience. 2001b;27:4498–4504. doi: 10.1523/JNEUROSCI.21-12-04498.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA. Gating of information flow within the limbic systema nd the pathophysiology of schizophrenia. Brain Research Reviews. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Hernandez A, Galarraga E, Tapia D, Laville A, Vergara R, Aceves J, Bargas J. Dopaminergic modulation of axon collaterals interconnecting spiny neurons of the rat striatum. The Journal of Neuroscience. 2003;23:8931–8940. doi: 10.1523/JNEUROSCI.23-26-08931.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Echeagaray E, Starling AJ, Cepeda C, Levine MS. Modulation of AMPA currents by D2 dopamine receptors in striatal medium-sized spiny neurons: Are dendrites necessary? European Journal of Neuroscience. 2004;19:2455–2463. doi: 10.1111/j.0953-816X.2004.03344.x. [DOI] [PubMed] [Google Scholar]
- Huang T, Yang L, Gitzen J, Kissinger PT, Vreeke M, Heller A. Detection of basal acetylcholine in rat brain microdialysate. Journal of Chromotography. 1995;670:323–327. doi: 10.1016/0378-4347(95)00181-6. [DOI] [PubMed] [Google Scholar]
- Hsu KS, Huang CC, Yang CH, Gean PW. Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Research. 1995;690:264–268. doi: 10.1016/0006-8993(95)00734-8. [DOI] [PubMed] [Google Scholar]
- Kozak R, Bruno JP, Sarter M. Augmented prefrontal acetylcholine release During Challenged Attentional Performance. Cerebral Cortex. 2006;16:9–17. doi: 10.1093/cercor/bhi079. [DOI] [PubMed] [Google Scholar]
- Levant B, Grigoriadis DE, DeSouza EB. [3H] quinpirole binding to putative D2 and D3 dopamine receptors in rat brain and pituitary gland: A quantitative autoradiographic study. Journal of Pharmacology and Experimental Therapeutics. 1993;264:991–1001. [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to tress and to amphetamine after neonatal excitotoxic hippocampal damage. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR. To model a psychiatric disorder in animals: Schizophrenia as a reality test. Neuropsychopharmacology. 2002;23:223–239. doi: 10.1016/S0893-133X(00)00137-8. [DOI] [PubMed] [Google Scholar]
- Moore H, Stuckman S, Sarter M, Bruno JP. Stimulation of cortical acetylcholine efflux by FG-7142 measured with repeated microdialysis sampling. Synapse. 1995;21:324–331. doi: 10.1002/syn.890210407. [DOI] [PubMed] [Google Scholar]
- Neigh GN, Arnold HM, Rabenstein RL, Sarter M, Bruno JP. Neuronal activity in the nucleus accumbens is necessary for performance-related increases in cortical acetylcholine release. Neuroscience. 2004;123:635–645. doi: 10.1016/j.neuroscience.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Nelson CL, Sarter M, Bruno JP. Prefrontal cortical modulation of acetylcholine release in posterior parietal cortex. Neuroscience. 2005;132:347–359. doi: 10.1016/j.neuroscience.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Yun IA, Wakabayashi KT, Fields HL. Cue-evoked firing of nucleus accumbens neurons encodes motivational significance during A discriminative stimulus task. Journal of Neurophysiology. 2004;91:1840–1865. doi: 10.1152/jn.00657.2003. [DOI] [PubMed] [Google Scholar]
- O’Donnell P. Dopamine gating of forebrain neural ensembles. European Journal of Neuroscience. 2003;17:429–435. doi: 10.1046/j.1460-9568.2003.02463.x. [DOI] [PubMed] [Google Scholar]
- O’Donnell P. Ensemble encoding in the nucleus accumbens. Psychobiology. 1999;27:187–197. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; New York: 1998. [DOI] [PubMed] [Google Scholar]
- Pennartz CMA, Groenewegen HJ, Lopes da Silva FH. Nucleus accumbens as a complex of functionally distinct neuron al ensembles: An integration of behavioral, electrophysiological and anatomical data. Progress in Neurobiology. 1994;42:719–761. doi: 10.1016/0301-0082(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Potter PE, Meek JL, Neff NH. Acetylcholine and choline in neuronal tissue measured by HPLC with electrochemical detection. Journal of Neurochemistry. 1983;41:188–194. doi: 10.1111/j.1471-4159.1983.tb13668.x. [DOI] [PubMed] [Google Scholar]
- Roberts C, Cummins R, Gnoffo Z, Kew JN. Dopamine D3 receptor modulation of dopamine efflux in the rat nucleus accumbens. European Journal of Pharmacology. 2006;534:108–114. doi: 10.1016/j.ejphar.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Sarter M, Givens B, Bruno JP. The cognitive neuroscience of sustained attention: Where top-down meets bottom-up. Brain Research Reviews. 2001;35:146–160. doi: 10.1016/s0165-0173(01)00044-3. [DOI] [PubMed] [Google Scholar]
- Sarter M, Nelson CL, Bruno JP. Cortical cholinergic transmission and cortical information processing in schizophrenia. Schizophrenia Bulletin. 2005;31:117–138. doi: 10.1093/schbul/sbi006. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Pickle VM. In the rat medial nucleus accumbens, hippocampal and catecholaminergic terminals converge on spiny neurons and are in oppostition to each other. Brain Research. 1990;527:266–279. doi: 10.1016/0006-8993(90)91146-8. [DOI] [PubMed] [Google Scholar]
- Shilliam CS, Heidbreder CA. Gradient of dopamine responsiveness to dopamine receptor agonists in subregions of the rat nucleus accumbens. European Journal of Pharmacology. 2003;477:113–122. doi: 10.1016/j.ejphar.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDA)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. The Journal of Neuroscience. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Amin F, Lewis BL, O’Donnell P. Altered prefrontal cortical metabolic response to mesocortical activation in adult animals with a neonatal ventral hippocampal lesion. Biological Psychiatry. 2006;60:585–590. doi: 10.1016/j.biopsych.2006.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasey MW, Thayer JF. The continuing problem of false oositives in repeated measures ANOVA in psychophysiology: A multivariate solution. Psychopysiology. 1987;24:479–486. doi: 10.1111/j.1469-8986.1987.tb00324.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Dopamine D2 receptors are present in prefrontal cortical afferents and their targets in patches of the rat caudate-putamen nucleus. Journal of Comparative Neurology. 2002;442:392–404. doi: 10.1002/cne.10086. [DOI] [PubMed] [Google Scholar]
- West AR, Floresco SB, Charara A, Rosenkranz JA, Grace AA. Electrophysiological interactions between striatal glutamatergic and dopaminergic systems. Annals of the New York Academy of Sciences. 2003;1003:53–74. doi: 10.1196/annals.1300.004. [DOI] [PubMed] [Google Scholar]
- Yun IA, Nicola SM, Fields HL. Contrasting effects of dopamine and glutamate receptor antagonist injection in the nucleus accumbens suggest a neural mechanism underlying cue-evoked goal-directed behavior. European Journal of Neuroscience. 2004a;20:249–263. doi: 10.1111/j.1460-9568.2004.03476.x. [DOI] [PubMed] [Google Scholar]
- Yun IA, Wakabayashi KT, Fields HL, Nicola SM. The ventral tegmental area is required for the behavioral and nucleus accumbens neuronal firing responses to incentive cues. The Journal of Neuroscience. 2004b;24:2923–2933. doi: 10.1523/JNEUROSCI.5282-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Gaykema RP, Swanson DJ, Cullinan WE. Cortical input to the basal forebrain. Neuroscience. 1997;79:1051–1078. doi: 10.1016/s0306-4522(97)00049-3. [DOI] [PubMed] [Google Scholar]
- Zmarowski AL, Sarter M, Bruno JP. NMDA and Dopamine Interactions in the Nucleus Accumbens Modulate Cortical Acetylcholine Release. European Journal of Neuroscience. 2005;22:1731–1740. doi: 10.1111/j.1460-9568.2005.04333.x. [DOI] [PubMed] [Google Scholar]